Introduction
Neurofibromatosis type 1 (NF1) is one of the most
commonly inherited autosomal dominant human genetic disorder with
an incidence of approximately 1 in 3,000–3,500 individuals
worldwide (1–3). NF1 has been reported to be caused by
de novo mutations in approximately 30–50% of patients
(2,3). NF1 is characterized by extremely
variable phenotypic features including multiple café-au-lait (CAL)
spots, multiple cutaneous neurofibromas, and soft-tissue tumors and
has a very poor prognosis (2,3).
NF1 is caused by loss-of-function mutations in the NF1 gene
encoding neurofibromin, a GTPase-activating protein (GAP) which is
one of the major GAP proteins that regulates the small GTPase Ras
signaling pathway by converting the active GTP-Ras to an inactive
GDP-Ras (4).
Malignant peripheral nerve sheath tumor (MPNST),
also referred to as malignant schwannoma or neurofibrosarcoma, is
the most frequent malignant neoplasm associated with NF1. MPNST
represents a major cause of mortality in patients with NF1 because
of its particularly aggressive course (5,6).
NF1 is notable for the malignant transformation of benign tumor
tissues to MPNSTs. Approximately one half of NF1 patients have
benign plexiform neurofibromas (PNs) (2,5).
Malignant transformation of benign neurofibromas and PNs to MPNSTs
was observed in 2–5% of these patients (2). The lifetime risk for developing
MPNSTs in patients with NF1 has been estimated at 8–13% (7).
Although the surgical approach is the mainstay of
treatment for NF1, because of very low 5- and 10-year survival
rates particularly in male patients with severely malignant
NF1-associated MPNSTs characterized by invasive growth, higher
propensity to metastasize and limited sensitivity to radiation
(7,8), alternative therapeutic approaches
have been developed. Recently, there have been several chemotherapy
studies on thalidomide (9),
interferon α (10), pirfenidone
(11), and farnesyltransferase
inhibitors (FTI) R11577 (12) for
PNs. Phase II clinical trial of pirfenidone in patients with NF1
were expected to but did not achieve good results (11). A phase I clinical trial of R11577
in patients with NF1-related PNs revealed a limited efficacy of
this drug in NF1 (12). In
addition, as a clinical therapy for NF1-associated MPNSTs, a
combination of chemotherapeutic agents, carboplatin/etoposide
(13), cisplatin/adriamycin
(14), and ifosfamide/doxorubicin
(15,16), have been tested in patients with
NF1-associated MPNSTs, after surgical resection and radiation
treatments. Long-term investigations using a multimodel therapeutic
strategy demonstrated that patients with NF1-associated MPNSTs
showed a significantly lower response rate to chemotherapy compared
to patients with MPNSTs not associated with NF1 (17), indicating that there are still
many hurdles to overcome in chemotherapy for the NF1-associated
MPNSTs.
The limited successful results of these clinical
studies have led to the discovery of new drugs that mainly target
the proteins involved in the Ras-signaling pathway (18). In a recent study, preclinical
in vivo evaluation of rapamycin (Sirolimus) or its
derivative RAD001 (Everolimus) demonstrated the inhibitory effect
of rapamycin on MPNSTs in a xenograft mouse model (19). In addition, B-Raf inhibitor,
sorafenib, EGFR inhibitor, erlotinib and R11577/lovastatin mediated
the inhibition of cell proliferation in the MPNST cells (20–22).
In this study, we aimed to find new target molecules
and/or drugs in order to improve chemotherapy approaches effective
in the treatment of the NF1-associated MPNSTs. By comparison
analysis between the benign neurofibroma cell line and MPNST cell
line and primary normal cells and MPNST cells that bear an
identical germ line mutation derived from an NF1 patient, we found
overexpression of anti-apoptotic Bcl-xL protein in the MPNST cells,
which is responsible for the anticancer drug resistance of the
NF1-associated MPNST cells. This finding presented an opportunity
to develop new strategies for targeted chemotherapy in the NF1
patients with MPNSTs.
Materials and methods
Drugs
Doxorubicin, cisplatin and etoposide were purchased
from Sigma-Aldrich Co. ABT-737 was purchased from Santa Cruz
Biotechnology, Inc.
Cell lines
The neurofibromin-deficient MPNST cell line,
sNF02.2, and benign the neurofibroma cell line, Hs 53.T, were
purchased from the American Type Culture Collection and grown in
DMEM media (Hyclone Laboratories) supplemented with 10% FBS
(Hyclone Laboratories), penicillin (100 U/ml), and streptomycin
(100 μg/ml). All cultured cells were incubated at 37°C in a
humidified atmosphere containing 5% CO2.
Patient and tissue samples
Normal tissue and tumor tissue specimens were
obtained by skin biopsy and surgical resection, respectively, from
a 24-year-old male patient with NF1 (23,24). The patient having a NF1
nonsense mutation Y2264X (c.6792C>G) in the NF1 gene
presented the clinical features of NF1 with CAL spots, scoliosis,
cutaneous neurofibromas, subcutaneous neurofibromas, PNs and MPNSTs
(23,24). After surgical resection at 24
years of age, the patient died at the age of 25. Three types of
tissues, normal phenotypic tissues, PNs and MPNSTs, were
pathologically evaluated by routine light microscopy after staining
with hematoxylin and eosin (H&E) as previously described
(24). The study was approved by
the Institutional Review Board Committee of the Ajou University
School of Medicine.
Primary tissue culture
Primary tissue culture was performed by primary
explant technique. The dissected tissues were finely chopped,
rinsed with PBS, and the pieces were seeded onto the surface of a
tissue culture flask in 1 ml of DMEM supplemented with a high
concentration (40%) of FBS. After an overnight incubation at 37°C,
the medium volume was made up to 5 ml and then changed weekly until
a substantial outgrowth of cells was observed. Cells were then
grown in DMEM media supplemented with 15% FBS. Cells were used from
passages 5 through 10. The primary cells from the three types of
tissues, normal phenotypic tissues, PNs and MPNSTs, demonstrated
their distinct cellular characteristics by western blotting with
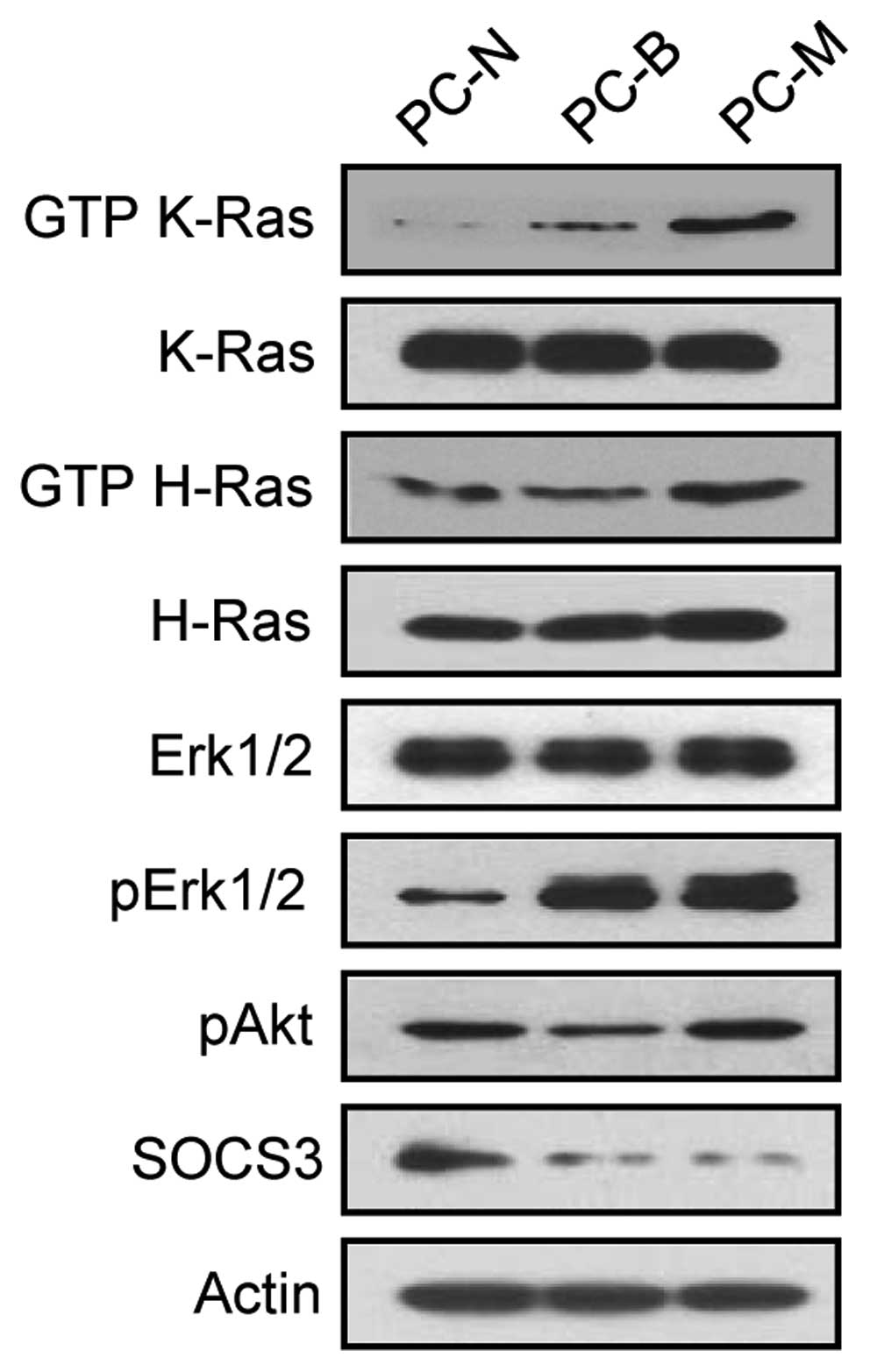
antibodies against GTP-Ras and its downstream effectors (Fig. 1).
Cell viability assay
The cell viability assay was performed by using the
EZ-Cytox Cell Viability Assay kit (Daeil Lab Service, Korea). The
cultured cells were plated at a density of 4×103 in a
96-well flat-bottom tissue-culture plate, incubated overnight, and
were treated with the indicated concentrations of drugs. After 24 h
incubation, 10 μl of Ez-Cytox reagent was added, and the cells were
incubated for another 2 h and then absorbance was measured at a
wavelength of 450 nm with an ELISA microplate reader (Model 680;
Bio-Rad).
Gene silencing
The target sequences for the small interfering RNAs
(siRNAs) (Genolution, Korea) were as follows: 5′-CAG
TGAACGTAAGGGTTCT-3′ for the NF1 gene,
5′-CAGGGACAGCATATCAGAG-3′ for the BCL2L1 (Bcl-xL) gene and
5′-CCTACGCCACCAATTTCGT-3′ for the nonspecific negative control. The
siRNAs were diluted in serum-free Opti-MEM (Invitrogen) and
transfected into cells using Lipofectamine™ RNAiMax
(Invitrogen).
Real-time reverse
transcription-polymerase chain reaction (real-time RT-PCR)
Total-RNAs were isolated from cultured cells using
TRIzol reagent (Invitrogen), treated with RNase-free DNase I
(Invitrogen) to avoid amplification of genomic DNA, and were
subsequently reverse transcribed by the RevertAid™ H Minus First
Strand cDNA Synthesis kit (Fermentas) with
oligo(dt)15–18 primer. Real-time RT-PCR was performed
using the SYBR-Green I qPCR kit (Takara, Japan). The specific
primers used were as follows: 5′-GTCGGATCG CAGCTTGGATGGCCAC-3′ and
5′-CGTCAGGAACCAG CGGTTGAAGCGT-3′ for BCL2L1, P238284 primer
set (Bioneer, Korea) for NF1, and 5′-TGTTGCCATCAATGA
CCCCTT-3′ and 5′-CTCCACGACGTACTCAGCG-3′ for the GAPDH gene
(a relative quantification standard). All real-time RT-PCR
measurements were performed using the ABI PRISM 7000 Sequence
Detection System (Applied Biosystems).
Western blot analysis
Cultured cells were lysed in RIPA buffer (150 mM
NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50
mM Tris buffer, pH 8.0). Proteins were heated at 95°C for 5 min and
analyzed by SDS-PAGE on 8–12% polyacrylamide gels. The proteins
were electroblotted onto a PVDF membrane (Millipore). The membrane
blots were blocked with 5% (w/v) nonfat dried milk, incubated with
primary and secondary antibodies, and then were visualized by the
ECL western blotting detection system (WEST-ZOL plus; Intron
Biotechnology, Korea). The following antibodies were used:
anti-Bcl-xL, anti-caspase-3 anti-Bcl-2, anti-Bax,
anti-phosphorylated Akt, anti-Erk1/2, anti-phosphorylated Erk1/2,
and anti-Ras antibodies (Cell Signaling Technology);
anti-p120RasGAP (BD Transduction Laboratories); anti-neurofibromin,
anti-actin, anti-p53, anti-Mcl-1, anti-K-Ras, anti-H-Ras,
anti-SOCS3, HRP-conjugated goat anti-rabbit IgG and HRP-conjugated
goat anti-mouse IgG antibodies (Santa Cruz Biotechnology,
Inc.).
Ras activation assay
Ras-GTP was detected by using a Ras-activation assay
kit (Upstate Biotechnology). Briefly, active Ras was precipitated
by a GST fusion protein containing the Ras-binding domain of Raf
(GST-Raf-RBD). Cells were lysed in lysis buffer (25 mM HEPES pH
7.5, 150 mM NaCl, 1% Igepal CA-630, 10 mM MgCl2, 1 mM
EDTA, and 2% glycerol). A total of 300 μg cellular lysate was
incubated with Raf-1 RBD agarose at 4°C for 1 h. Agarose beads were
washed three times with 1 ml of ice-cold lysis buffer, boiled with
a 2X Laemmli sample buffer, and separated on SDS-PAGE gels,
followed by western blot analysis using an anti-Ras antibody.
Statistical analysis
Results are expressed as the mean ± SD. All
experiments were repeated at least three times. Statistical
significance between the groups was calculated by a Student’s
t-test. Probability values <0.05 (P<0.05) were considered
statistically significant.
Results
Bcl-xL is overexpressed in the MPNST
cells harboring resistance to anticancer drugs inducing
apoptosis
Understanding the mechanism of drug resistance is
crucial for developing new strategies for targeted chemotherapy. To
examine whether the chemosensitivity to anticancer drugs between
the benign neurofibroma and NF1-associated malignant MPNST cells
was different, we firstly investigated the cytotoxic sensitivity to
the representative anticancer drugs inducing apoptosis in the
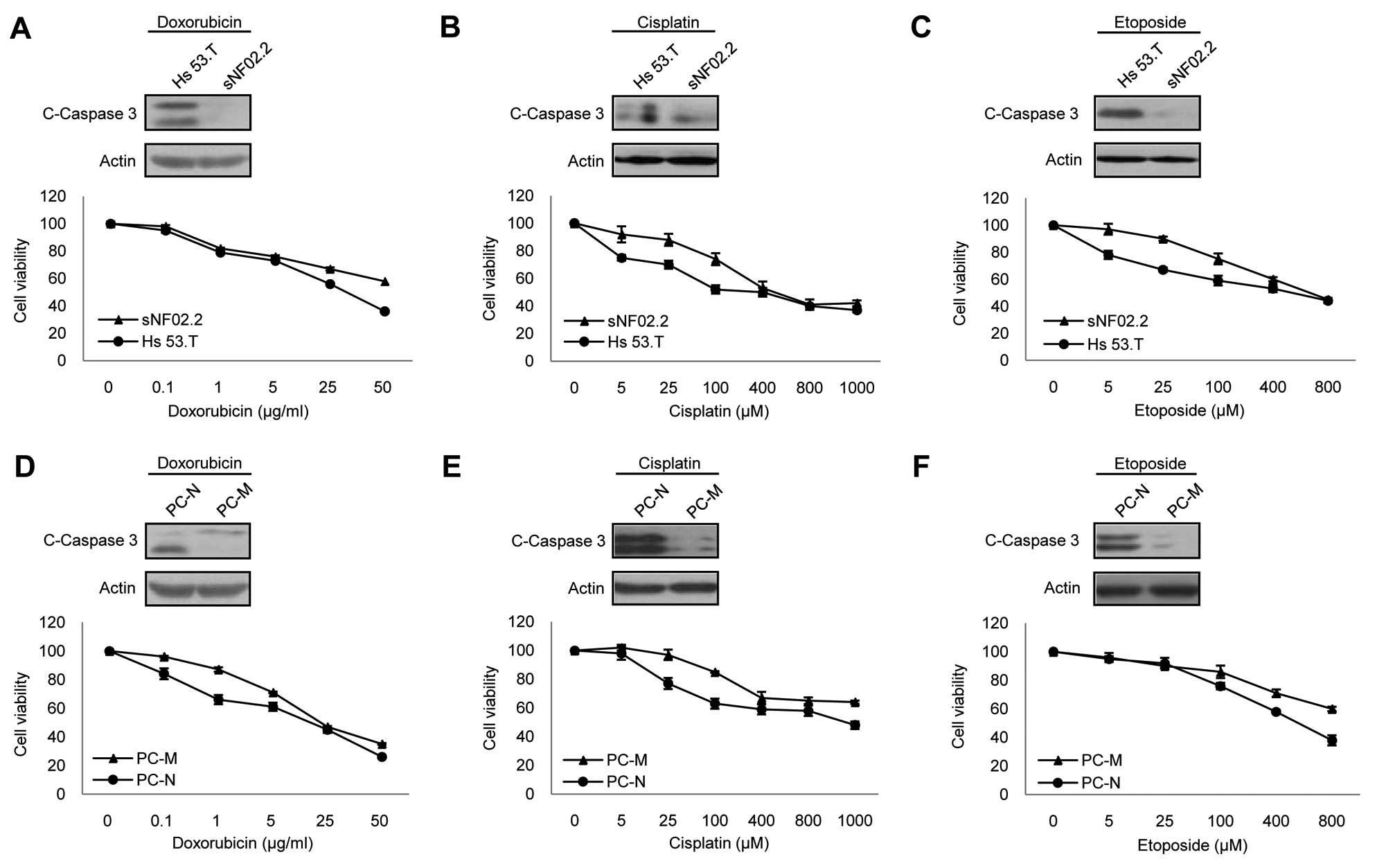
established cell lines, Hs 53.T and sNF02.2 (25). Since doxorubicin, cisplatin and
etoposide have been studied in the patients with NF1-associated
MPNSTs (13–16), we used these three anticancer
drugs in this study. The cell viability and caspase-3 cleavage
assay results showed that the MPNST sNF02.2 cells were more
resistant to all three drugs than benign Hs 53.T cells (Fig. 2A–C).
Next, we tried to confirm this result in the
NF1-associated primary cells. We performed primary tissue culture
of the three types of pathologically evaluated tissues; normal
phenotypic tissues (PC-N), benign PNs (PC-B) and malignant MPNSTs
(PC-M), derived from a patient with NF1, and demonstrated their
distinct cellular characteristics by comparing the levels of
GTP-Ras and its downstream effectors, phosphorylated Erk1/2 and
phosphorylated Akt, and by the expression level of SOCS3,
suppressor of cytokine signaling (Fig. 1). Due to poor proliferation in the
primary benign PN cells after 5 passages, the primary normal cells
and MPNSTs cells were used in this study. As observed in the cell
lines, the primary neurofibromin-deficient MPNST cells were more
resistant to all three drugs than the primary
neurofibromin-deficient normal phenotypic cells (Fig. 2D–F). In order to understand the
reason for the difference in drug resistance between the MPNST
cells and benign/normal cells, we further investigated the
expression levels of apoptosis-related proteins in these cells.
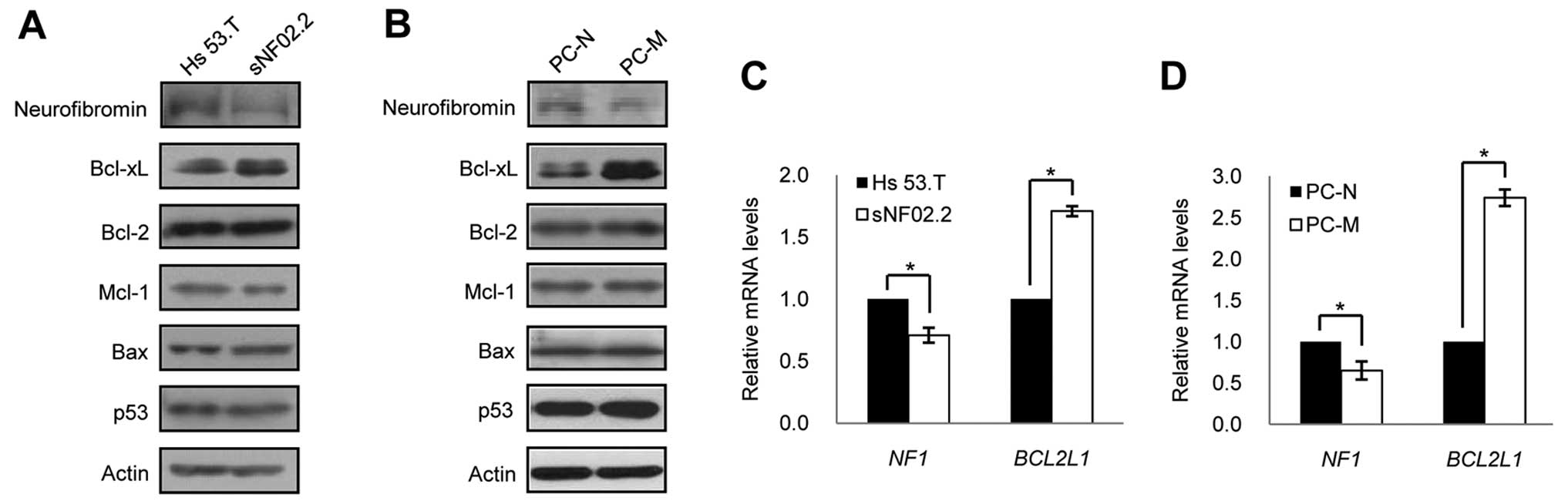
Interestingly, we found that the basal expression level of Bcl-xL
was significantly increased in both the MPNST cell line and primary
MPNST cells compared to the benign cell line and primary normal
cells, while none of the expression levels of other anti-apoptotic
proteins Bcl-2 and Mcl-l were found to be different (Fig. 3A and B) in the MPNST cells
compared to that in benign/normal cells.
Overexpression of Bcl-xL in the MPNST
cells is caused by the decreased expression of NF1
Activated cell survival signaling linked with
apoptosis prevention in the NF1-associated MPNSTs is implicated in
the activation of the Ras-signaling pathway (4,6).
Since the reduced expression of neurofibromin, a negative regulator
of this signaling pathway, in the MPNSTs has been reported
(26), we firstly investigated
the basal expression levels of neurofibromin and observed that the
neurofibromin expression levels were significantly lower in both
the MPNST cell line and primary MPNST cells compored to the
benign/normal cells (Fig. 3A and
B). Quantitative RT-PCR revealed that the different expression
patterns of neurofibromin and Bcl-xL proteins between the MPNST
cells and benign/normal cells originated from the difference in the
transcriptional expression of the NF1 and BCL2L1
genes (Fig. 3C and D). To
elucidate whether the decreased expression level of neurofibromin
in the MPNST cells was caused by an additional mutation in the
intact NF1 allele, we performed molecular analysis of the
NF1 gene in the primary MPNST cells. The presence of the
normal NF1 allele besides the mutated NF1 allele in
trans-chromosomes was confirmed in the primary MPNST cells
(data not shown).
Downregulation of neurofibromin
expression in the benign neurofibroma cell line/primary normal
cells induces an increase in Bcl-xL expression and a decrease in
the sensitivity to apoptosis
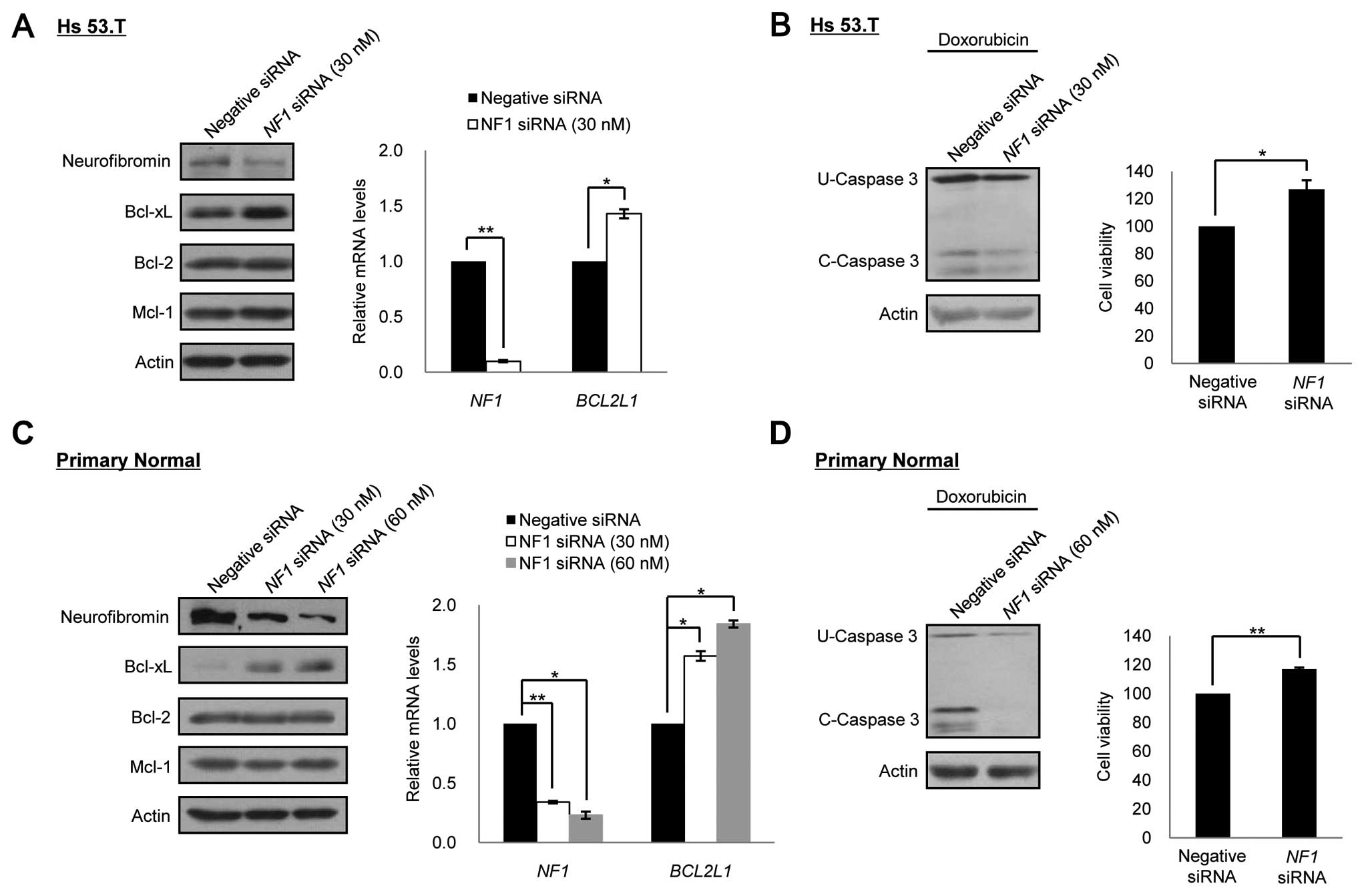
In order to determine if the expression level of
Bcl-xL were dependent on the expression levels of the NF1
gene, we performed the NF1 gene silencing experiment in the
benign/normal cells. Depletion of neurofibromin expression by siRNA
treatment for the NF1 gene caused an increase in the Bcl-xL
expression in a dose-dependent manner in both the benign and normal
cells, but it did not have any effect on other anti-apoptotic
proteins, Bcl-2 and Mcl-1. Quantitative RT-PCR analysis
demonstrated that the knockdown of NF1 by RNAi led to the
increase in the mRNA level of the BCL2L1 gene (Fig. 4A and C). We next examined whether
the depletion of neurofibromin in the benign/normal cells had an
influence on the resistance to anticancer drugs. When doxorubicin
was co-treated for 24 after 72 h of NF1 siRNA treatment, the
neurofibromin-depleted cells showed increased cell viability and
decreased caspase-3 cleavage activity in both the benign and normal
cells compared to control RNAi cells (Fig. 4B and D).
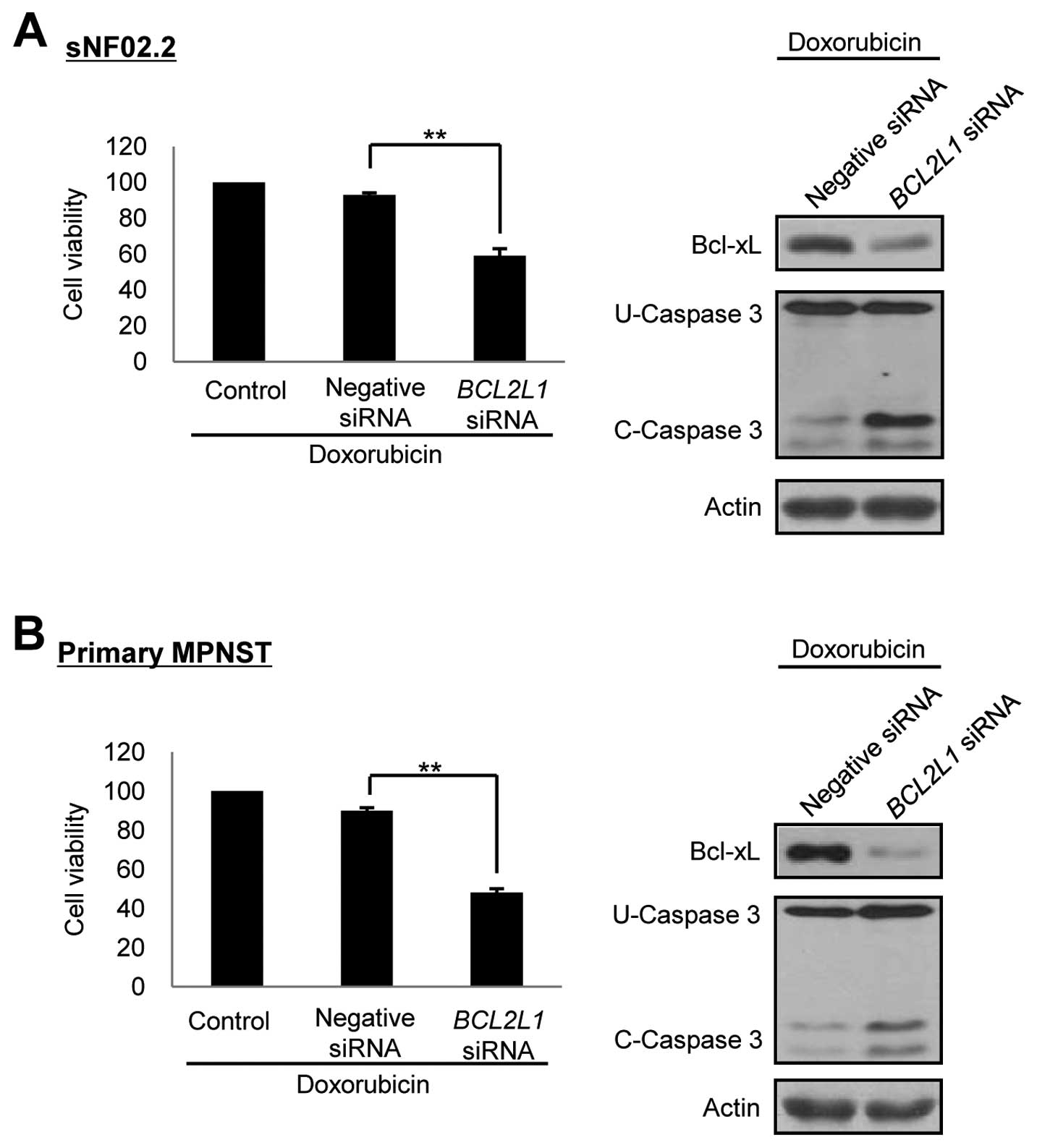
Downregulation of Bcl-xL expression by
siRNA enhances doxorubicin-induced apoptotic cell death in the
MPNST cells
Our results demonstrated that resistance of the
MPNST cells to anticancer drugs was caused by increased Bcl-xL
expression. Therefore, our study focused on the manipulation of the
Bcl-xL expression and the siRNAs targeted against downregulation of
Bcl-xL in the MPNST cells. The BCL2L1 siRNAs per se did not
have any effect on cell toxicity in both the MPNST cell line and
primary MPNST cells (data not shown). However, co-treatment for 24
h with doxorubicin after 72 h of BCL2L1 siRNA treatment
dramatically reduced the cell viability and increased the caspase-3
cleavage activity in both the MPNST cell line and primary MPNST
cells. There was no effect when the negative control siRNAs were
co-treated with doxorubicin (Fig.
5). These results indicated that the downregulation of Bcl-xL
by RNAi enhanced chemosensitivity of the MPNST cells to the
anticancer drug doxorubicin.
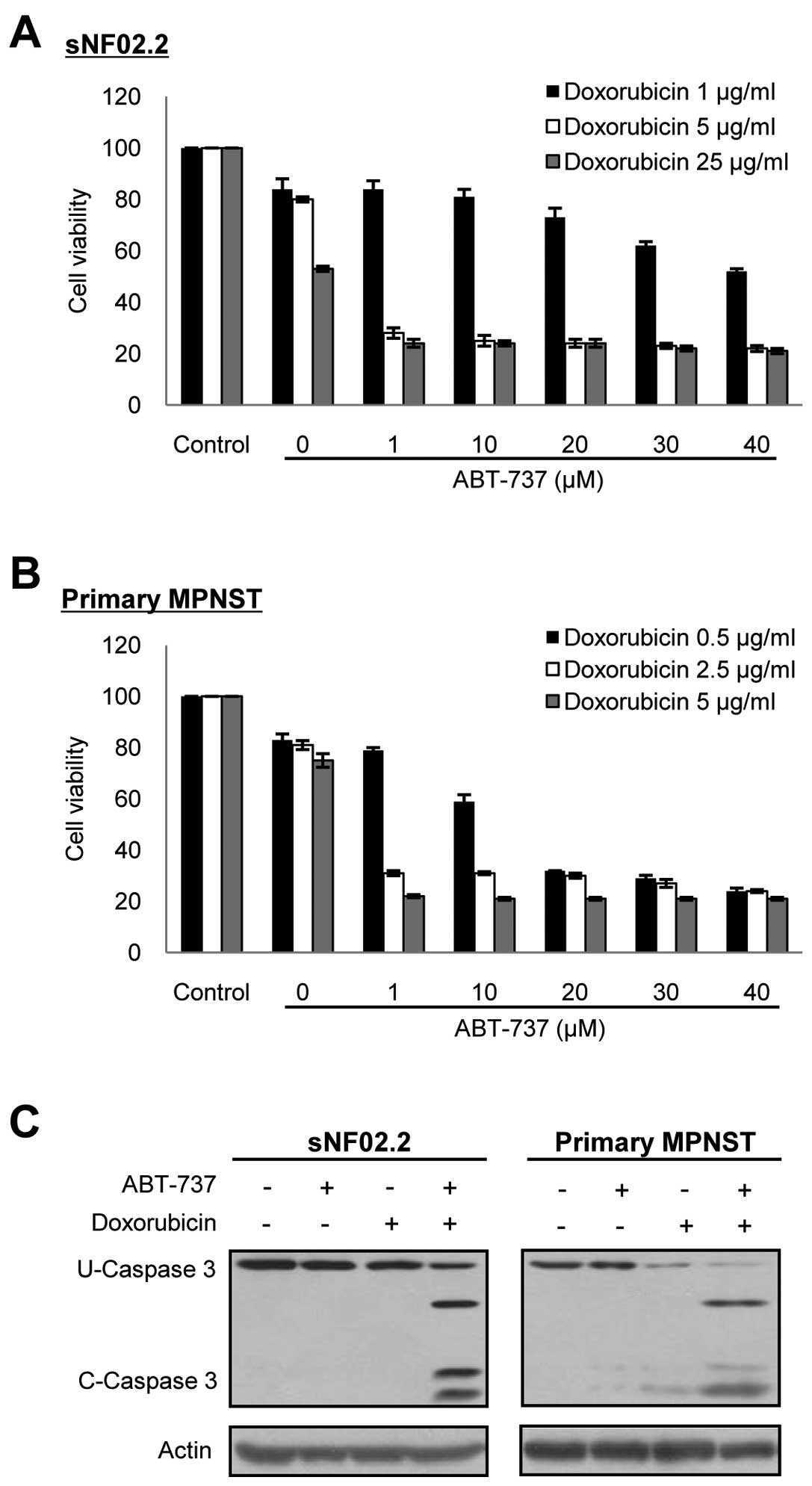
ABT-737 in combination with cytotoxic
drugs enhances chemotherapy sensitivity in the MPNST cells
We next investigated whether ABT-737 (27), a recently developed Bcl-2 family
protein specific inhibitor, had an inhibitory effect against Bcl-xL
in the MPNST cells, and whether ABT-737 displayed a synergistic
cytotoxicity with anticancer chemotherapeutic agents as in the
co-treatment of BCL2L1 siRNA. The concentration of
doxorubicin (0.5–5 μg/ml) required for effective apoptosis was
estimated from the results of Figs.
2 and 5. ABT-737 alone did
not have an effect on cell toxicity in both the MPNST cell line and
primary MPNST cells at the concentrations tested (1–80 μM) (data
not shown). However, when the cells were co-treated with
doxorubicin, ABT-737 effectively enhanced apoptotic cell death in a
dose-dependent manner in both the MPNST cell line and primary MPNST
cells, compared to cells with single treatment of doxorubicin
(Fig. 6). Notably, ABT-737 in
combination with doxorubicin dramatically enhanced chemotherapy
sensitivity in the MPNST cells. Furthermore, ABT-737 effectively
reduced the dosage of doxorubicin required for efficacious MPNST
cell death. The concentrations of both ABT-737 and doxorubicin
required for causing approximately 80% cytotoxicity in the
established and primary tissue cultured NF1-associated MPNST cells
were calculated as; 1 μM of ABT-737 plus 2.5–5 μg/ml of
doxorubicin.
Discussion
Since haploinsufficiency of neurofibromin activity
by NF1 mutation exists in all the cells of the NF1 patient,
even in the normally functional cells, it has been suggested that
the additional genetic or epigenetic changes may participate in
malignant development of benign tumor tissues to MPNSTs as well as
in tumorigenesis of NF1 (4).
Somatic loss of heterozygosity (LOH) at the NF1 locus and
genomic imbalances in chromosomes 17, 19 and 22q are responsible
for this tumor development (28,29). Furthermore, mutations and/or gene
expression changes in many genes such as CD44,
CDKN2A, EGFR, PTEN, RB1, SOX9
and TP53, have also been reported (4,5).
Recently, genome-wide transcriptome analyses revealed that p53
inactivation mediated loss of miR-34a expression in MPNSTs
(30). However, the exact
molecular pathogenesis of malignant transformation of benign tumor
tissues to MPNSTs in NF1 patients has not yet been elucidated.
In this study, we demonstrated that Bcl-xL was
overexpressed in the established and primary tissue cultured
NF1-associated MPNST cells compared to the benign neurofibroma cell
line and primary normal cells. This originated from the decreased
transcriptional expression of the NF1 gene, despite no
additional mutations in the normal NF1 allele besides the
mutant NF1 allele. The reduced expression of neurofibromin,
a negative-regulator for the Ras-signaling pathway, in the MPNSTs
has been reported (26). However,
no frequent somatic mutations in the NF1 gene or
hypermethylation of the NF1 gene have been detected in
MPNSTs (31). We also
investigated the methylation levels in the promoter region of the
NF1 gene in the primary MPNST and normal cells by DNA
methylation chip analysis using the GoldenGate Methylation Cancer
Panel I (Illumina) but did not detect hypermethylation in both the
cell types (data not shown). Overexpression of the BCL2L1
mRNA in MPNSTs can be explained by the decreased
NF1/neurofibromin expression. Since Ets and NF-κB, the
downstream proteins in the Ras-signaling pathway, are well known as
the main transcriptional factors for the BCL2L1 gene
(32), deficiency in
neurofibromin can cause hyperactivation of Ras-signaling due to the
promotion of Ets and NF-κB expression. These results suggest that
alteration in Bcl-xL expression level is caused by somatic
expression changes in the intact NF1 locus and not by
somatic NF1 mutation. The role of Ras-signaling pathway has
been closely implicated in malignant transformation and drug
resistance in many types of cancers (18). Although the molecular mechanisms
for somatic loss of NF1 in the MPNST cells have not been
elucidated, loss of neurofibromin may directly contribute to Bcl-xL
overexpression through the activation of the pathway and may
further contribute to malignant development of benign tumor tissues
or normal tissues to MPNSTs.
Chemoresistance in NF1-associated MPNSTs has been
poorly discussed so far. Our results have demonstrated that
overexpression of Bcl-xL is a principal cause for drug resistance
of the MPNST cells to anticancer drugs. Apoptotic Bcl-2 family
proteins, Bcl-2, Bcl-xL, Mcl-l and Bcl-w, which contribute to
tumorigenesis, tumor progression and tumor chemoresistance are
known to be overexpressed in many cancers (33). Particularly, Bcl-xL overexpression
is involved in resistance to a large number of cytotoxic agents in
human cancer cell lines (34).
Therefore, Bcl-xL is considered one of the promising targets for
overcoming drug resistance by enhancing apoptosis in malignant
tumor cells. Inhibition of Bcl-xL by antisense olignucleotides or
siRNAs significantly enhanced chemosensitivity to cisplatin
(35,36). In addition, various non-peptidic
small molecule inhibitors against Bcl-2 family proteins have been
developed and preclinical or clinical trials for various cancer
therapies have were performed (37). Among them, ABT-737, a mimetic of
the BH3-only protein BAD, and its modified form ABT-263 are drawing
attention as good candidates to selectively target cancer cells and
are in the phase I/II of clinical trials for various cancer
therapies (38). ABT-737
selectively inhibits Bcl-2, Bcl-xL and Bcl-w and synergizes with
conventional chemotherapeutic drugs to promote apoptosis in
multiple cancer types (38,39). Our results showed that either
depletion of Bcl-xL expression by RNAi or inactivation of Bcl-xL by
ABT-737 enhanced doxorubicin-induced apoptosis in MPNST cells
(Figs. 5 and 6). However, regarding the possibilities
and limitations of its clinical application, ABT-737 is considered
more effective than BCL2L1 siRNA. Single treatment with
ABT-737 did not exert a cytotoxic effect in the MPNST cells as
previously reported in other types of cells (38), but, a low concentration (1 μM) of
ABT-737 could significantly enhance the cytotoxic effect of
doxorubicin, when used as combination therapy in our study.
Previously, many cancer cell types were shown to be refractory to
ABT-737 because of high expression of Mcl-1 (40,41). However, our MPNST cells did not
exhibit higher Mcl-1 expression levels compared to the benign and
normal cells (Fig. 2), thereby
suggesting a beneficial effect of ABT-737 in NF1-associated MPNSTs.
Doxorubicin is a clinically used anticancer drug that functions as
topoisomerase II inhibitor and forms covalent DNA adducts. In fact,
doxorubicin recently studied as a chemotherapeutic agent in
combination with ifosfamide in patients with NF1-associated MPNSTs
(15,16) and in combination with ABT-737 in
promyelocytic leukemia cells and chondrosarcoma cell (42,43). Our results suggest that the
combination of ABT-737 and doxorubicin is very effective in
enhancing chemotherapy sensitivity in the NF1-associated MPNST
cells.
In conclusion, to the best of our knowledge, this is
the first study to demonstrate that overexpression of Bcl-xL caused
by downregulation of NF1 is closely associated with drug
resistance in the NF1-associated MPNST cells and suggests that
Bcl-xL inhibition by ABT-737 in combination with doxorubicin can be
a potential therapeutic strategy for the treatment of the
NF1-associated MPNSTs. Further studies are necessary to evaluate
this combination therapy in preclinical models of MPNSTs. We
believe that this study will have a significant impact in the field
of cancer and will be helpful to those studying chemotherapy and
molecular mechanisms involved in the pathogenesis of NF1-associated
MPNSTs.
Acknowledgements
This study was supported by the Basic
Science Research Program through the National Research Foundation
of Korea (NRF) funded by the Ministry of Education, Science and
Technology (2007-0054214, 2009-0093189 and 2009-0075599)
References
|
1.
|
AI McClatcheyNeurofibromatosisAnnu Rev
Pathol2191216200710.1146/annurev.pathol.2.010506.091940
|
|
2.
|
K JettJM FriedmanClinical and genetic
aspects of neurofibromatosis 1Genet
Med12111201010.1097/GIM.0b013e3181bf15e3
|
|
3.
|
A SavarDM CestariNeurofibromatosis type I:
genetics and clinical manifestationsSemin
Ophthalmol234551200810.1080/0882053070174522318214791
|
|
4.
|
B DasguptaDH GutmannNeurofibromatosis 1:
closing the GAP between mice and menCurr Opin Genet
Dev132027200310.1016/S0959-437X(02)00015-112573431
|
|
5.
|
SR GrobmyerJD ReithA ShahlaeeCH BushSN
HochwaldMalignant peripheral nerve sheath tumor: molecular
pathogenesis and current management considerationsJ Surg
Oncol97340349200810.1002/jso.20971
|
|
6.
|
D KatzA LazarD LevMalignant peripheral
nerve sheath tumour (MPNST): the clinical implications of cellular
signalling pathwaysExpert Rev Mol
Med11e30200910.1017/S146239940900122719835664
|
|
7.
|
DG EvansME BaserJ McGaughranS SharifE
HowardA MoranMalignant peripheral nerve sheath tumours in
neurofibromatosis 1J Med
Genet39311314200210.1136/jmg.39.5.31112011145
|
|
8.
|
D MuirD NeubauerIT LimAT YachnisMR
WallaceTumorigenic properties of neurofibromin-deficient
neurofibroma Schwann cellsAm J
Pathol158501513200110.1016/S0002-9440(10)63992-211159187
|
|
9.
|
A GuptaBH CohenP RuggieriRJ PackerPC
PhillipsPhase I study of thalidomide for the treatment of plexiform
neurofibroma in neurofibromatosis
1Neurology60130132200310.1212/01.WNL.0000042321.94839.7812525736
|
|
10.
|
EC CitakA OguzC KaradenizA OkurL MemisO
BoyunagaManagement of plexiform neurofibroma with inter-feron
alphaPediatr Hematol Oncol25673678200810.1080/08880010802315983
|
|
11.
|
D Babovic-VuksanovicK BallmanV
MichelsPhase II trial of pirfenidone in adults with
neurofibromatosis type
1Neurology6718601862200610.1212/01.wnl.0000243231.12248.6717035676
|
|
12.
|
BC WidemannWL SalzerRJ ArceciPhase I trial
and pharmacokinetic study of the farnesyltransferase inhibitor
tipifarnib in children with refractory solid tumors or
neurofibromatosis type I and plexiform neurofibromasJ Clin
Oncol24507516200610.1200/JCO.2005.03.8638
|
|
13.
|
Y KinebuchiW NoguchiY IgawaO
NishizawaRecurrent retroperitoneal malignant nerve sheath tumor
associated with neurofibromatosis type 1 responding to carboplatin
and etoposide combined chemotherapyInt J Clin
Oncol10353356200510.1007/s10147-005-0495-8
|
|
14.
|
H LandyL FeunA MarkoeExtended remission of
a recurrent median nerve malignant peripheral nerve sheath tumor
after multimodal treatment. Case reportJ
Neurosurg103760763200510.3171/jns.2005.103.4.0760
|
|
15.
|
VM MorettiEA CrawfordAP StaddonRD
LackmanCM OgilvieEarly outcomes for malignant peripheral nerve
sheath tumor treated with chemotherapyAm J Clin
Oncol34417421201110.1097/COC.0b013e3181e9c08a20838322
|
|
16.
|
JR KroepM OualiH GelderblomFirst-line
chemotherapy for malignant peripheral nerve sheath tumor (MPNST)
versus other histological soft tissue sarcoma subtypes and as a
prognostic factor for MPNST: an EORTC soft tissue and bone sarcoma
group studyAnn Oncol22207214201110.1093/annonc/mdq338
|
|
17.
|
A FerrariR MiceliA ReyNon-metastatic
unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas:
results of a pooled analysis from United States and European
groupsEur J
Cancer47724731201110.1016/j.ejca.2010.11.01321145727
|
|
18.
|
JA McCubreyLS SteelmanSL AbramsRoles of
the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant
transformation and drug resistanceAdv Enzyme
Regul46249279200610.1016/j.advenzreg.2006.01.00416854453
|
|
19.
|
P BholaS BanerjeeJ MukherjeePreclinical in
vivo evaluation of rapamycin in human malignant peripheral nerve
sheath explant xenograftInt J
Cancer126563571201010.1002/ijc.2478319634141
|
|
20.
|
G AmbrosiniHS CheemaS SeelmanSorafenib
inhibits growth and mitogen-activated protein kinase signaling in
malignant peripheral nerve sheath cellsMol Cancer
Ther7890896200810.1158/1535-7163.MCT-07-051818413802
|
|
21.
|
N HoltkampE MalzerJ ZietschEGFR and erbB2
in malignant peripheral nerve sheath tumors and implications for
targeted therapyNeuro
Oncol10946957200810.1215/15228517-2008-05318650488
|
|
22.
|
JW WojtkowiakF FouadDT LaLondeInduction of
apoptosis in neurofibromatosis type 1 malignant peripheral nerve
sheath tumor cell lines by a combination of novel farnesyl
transferase inhibitors and lovastatinJ Pharmacol Exp
Ther326111200810.1124/jpet.107.135830
|
|
23.
|
SY JeongSJ ParkHJ KimThe spectrum of NF1
mutations in Korean patients with neurofibromatosis type 1J Korean
Med Sci21107112200610.3346/jkms.2006.21.1.10716479075
|
|
24.
|
SY JeongJH HanYY ParkHJ KimIdentification
of differentially expressed genes related to NF1-associated
malignant transformation from a patient with neurofibromatosis type
1Genes Genomics304074182008
|
|
25.
|
Y LiPK RaoR WenNotch and Schwann cell
transformationOncogene2311461152200410.1038/sj.onc.120706814762442
|
|
26.
|
TN BasuDH GutmannJA FletcherTW GloverFS
CollinsJ DownwardAberrant regulation of ras proteins in malignant
tumour cells from type 1 neurofibromatosis
patientsNature356713715199210.1038/356713a01570015
|
|
27.
|
T OltersdorfSW ElmoreAR ShoemakerAn
inhibitor of Bcl-2 family proteins induces regression of solid
tumoursNature435677681200510.1038/nature0357915902208
|
|
28.
|
E LegiusDA MarchukFS CollinsTW
GloverSomatic deletion of the neurofibromatosis type 1 gene in a
neurofibrosarcoma supports a tumour suppressor gene hypothesisNat
Genet3122126199310.1038/ng0293-1228499945
|
|
29.
|
T KogaH IwasakiM IshiguroA MatsuzakiM
KikuchiFrequent genomic imbalances in chromosomes 17, 19, and 22q
in peripheral nerve sheath tumours detected by comparative genomic
hybridization analysisJ
Pathol19798107200210.1002/path.110112081210
|
|
30.
|
S SubramanianV ThayanithyRB
WestGenome-wide transcriptome analyses reveal p53 inactivation
mediated loss of miR-34a expression in malignant peripheral nerve
sheath tumoursJ Pathol2205870201010.1002/path.2633
|
|
31.
|
A HarderM RoscheDE ReussMethylation
analysis of the neurofibromatosis type 1 (NF1) promoter in
peripheral nerve sheath tumoursEur J
Cancer4028202828200410.1016/j.ejca.2004.07.02115571966
|
|
32.
|
L SevillaA ZaldumbideP PognonecKE
BoulukosTranscriptional regulation of the bcl-x gene encoding the
anti-apoptotic Bcl-xL protein by Ets, Rel/NFkappaB, STAT and AP1
transcription factor familiesHistol
Histopathol16595601200111332715
|
|
33.
|
V KirkinS JoosM ZornigThe role of Bcl-2
family members in tumorigenesisBiochim Biophys
Acta1644229249200410.1016/j.bbamcr.2003.08.00914996506
|
|
34.
|
SA AmundsonTG MyersD ScudieroS KitadaJC
ReedAJ Fornace JrAn informatics approach identifying markers of
chemosensitivity in human cancer cell linesCancer
Res6061016110200011085534
|
|
35.
|
JE LittlejohnX CaoSD MillerBcl-xL
antisense oligonucleotide and cisplatin combination therapy extends
survival in SCID mice with established mesothelioma xenograftsInt J
Cancer123202208200810.1002/ijc.23452
|
|
36.
|
E BrotinM Meryet-FiguiereK SimoninBcl-XL
and MCL-1 constitute pertinent targets in ovarian carcinoma and
their concomitant inhibition is sufficient to induce apoptosisInt J
Cancer1268858952010
|
|
37.
|
AS AzmiRM MohammadNon-peptidic small
molecule inhibitors against Bcl-2 for cancer therapyJ Cell
Physiol2181321200910.1002/jcp.2156718767026
|
|
38.
|
A RichardsonSB KayePharmacological
inhibition of the Bcl-2 family of apoptosis regulators as cancer
therapyCurr Mol
Pharmacol1244254200810.2174/187446721080103024420021437
|
|
39.
|
D ReynosoLK NoldenD YangSynergistic
induction of apoptosis by the Bcl-2 inhibitor ABT-737 and imatinib
mesylate in gastrointestinal stromal tumor cellsMol
Oncol593104201110.1016/j.molonc.2010.10.00321115411
|
|
40.
|
MF van DelftAH WeiKD MasonThe BH3 mimetic
ABT-737 targets selective Bcl-2 proteins and efficiently induces
apoptosis via Bak/Bax if Mcl-1 is neutralizedCancer
Cell10389399200617097561
|
|
41.
|
C TouzeauC DoussetL BodetABT-737 induces
apoptosis in mantle cell lymphoma cells with a Bcl-2high/Mcl-1low
profile and synergizes with other antineoplastic agentsClin Cancer
Res1759735981201110.1158/1078-0432.CCR-11-095521821698
|
|
42.
|
M UgarenkoA NudelmanA RephaeliK KimuraDR
PhillipsSM CuttsABT-737 overcomes Bcl-2 mediated resistance to
doxorubicin-DNA adductsBiochem
Pharmacol79339349201010.1016/j.bcp.2009.09.00419737541
|
|
43.
|
D LimJ MuirImatinib for chronic myeloid
leukaemia: a NICE
messLancet3581903200110.1016/S0140-6736(01)06903-311741656
|