Introduction
Oxidized low-density lipoprotein (ox-LDL) has been
demonstrated to be a key molecule in the initiation and development
of atherosclerotic plaque (1),
which is abundant in atherosclerotic arterial walls and has
numerous detrimental effects on endothelial cell function (2). Recent clinical research has shown
the association between circulating ox-LDL and preclinical
atherosclerosis, coronary and peripheral artery atherosclerosis,
and acute coronary syndromes (ACS) (3). The level of ox-LDL in circulation is
a marker of the severity and course of ACS (4). Moreover, ox-LDL levels are
independently associated with higher risk of progression in lacunar
strokes (5). Recent studies have
identified several mechanisms by which ox-LDL exerts its
atherogenic effects, including activation of endothelial cell
arginase II, which decreases endothelial nitric oxide (NO)
production (6) and upregulation
of inflammatory genes such as tumor necrosis factor (TNF)-α
(7), adhesive factors, monocyte
chemotactic agents (8) and
metalloproteinases (9,10). Through these mechanisms, ox-LDL
facilitates endothelial dysfunction, platelet aggregation and
thrombus formation, leading to augmentation of the inflammatory
reaction and destabilization of the atherosclerotic plaques
(11). Interestingly, low
concentrations of ox-LDL appear to have an opposite effect, as
in vitro studies have shown that ox-LDL at low concentration
can promote angiogenesis and activate nitric oxide synthesis in
human coronary artery endothelial cells (12). These contradictory data show us
that the mechanism of ox-LDL activity on endothelial cells is far
beyond our current understanding.
Krüppel-like factors (KLF) are a subclass of
evolutionarily conserved zinc finger-containing transcription
factors. The expression of KLF2 and KLF4 has been documented in
endothelial cells and the overexpression of these factors induces
expression of multiple anti-inflammatory and anti-thrombotic
factors, including endothelial nitric oxide synthase (NOS) and
thrombomodulin. KLF2 and KLF4 are also novel regulators of
endothelial activation in response to pro-inflammatory stimuli
(13–15); however, no studies have been
performed on the role of other KLF family members in endothelial
function. Because ox-LDL is one of the initial triggers of
atherosclerosis, these studies led us to the question whether
ox-LDL exerts its atherogenic effects by signaling through the KLF
family members. KLF2 and KLF4 showed opposing effects on some
factors compared with ox-LDL, including eNOS and thrombomodulin, so
we hypothesized that ox-LDL might reduce KLF production in
endothelial cells.
Atorvastatin is a synthetic
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor
and is used to treat patients with high cholesterol. In general,
statins regulate endothelial function, inhibit macrophage
activities, modulate several inflammatory mechanisms involved in
the atherosclerotic process and inhibit migration and proliferation
of endothelial smooth muscle cells. Studies have shown that
atorvastatin decreases the amount of circulating ox-LDL (16) and increases the expression of
atheroprotective genes such as KLF2 (17) and its downstream targets, namely,
endothelial nitric oxide synthase (eNOS) and thrombomodulin
(15), so we hypothesized that
atorvastatin may also modulate other KLF family members.
This study aims to investigate the effect of ox-LDL
on KLF expression and the role of atorvastatin in the regulation of
KLF expression with or without ox-LDL induction. To conduct our
experiments, we selected EA.hy926 cells as our model of
macrovascular endothelial cells.
Materials and methods
Materials
The human umbilical vein endothelial cell line
EA.hy926 was purchased from the cell bank of Institute of Cellular
Biology (Chinese Academy of Science, Shanghai, China). Cell culture
materials were from Costar (Corning Incorporation, Corning, NY,
USA). Atorvastatin was purchased from the National Institute for
the Control of Pharmaceutical and Biological Products (China).
Other reagents are indicated in the text.
Cell culture and reagent preparation
The EA.hy926 endothelial cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine
serum (FBS) (Gibco-BRL, Gaithersburg, MD, USA) and 1%
penicillin/streptomycin at 37°C in a humidified atmosphere
containing 95% O2 and 5% CO2. Experiments were performed
with cells grown to a confluency of 80%. EA.hy926 cells at passages
3–5 were used in this study. Atorvastatin was dissolved in dimethyl
sulfoxide (DMSO; Sigma, St. Louis, MO, USA) (stock 10 mM) and added
to the cells at the indicated concentrations for the entire
incubation period. The equivalent amount of DMSO was added to the
control samples. The final concentration of DMSO never exceeded
0.1% and did not affect cell viability (data not shown).
DNA microarray
After EA.hy926 cells were treated with atorvastatin
(10 μM) or control (DMSO) for 24 h, gene expression profiles
were determined with Affymetrix U133A plus 2.0 genechip (CapitalBio
Corporation, Beijing, China), which included 47,000 characterized
human genes. Each group had 3 biological repeats.
Total-RNA was isolated using TRIzol®
(Invitrogen, Inc., Grand Island, NY, USA) and purified using RNeasy
spin columns (Qiagen, Hilden, Germany). Total-RNA (7 μg) was
used to synthesize cDNA by using the Superscript II double-stranded
cDNA synthesis kit (Applied Biosystems, Carlsbad, CA, USA) and the
cDNA was then used for in vitro transcription in the
presence of biotin-labeled ribonucleotides (biotin-11-CTPs und
biotin-16-UTPs) to yield biotin-labeled cRNA. The biotin-labeled
RNA fragments were hybridized to the probe array during 16 h of
incubation, and then the array was stained with
streptavidin-phycoerythrin conjugate and scanned by the
GeneChip® Scanner 3000. Raw data were analyzed using
Affymetrix® GeneChip® Operating Software
Version 1.4.
Semi-quantitative real-time polymerase
chain reaction (PCR)
RNA was extracted using TRIzol and quantified by
measuring the absorbance at 260 nm. Reverse transcription was
performed using the One Step RT-PCR kit (Promega Corporation,
Madison, WI, USA), according to the manufacturer’s instructions.
cDNA samples (2 μl) were amplified in 20 μl of 1X
SYBR-Green PCR master mix (Applied Biosystems) and real-time PCR
was performed on duplicate samples by using the Applied Biosystems
ABI PRISM® 7300 Real-Time PCR System with the following
cycling parameters: initial denaturation at 95°C for 10 min,
followed by 40 cycles of denaturation at 95°C for 15 sec and
annealing/extension at 61°C for 31 sec. Data were normalized to
human GAPDH mRNA levels as an endogenous control and were expressed
relative to the DMSO-treated control using the 2-ΔΔCt method. The
primers were designed using Primer Express® Primer
Design Software V3.0 (Applied Biosystems) (Table I).
 | Table IPrimers for real-time quantitative
RT-PCR. |
Table I
Primers for real-time quantitative
RT-PCR.
| Gene name | Primer
sequence | Amplicon size
(bp) |
|---|
| GAPDH | F:
TGCGCAGAAAACAAGATGAG
R: CACCTTCACCGTTCCAGTTT | 114 |
| KLF2 | F:
GCAAACGCACCGCCACTCACACCT
R: CTTCCAGCCGCAGCCGTCCCAGTT | 140 |
| KLF3 | F:
GTGATTATGATGGATGCAACAAA
R: TTCATCAGACCGAGCAAACTT | 134 |
| KLF4 | F:
GCGGGCTGCGGCAAAACCTACAC
R: CATCCACAGCCGTCCCAGTCACAG | 104 |
| KLF6 | F:
AGCTCCTCTGTCACCTCCAC
R: CAGCTCCCCGGGCACGCAA | 87 |
| KLF7 | F:
GTTTTGCACGAAGCGATGAG
R: ATGTGGAGGGCAAGATGGTC | 118 |
| KLF9 | F:
GAAACACGCCTCCGAAAAG
R: TCACCTGTATGCACTCTGTAATGG | 105 |
| KLF13 | F:
TCGGGAGAATACAGCTCCGATTTCT
R: TGTCCATAAAGGTACTGAAGCTG | 112 |
Western blot analysis
Following treatment, cells were washed with ice-cold
phosphate-buffered saline (PBS) and lysed with NucBuster™ protein
extraction kit (Calbiochem Merck, Co., Darmstadt, Germany). Cell
lysates were separated on SDS-PAGE and transferred to Whatman
nitrocellulose membranes. The membranes were blocked with
Blotto-Tween (5% nonfat milk and 0.05% Tween-20 in PBS) and
incubated with primary antibodies against KLF2, KLF3, KLF4, KLF6
and KLF7 for 2 h at room temperature. After washing, the membranes
were incubated with horseradish peroxidase-conjugated secondary
antibodies. The bands were detected by chemiluminesence detection
agents. The blots were subjected to densitometry and the bands were
analyzed by Gene Genius bio imaging system (Syngene, Frederick, MD,
USA).
Immunofluorescence and confocal
microscopy
To explore the relationship between ox-LDL and the
KLFs, we used immunofluorescence with specific primary antibodies
against the members of the KLF family. EA.hy926 cells were plated
onto glass coverslips, which were incubated in DMEM in the presence
of DMSO control (A), 10 μM atorvastatin (B), 100
μg/ml ox-LDL (C) or 10 μM atorvastatin + 100
μg/ml ox-LDL (D) for 24 h. After incubation, the cells were
fixed with 4% formaldehyde for 15 min, stained with anti-KLF
antibody [goat KLF2, goat KLF3 and rabbit KLF6 antibodies (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA), 1/500; mouse KLF4
antibody (ProMab Biotechnologies, Inc., Richmond, CA, USA), 1:800;
rabbit KLF7 antibody (Proteintech Group, Inc., Chicago, IL, USA),
1:400] at 4°C overnight, and then incubated for 30 min with
fluorescein-conjugated secondary antibodies (rabbit anti-goat
IgG/HRP, 1:40,000; goat anti-mouse IgG/HRP,1:80,000 and goat
anti-rabbit IgG/HRP, 1:40,000). Finally, immunostaining was
performed, visualized and photographed using a Leica TCS-SP
confocal laser-scanning microscope (11,12).
Statistical analysis
All experiments were performed in duplicate or
triplicate with at least 2 biological replicates. All data are
presented as mean ± standard deviation (SD). Comparisons among
groups were performed by one-way ANOVA followed by a posteriori
Tukey’s test. Differences were accepted as statistically
significant when P<0.05.
Results
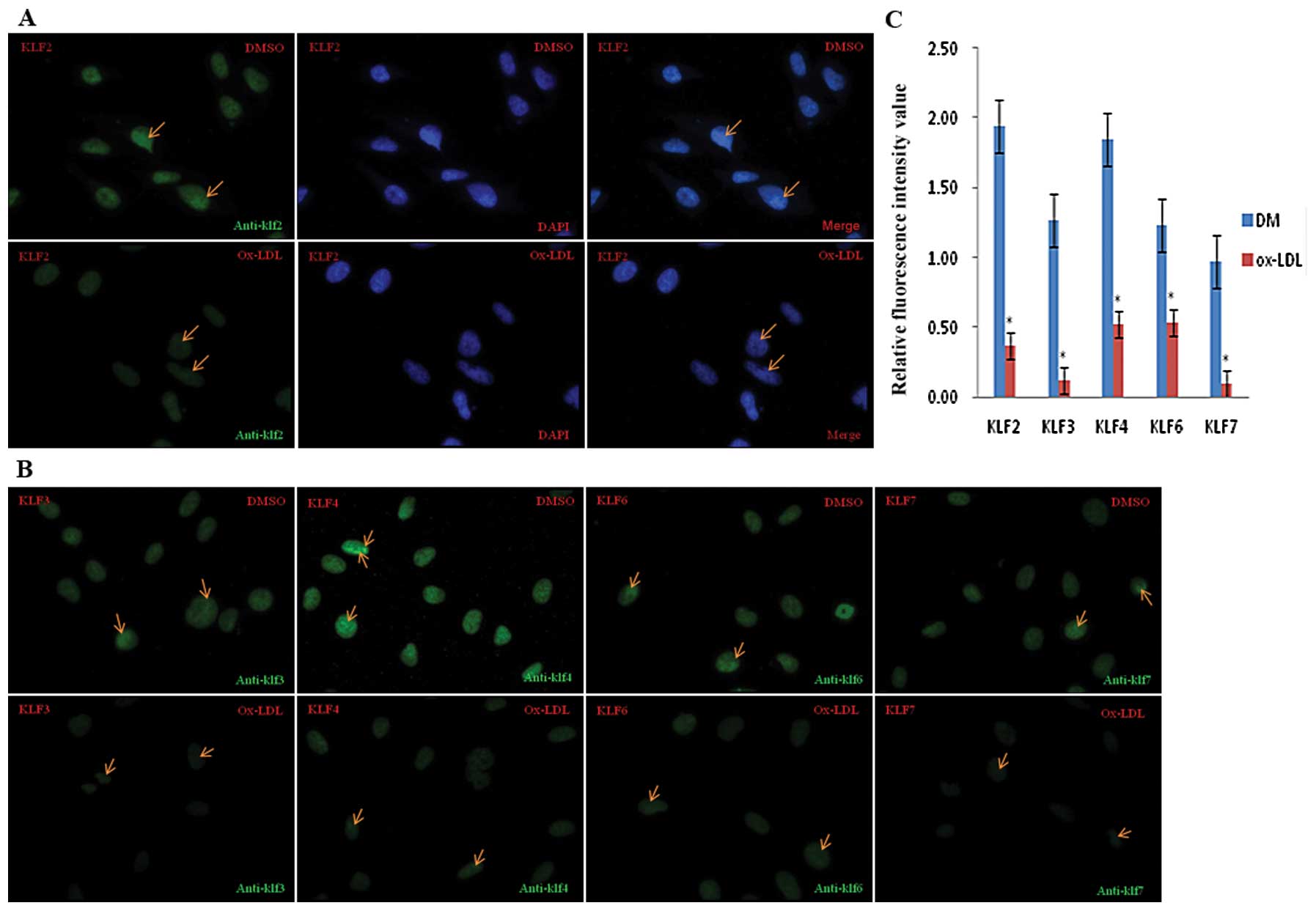
Ox-LDL downregulates KLF expression in
EA.hy926 cells
To investigate the effect of ox-LDL on KLF
expression, we used immunofluorescence and confocal microscopy to
assess KLF protein levels in EA.hy926 cells treated with ox-LDL. As
shown in Fig. 1A and B, KLF2,
KLF3, KLF4, KLF6 and KLF7 (green) were all expressed in the nuclei,
which are counter-stained with DAPI. KLF production was moderate
without ox-LDL stimulation; however, incubation with 100
μg/ml ox-LDL for 24 h led to a significant decrease in KLF
expression. The fluorescence intensity values of KLF2, KLF3, KLF4,
KLF6 and KLF7 gene expression before and after ox-LDL treatment
were quantitatively analyzed and the results indicate a >2-fold
decrease in KLF expression after ox-LDL treatment (Fig. 1C).
Atorvastatin upregulates KLF expression
in quiescent EA.hy926 cells
DNA microarray analysis reveals that
atorvastatin elevates the expression of KLF family members
To discover the molecular mechanism for the
pleiotropic effects of atorvastatin, we conducted a cell-based
microarray analysis to analyze the genome-wide transcriptional
changes occurring in EA.hy926 cells after exposure to 10 μM
atorvastatin for 24 h. Atorvastatin increased the expression of all
KLF genes by >2-fold compared to DMSO. KLF4 showed the maximum
change, with >4-fold upregulation (Table II).
| Table IIKLF expression in EA.hy926 cells
after 24-h atorvastatin treatment. |
Table II
KLF expression in EA.hy926 cells
after 24-h atorvastatin treatment.
| | Average gray values
in 3 biological repeats
| Fold change
(ranking)
|
|---|
| Accession
number | Gene name | DMSO | ATa | AT/DMSO |
|---|
| NM_016270 | Kruppel-like factor
2 (lung) (KLF2) | 4122.30 | 1504.64 | 2.74 (72) |
| NM_016531 | Kruppel-like factor
3 (basic) (KLF3) | 351.96 | 841.05 | 2.38 (131) |
| NM_004235 | Kruppel-like factor
4 (gut) (KLF4) | 101.20 | 433.28 | 4.27 (14) |
| NM_001160124 | Kruppel-like factor
6 (KLF6) | 960.31 | 2044.95 | 2.13 (225) |
| NM_003709 | Kruppel-like factor
7 (KLF7) | 211.80 | 451.06 | 2.14 (215) |
| NM_001206 | Kruppel-like factor
9 (KLF9) | 49.81 | 104.61 | 2.03 (276) |
| NM_015995 | Kruppel-like factor
13 (KLF13) | 205.64 | 460.73 | 2.25 (170) |
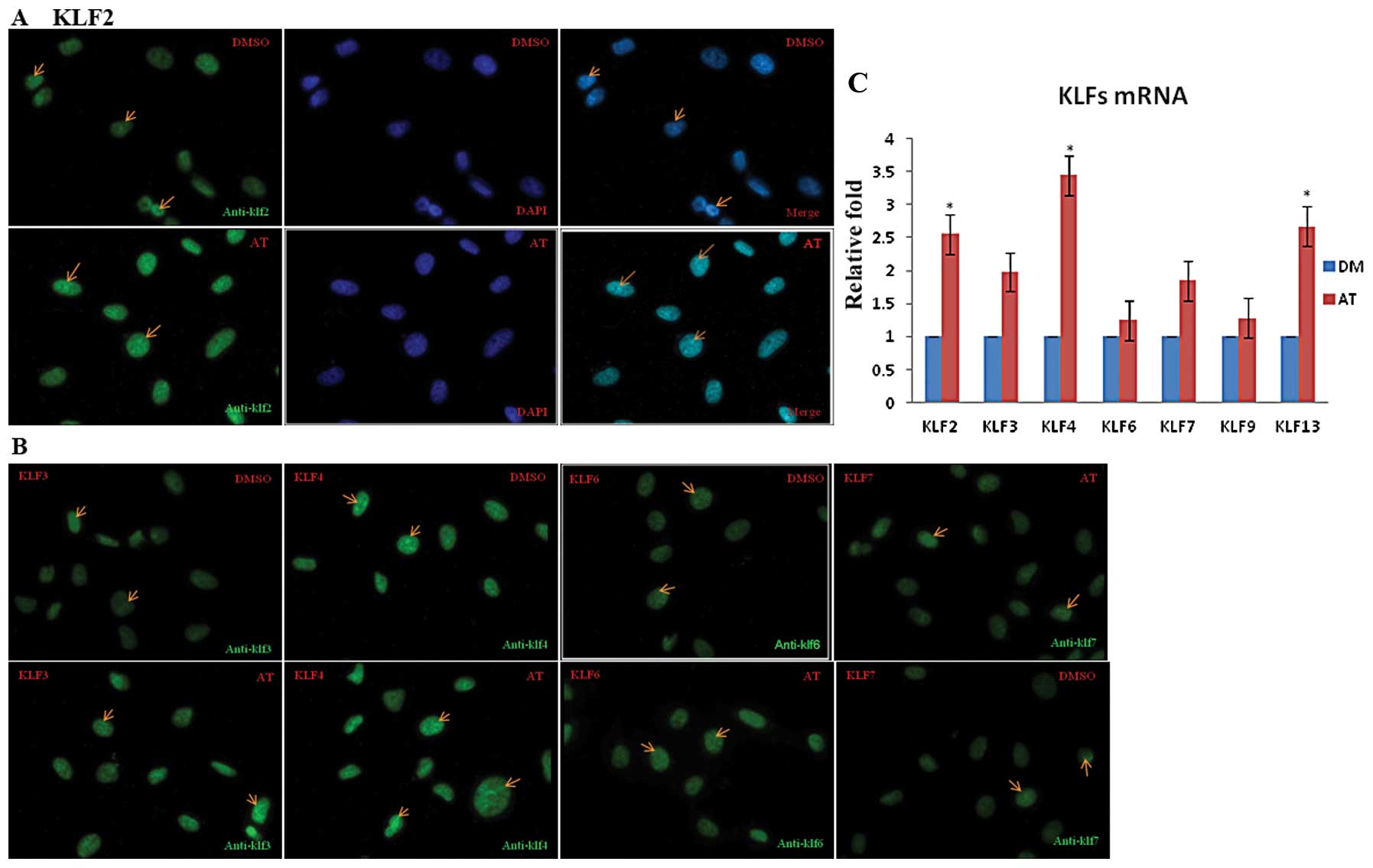
Real-time PCR analysis validates the
effect of atorvastatin on KLF gene expression
To validate the effect of atorvastatin on KLF gene
expression, we used real-time PCR to measure mRNA levels in the
EA.hy926 cells treated with atorvastatin for 24 h. Under control
(DMSO) conditions, KLF mRNA levels were low. Stimulation of
EA.hy926 cells with 0.1–10 μM atorvastatin led to a
concentration- and time-dependent increase in KLF mRNA expression
that peaked 24 h after treatment with 10 μM atorvastatin
(data not shown). After 24 h, the levels of KLF2, KLF4 and KLF13
mRNA were significantly higher in these cells than in the control
cells (P<0.05) (as shown in Fig.
3C)
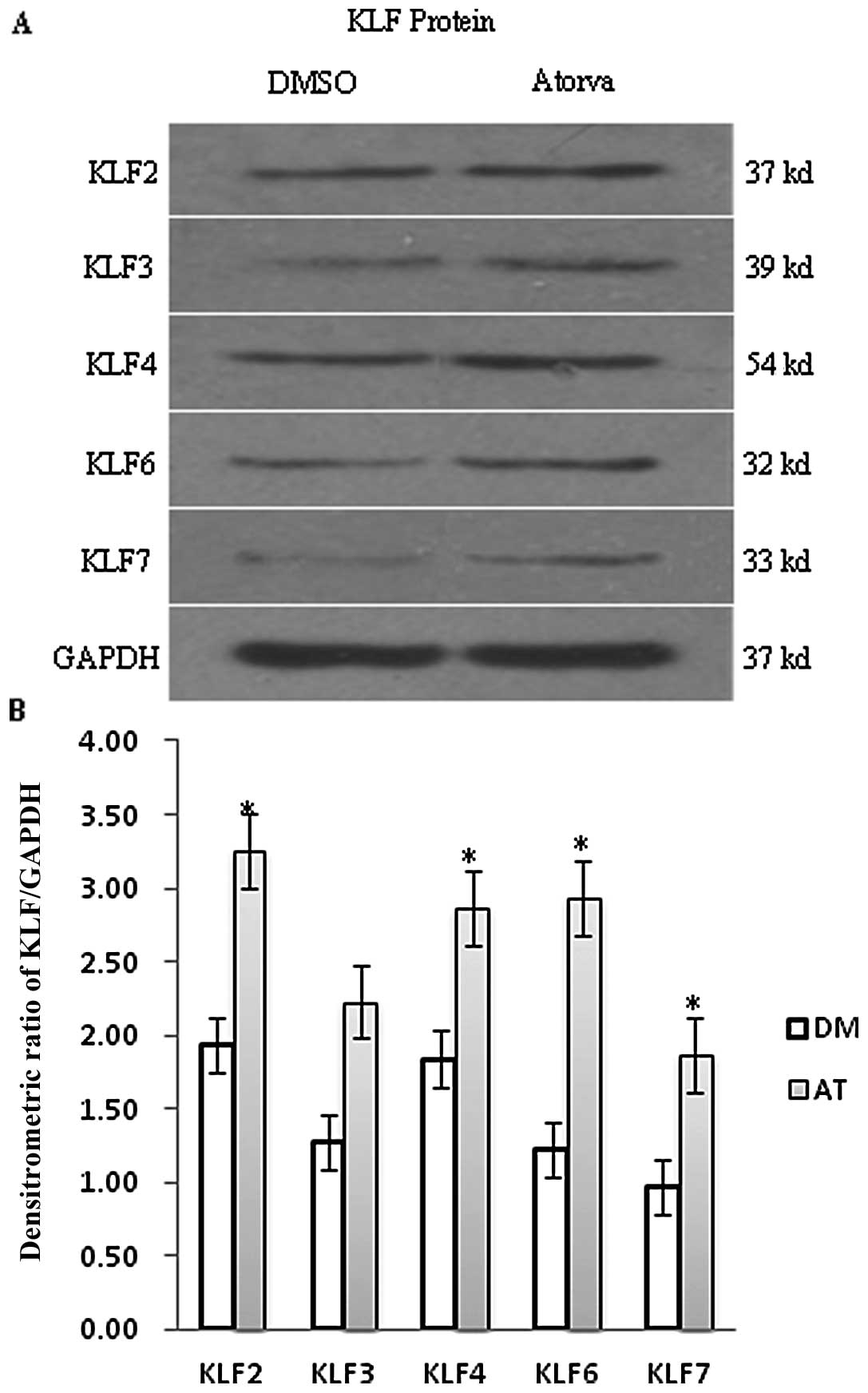
Atorvastatin promotes the expression
of KLF protein in quiescent EA.hy926 cells
To confirm whether the change in KLF protein levels
was consistent with PCR and DNA microarray results, KLF protein was
analyzed and quantified by immunofluorescence and western blot
analyses. As shown in Fig. 2, the
expression of KLF2 and KLF4 protein in quiescent EA.hy924 cells was
significantly higher than that of KLF3, KLF6 and KLF7. After
incubation with 10 μM atorvastatin for 24 h, KLF2, KLF3,
KFL4, KLF6 and KLF7 were all increased compared with the DMSO
control, but protein levels were not altered to the same degree as
the mRNA levels. The increase in KLF2 protein expression was
<2-fold over that of the control and we found the same result by
immunofluorescence analysis. Green fluorescence intensity values
based on the binding intensity of the KLF antibodies were enhanced
in the atorvastatin group compared to the DMSO group, but the
change was <2-fold (Fig. 3A and
B).
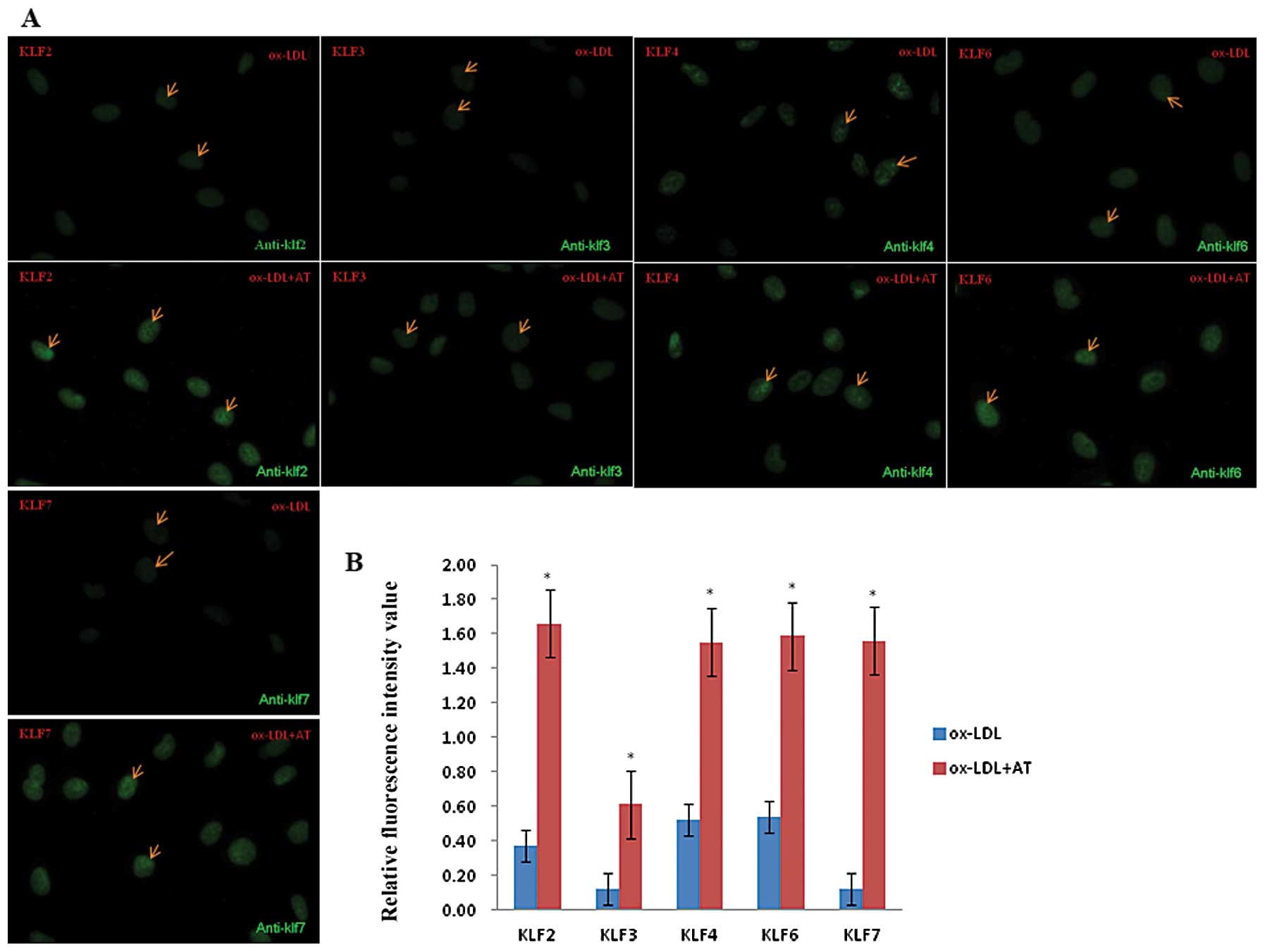
Atorvastatin counteracts
ox-LDL-induced downregulation of KLF expression in EA.hy926
cells
As mentioned above, ox-LDL sharply decreased the
levels of KLF protein, while atorvastatin upregulated the KLF
expression at both the mRNA and protein levels. We further examined
the effects of atorvastatin on ox-LDL-induced KLF expression by
using immunofluorescence. In the nuclei of these cells, we observed
a >2-fold decrease in KLF protein in the 100 μg/ml ox-LDL
group compared to the DMSO control group (Fig. 1A and B), while the fluorescence
intensity values of KLF2, KLF3, KLF4, KLF6 and KLF7 in ox-LDL +
atorvastatin group were all elevated >3-fold over the ox-LDL
alone group. These results demonstrate that atorvastatin
counteracts the inhibitory effect induced by ox-LDL on KLF
expression (Fig. 4).
Discussion
KLFs are members of the zinc finger family of
transcription factors and previous studies have implicated these
transcription factors as the key regulators of the endothelial
pathways in vascular biology (18). KLF2 is the most widely studied KLF
in endothelial cells and it is known as a ‘molecular switch’ that
regulates the important aspects of vascular function and disease.
KLF2 regulates endothelial thrombotic function by inducing the
expression of potent anti-thrombotic and anti-inflammatory genes
such as eNOS, thrombomodulin and plasminogen activator inhibitor 1
(PAI-1). These results were further substantiated by siRNA
knockdown experiments (8). In
addition, KLF2 blocks endothelial cell activation by inhibiting
interleukin (IL)-1β (19) and
TNF-α (20), and inhibits
angiogenesis by reducing endothelial cell proliferation and
migration (21).
Both KLF4 and KLF10 are novel regulators of the
inflammatory response (22,23). KLF10 plays a central role in the
anti-proliferative response via the TGFβ-Smad pathway, which
explains why KLF10 knockout mice develop cardiac hypertrophy
(24). KLF4, which acts as a
novel regulator of macrophage polarization, was robustly induced in
M2 macrophages, whereas it was strongly reduced in M1 macrophages
(25). KLF6 plays a key role in
vascular development, remodeling and response to injury (12). KLF7 was recently reported to be a
novel candidate for conferring susceptibility to type 2 diabetes
(26). However, the underlying
mechanisms responsible for endothelial cell injury with decreased
KLF expression, remain to be elucidated.
In atherosclerosis, ox-LDL accumulates in the vessel
walls and causes endothelial dysfunction, leading to the initiation
and progression of atherosclerosis. However, the association
between ox-LDL accumulation and KLF expression, as well as the
effect of atorvastatin on ox-LDL-induced KLF expression, has not
been determined. Therefore, we examined the effect of atorvastatin
on ox-LDL-induced regulation of endothelial cell KLF
expression.
Several studies have demonstrated that KLF2 is a
novel nuclear mediator of the effects of statin in endothelial
cells (27–30). In the present study, we found that
ox-LDL can significantly decrease the mRNA expression of KLF2,
KLF3, KLF4, KLF6, KLF7, KLF9 and KLF13. We further demonstrated
that the downregulation of KLF expression by ox-LDL was
counteracted by treatment with atorvastatin. These findings suggest
that other KLFs, in addition to KLF2 and KLF4, play a role in the
pathogenesis of atherosclerosis (13,31). Downregulation of KLF expression by
ox-LDL might be an effective target of atorvastatin in
atherosclerosis. The signaling mechanisms orchestrating endothelial
KLF2 expression as well as the expression of other KLFs in the
vascular endothelium deserve further investigation.
Lectin-like oxidized low-density lipoprotein
receptor-1 (LOX-1) is a major receptor for ox-LDL in the
endothelial cells and is important for tumor growth, suggesting a
molecular connection between atherogenesis and tumorigenesis
(32–34). Recent studies show a positive
correlation between increased serum ox-LDL levels and an increased
risk of colon, breast, ovarian and esophageal cancers (35–37). Studies have revealed that KLF4,
KLF9 and KLF10 have important tumor suppressor functions (23,26,38–40). Based on our results that ox-LDL
downregulates KLF expression, which would increase the risk of
cancer, we hypothesized that KLFs were the targets of the
pro-carcinogenic effect of ox-LDL and the anti-carcinogenic effect
of atorvastatin.
Maintenance of the cellular participants of
inflammatory reactions in a quiescent or inflammatory state is
equally important. Therefore, we investigated these two states.
Atorvastatin upregulated KLF expression in both the quiescent and
ox-LDL-induced inflammatory states. This suggests that KLFs are
involved in the control of cell proliferation and differentiation
in normal as well as in pathological situations.
Although the mechanism of KLF reduction by ox-LDL is
not well understood in EA.hy926 endothelial cells, our observations
suggest that KLFs regulate the endothelial phenotype under both
basal and inflammatory conditions. The evidence suggests that KLFs
play an important role in endothelial dysfunction. Moreover,
statin-induced upregulation of KLF expression may play an important
role in the pharmacological and clinical effects of statins
observed in cardiovascular diseases and carcinoma. Downregulation
of KLFs in patient plasma might occur during the early stages of
cardiovascular disease and cancer, and this may be correlated with
disease severity, because ox-LDL levels are related to both
atherosclerosis and an increased risk of cancer (33). Further clinical research is
required to provide more information on the potential benefits of
using statins to treat atherosclerotic diseases at an earlier
stage.
Acknowledgements
This study was supported by the
National Science and Technology Support Program (no.
2009BAI86B04).
References
|
1.
|
RA BoonJO FledderusOL VolgerKLF2
suppresses TGF-beta signaling in endothelium through induction of
Smad7 and inhibition of AP-1Arterioscler Thromb Vasc
Biol27532539200710.1161/01.ATV.0000256466.65450.ce17194892
|
|
2.
|
S MitraT GoyalJL MehtaOxidized LDL, LOX-1
and atherosclerosisCardiovasc Drugs
Ther25419429201110.1007/s10557-011-6341-521947818
|
|
3.
|
S EharaM UedaT NarukoElevated levels of
oxidized low density lipoprotein show a positive relationship with
the severity of acute coronary
syndromesCirculation10319551960200110.1161/01.CIR.103.15.195511306523
|
|
4.
|
S TsimikasC BergmarkRW BeyerTemporal
increases in plasma markers of oxidized low-density lipoprotein
strongly reflect the presence of acute coronary syndromesJ Am Coll
Cardiol41360370200310.1016/S0735-1097(02)02769-912575961
|
|
5.
|
E Cuadrado-GodiaA OisE
Garcia-RamalloBiomarkers to predict clinical progression in small
vessel disease strokes: prognostic role of albuminuria and oxidized
LDL
cholesterolAtherosclerosis219368372201110.1016/j.atherosclerosis.2011.07.11421862014
|
|
6.
|
S RyooA BhuniaF ChangA ShoukasDE
BerkowitzLH RomerOxLDL-dependent activation of arginase II is
dependent on the LOX-1 receptor and downstream RhoA
signalingAtherosclerosis214279287201110.1016/j.atherosclerosis.2010.10.04421130456
|
|
7.
|
D MohtyP PibarotJP DespresAssociation
between plasma LDL particle size, valvular accumulation of oxidized
LDL, and inflammation in patients with aortic stenosisArterioscler
Thromb Vasc Biol28187193200810.1161/ATVBAHA.107.15498917975118
|
|
8.
|
GT OhJH ChoiJJ HongDietary hematein
ameliorates fatty streak lesions in the rabbit by the possible
mechanism of reducing VCAM-1 and MCP-1
expressionAtherosclerosis1591726200110.1016/S0021-9150(01)00464-611689202
|
|
9.
|
B ZhaoX LuoH ShiD MaTissue factor pathway
inhibitor-2 is downregulated by ox-LDL and inhibits ox-LDL induced
vascular smooth muscle cells proliferation and migrationThromb
Res128179185201110.1016/j.thromres.2011.02.02521458846
|
|
10.
|
HH WangHL HsiehCY WuCM YangOxidized
low-density lipoprotein-induced matrix metalloproteinase-9
expression via PKC-delta/p42/p44 MAPK/Elk-1 cascade in brain
astrocytesNeurotox
Res175065201010.1007/s12640-009-9077-219554388
|
|
11.
|
BL YuSP ZhaoXS HuangOxidized low-density
lipoprotein: a double-edged sword on atherosclerosisMed
Hypotheses69553556200710.1016/j.mehy.2007.01.04317368957
|
|
12.
|
S YuSL WongCW LauY HuangCM YuOxidized LDL
at low concentration promotes in-vitro angiogenesis and activates
nitric oxide synthase through PI3K/Akt/eNOS pathway in human
coronary artery endothelial cellsBiochem Biophys Res
Commun4074448201110.1016/j.bbrc.2011.02.096
|
|
13.
|
FF YanYF LiuY LiuYX ZhaoKLF4: a novel
target for the treatment of atherosclerosisMed
Hypotheses70845847200810.1016/j.mehy.2007.07.03117869009
|
|
14.
|
G Villarreal JrY ZhangHB LarmanJ
Gracia-SanchoA KooG Garcia-CardenaDefining the regulation of KLF4
expression and its downstream transcriptional targets in vascular
endothelial cellsBiochem Biophys Res
Commun391984989201010.1016/j.bbrc.2009.12.00219968965
|
|
15.
|
Z LinA KumarS SenBanerjeeKruppel-like
factor 2 (KLF2) regulates endothelial thrombotic functionCirc
Res96e48e57200510.1161/01.RES.0000159707.05637.a115718498
|
|
16.
|
RR AzarG BadaouiA SarkisEffect of
ezetimibe/atorvastatin combination on oxidized low density
lipoprotein cholesterol in patients with coronary artery disease or
coronary artery disease equivalentAm J
Cardiol106193197201010.1016/j.amjcard.2010.03.016
|
|
17.
|
F AliSS HamdulayAR
KinderlererStatin-mediated cytoprotection of human vascular
endothelial cells: a role for Kruppel-like factor 2-dependent
induction of heme oxygenase-1J Thromb
Haemost525372546200710.1111/j.1538-7836.2007.02787.x17927807
|
|
18.
|
H LiSB MiaoLH DongClinicopathological
correlation of Kruppel-like factor 5 and matrix metalloproteinase-9
expression and cartilage degeneration in human osteoarthritisPathol
Res Pract208914201110.1016/j.prp.2011.09.01522094285
|
|
19.
|
AC RaccaSA CamolottoME RidanoJL BoccoS
Genti-RaimondiGM Panzetta-DutariKruppel-like factor 6 expression
changes during trophoblast syncytialization and transactivates
sshCG and PSG placental genesPLoS
One6e22438201110.1371/journal.pone.002243821799854
|
|
20.
|
AB BialkowskaM CrispT
BannisterIdentification of small-molecule inhibitors of the
colorectal cancer oncogene Kruppel-like factor 5 expression by
ultrahigh-throughput screeningMol Cancer
Ther1020432051201110.1158/1535-7163.MCT-11-055021885866
|
|
21.
|
HJ KeeJS KwonS ShinY AhnMH JeongH
KookTrichostatin A prevents neointimal hyperplasia via activation
of Kruppel like factor 4Vascul
Pharmacol55127134201110.1016/j.vph.2011.07.00121763782
|
|
22.
|
DT DangX ChenJ FengM TorbensonLH DangVW
YangOverexpression of Kruppel-like factor 4 in the human colon
cancer cell line RKO leads to reduced
tumorigenecityOncogene2234243430200310.1038/sj.onc.120641312776194
|
|
23.
|
A MihailovaH MikazaneJ KlovinsL
Nikitina-ZakeAssociation of protein tyrosine phosphatase
non-receptor 22 (PTPN22) rs2476601 and Kruppel-like factor 12
(KLF12) rs1324913 single nucleotide polymorphisms with rheumatoid
arthritis in a Latvian populationScand J
Rheumatol40491492201110.3109/03009742.2011.608715
|
|
24.
|
S QinM LiuW NiuCL ZhangDysregulation of
Kruppel-like factor 4 during brain development leads to
hydrocephalus in miceProc Natl Acad Sci
USA1082111721121201110.1073/pnas.111235110922160720
|
|
25.
|
X LiaoN SharmaF KapadiaKruppel-like factor
4 regulates macrophage polarizationJ Clin
Invest12127362749201110.1172/JCI4544421670502
|
|
26.
|
Y KawamuraY TanakaR KawamoriS
MaedaOverexpression of Kruppel-like factor 7 regulates
adipocytokine gene expressions in human adipocytes and inhibits
glucose-induced insulin secretion in pancreatic beta-cell lineMol
Endocrinol20844856200610.1210/me.2005-013816339272
|
|
27.
|
BB McConnellSS KimK YuKruppel-like factor
5 is important for maintenance of crypt architecture and barrier
function in mouse
intestineGastroenterology14113021313201110.1053/j.gastro.2011.06.08621763241
|
|
28.
|
JL YoriDD SeachristE JohnsonKruppel-like
factor 4 inhibits tumorigenic progression and metastasis in a mouse
model of breast cancerNeoplasia13601610201121750654
|
|
29.
|
LK ChangPH HuangWT ShenSH YangWJ LiuCF
LoRole of Penaeus monodon Kruppel-like factor (PmKLF) in infection
by white spot syndrome virusDev Comp
Immunol36121129201210.1016/j.dci.2011.06.00821740926
|
|
30.
|
KM ParmarV NambudiriG DaiHB LarmanMA
Gimbrone JrG Garcia-CardenaStatins exert endothelial
atheroprotective effects via the KLF2 transcription factorJ Biol
Chem2802671426719200510.1074/jbc.C50014420015878865
|
|
31.
|
S SenBanerjeeZ LinGB AtkinsKLF2 Is a novel
transcriptional regulator of endothelial proinflammatory
activationJ Exp Med19913051315200410.1084/jem.2003113215136591
|
|
32.
|
J LuS MitraX WangM KhaidakovJL
MehtaOxidative stress and lectin-like ox-LDL-receptor LOX-1 in
atherogenesis and tumorigenesisAntioxid Redox
Signal1523012333201110.1089/ars.2010.379221338316
|
|
33.
|
M CaiazzoL Colucci-D’AmatoF
VolpicelliKruppel-like factor 7 is required for olfactory bulb
dopaminergic neuron developmentExp Cell
Res317464473201110.1016/j.yexcr.2010.11.00621093432
|
|
34.
|
L LebsonA GockeJ RosenzweigCutting edge:
The transcription factor Kruppel-like factor 4 regulates the
differentiation of Th17 cells independently of RORgammatJ
Immunol18571617164201010.4049/jimmunol.100275021076063
|
|
35.
|
D SivritasMU BecherT
EbrahimianAntiproliferative effect of estrogen in vascular smooth
muscle cells is mediated by Kruppel-like factor-4 and manganese
superoxide dismutaseBasic Res
Cardiol106563575201110.1007/s00395-011-0174-z21484412
|
|
36.
|
H LuX WangT LiIdentification of poly
(ADP-ribose) polymerase-1 (PARP-1) as a novel Kruppel-like factor
8-interacting and -regulating proteinJ Biol
Chem2862033520344201110.1074/jbc.M110.21563221518760
|
|
37.
|
JS MoonHE KimE KohKruppel-like factor 4
(KLF4) activates the transcription of the gene for the platelet
isoform of phosphofructokinase (PFKP) in breast cancerJ Biol
Chem2862380823816201110.1074/jbc.M111.23673721586797
|
|
38.
|
H GuanL XieF LeithauserKLF4 is a tumor
suppressor in B-cell non-Hodgkin lymphoma and in classic Hodgkin
lymphomaBlood11614691478201010.1182/blood-2009-12-25644620519630
|
|
39.
|
Y YangBG GoldsteinHH ChaoJP KatzKLF4 and
KLF5 regulate proliferation, apoptosis and invasion in esophageal
cancer cellsCancer Biol
Ther412161221200510.4161/cbt.4.11.209016357509
|
|
40.
|
C BureauN HanounJ TorrisaniJP VinelL
BuscailP CordelierExpression and function of Kruppel like-factors
(KLF) in carcinogenesisCurr
Genomics10353360200910.2174/13892020978892101020119532
|