Introduction
Congenital stationary night blindness (CSNB) is a
clinically and genetically heterogeneous group of inherited retinal
disorders characterized by nonprogressive impaired night vision and
sometimes accompanied with other ocular symptoms, including myopia,
nystagmus and strabismus (1).
Electroretinogram (ERG) recordings can classify CSNB into two
groups, complete CSNB (cCSNB or CSNB1) which show the complete
absence of rod pathway function and incomplete CSNB (icCSNB or
CSNB2) which is caused by abnormal rod and cone pathway function
(2). CSNB can be transmitted as
autosomal dominant (adCSNB), autosomal recessive (arCSNB), or
X-linked recessive traits (xlCSNB). To date, 12 genes have been
reported to be implicated in CSNB (RetNet, http://www.sph.uth.tmc.edu/retnet/), including RHO
(MIM 180380), GNAT1 (MIM 139330), PDE6B (MIM 180072), GRM6 (MIM
604096), TRPM1 (MIM 603576), SLC24A1 (MIM 603617), CABP4 (MIM
608965), CACNA2D4 (MIM 608171), SAG (MIM 181031), GRK1 (MIM
180381), NYX (MIM 300278) and CACNA1F (MIM 300110) (3–27).
Four of the 12 genes, TRPM1, GRM6, NYX and CACNA1F,
are involved in the signaling cascade from photoreceptors to
adjacent bipolar cells (1).
L-type voltage-dependent calcium channel α-1F subunit (encoded by
CACNA1F), locating in the rod synaptic terminal, regulates the
intracellular influx Ca2+ concentration, which influence
the glutamate release from rods to bipolar cells (28). Metabotropic glutamate receptor 6,
encoded by GRM6 (MGluR6), locating in a bipolar cell, receives the
glutamate released from rods and activates an intracellular cascade
that terminates in closure of TRPM1 (encoded by TRPM1) (4,29).
Nyctalopin (encoded by NYX) may interact with TRPM1 but the exact
function is yet to be identified (30–32). Any abnormality in the cascade will
lead to the signal transduction defect with clinical phenotype of
CSNB.
Mutations in the TRPM1, GRM6, NYX and CACNA1F genes
have been frequently studied in Caucasian or Japanese populations
(1,33). Mutation analysis of all these 4
genes at the same time are rare, especially in Chinese. In this
study, Sanger sequencing were used to analyze the coding exons and
their adjacent regions of the 4 genes in 24 unrelated Chinese
patients with CSNB.
Materials and methods
Patients
Twenty-four unrelated patients with CSNB were
collected from our Pediatric and Genetic Eye Clinic of the
Zhongshan Ophthalmic Center. Written informed consent conforming to
the tenets of the Declaration of Helsinki was obtained from each
participant or their guardians prior to the study. The
Institutional Review Board of Zhongshan Ophthalmic Center approved
this study. Genomic DNA was prepared from leukocytes of venous
blood samples as previously described (34).
Mutation screening
Eighty-six coding exons and their adjacent intronic
regions in the TRPM1, GRM6, NYX and CACNA1F genes were analyzed by
using Sanger dideoxy sequencing. Bioinformation of these 4 genes
(Table I) obtained from the
National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/). DNA fragments
encompassing individual exon was amplified by polymerase chain
reaction (PCR). The amplicons were analyzed with the ABI BigDye
Terminator cycle sequencing kit version 3.1 (Applied Biosystems,
Foster City, CA) using an ABI 3100 Genetic Analyzer (Applied
Biosystems). Sequencing results from the patients and the consensus
sequences from the NCBI Human Genome Database were compared using
the CLC Main Workbench program (http://www.clcbio.com/) (35). Each variation was initially
confirmed by bi-directional sequencing and then evaluated in 96
normal individuals. The description of the mutations follows the
recommendations of the Human Genomic Variation Society (HGVS,
http://www.hgvs.org/). The potential functional
effect of an amino acid substitution due to a mutation was
predicted using the PolyPhen-2 online tool (v2.0.23, http://genetics.bwh.harvard.edu/pph2/).
Sorting of the intolerant from tolerant (SIFT) was also used to
predict whether an amino acid substitution affects protein function
based on sequence homology and the physical properties of amino
acids (http://sift.jcvi.org/).
 | Table I.Genomic information of the four genes
studied. |
Table I.
Genomic information of the four genes
studied.
| Gene | Location | Genomic DNA | mRNA | Protein | Total number of
coding exons | Number of exons
analyzed |
|---|
| TRPM1 | 15q13.3 | NC_000015.9 | NM_002420.4 | NP_002411.3 | 26 | 26 |
| GRM6 | 5q35 | NC_000005.9 | NM_000843.3 | NP_000834.2 | 10 | 10 |
| NYX | Xp11.4 | NC_000023.10 | NM_022567.2 | NP_072089.1 | 2 | 2 |
| CACNA1F | Xp11.23 | NC_000023.10 | NM_005183.2 | NP_005174.2 | 48 | 48 |
Results
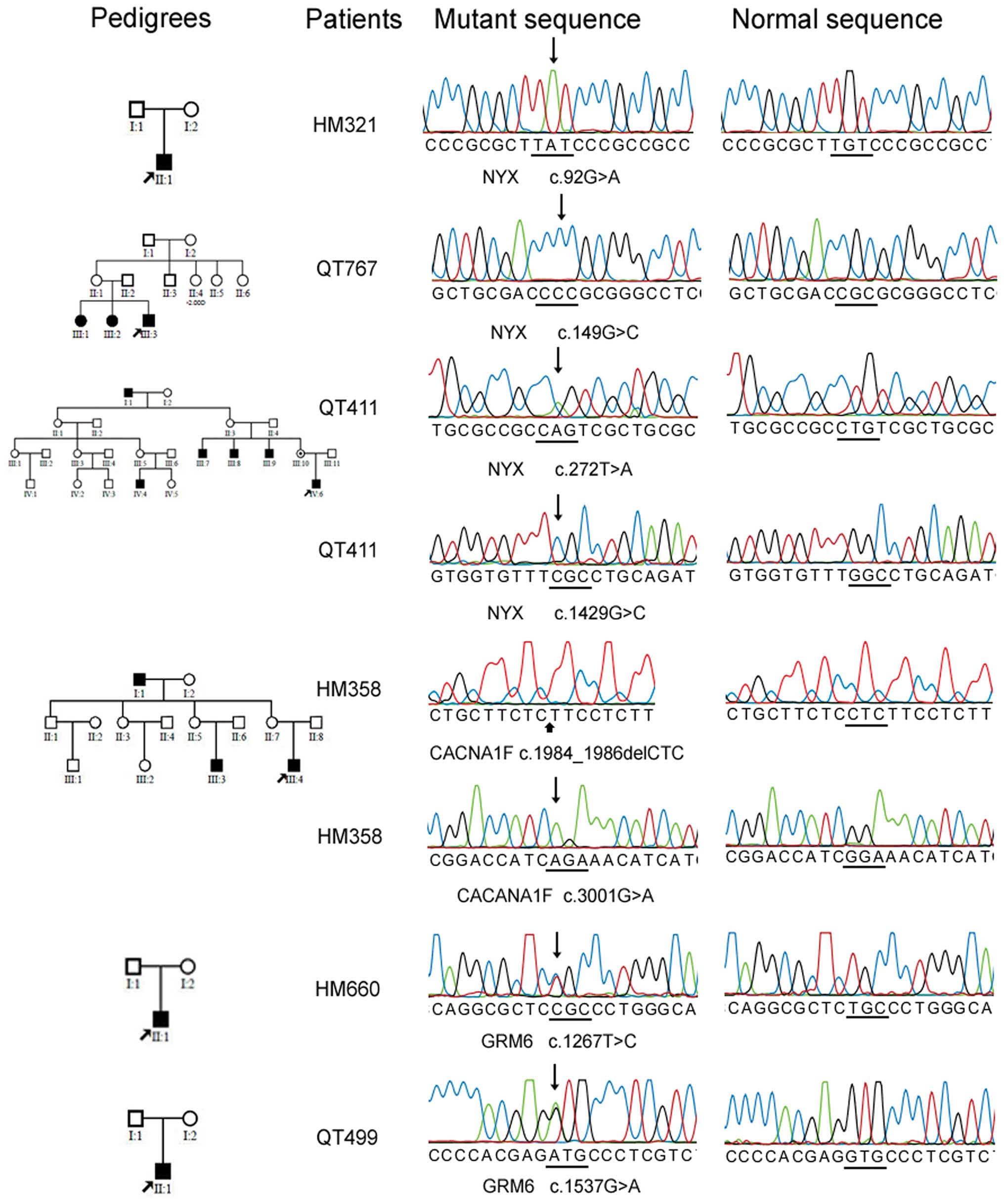
Mutations in the 4 genes were detected in six of the
24 families with CSNB (Table II
and Fig. 1), including 3 novel
mutations in NYX, 1 novel mutation in CACNA1F, and 2 heterozygous
mutations (one novel and one known) in GRM6. One mutation in NYX
and one mutation in CACNA1F were compound hemizygous mutations. The
mutations in each patient involve codons in which the encoded
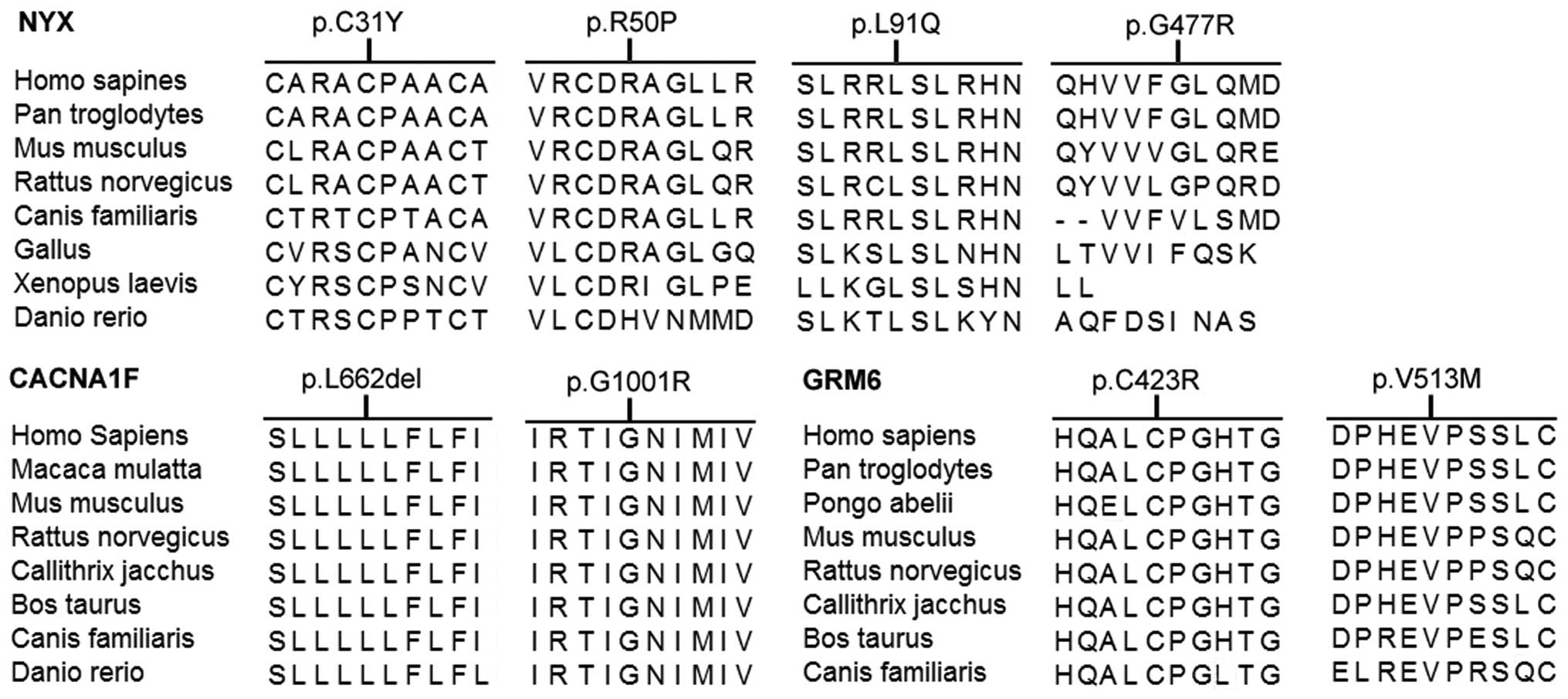
residues were well conserved (Fig.
2). These mutations were not detected in the 96 normal
individuals. No mutation was detected in TRPM1. Clinical
information of the patients with mutations are listed in Table III.
| Table II.Four CSNB genes mutations in Chinese
subjects. |
Table II.
Four CSNB genes mutations in Chinese
subjects.
| Gene | Exon | Patient | Nucleotide
change | Amino acid
change | BLOSUM 62
difference | PolyPhen-2 | SIFT | Note |
|---|
| NYX | 2 | HM321 | c.92G>A | p.Cys31Tyr | 9 to −2 | Unknown | Affect protein
function | Novel |
| NYX | 2 | QT767 | c.149G>C | p.Ary50Pro | 5 to −2 | Probably
damaging | Tolerated | Novel |
| NYX | 2 | QT411 |
c.[272T>A;1429G>C] |
p.[Leu91Gln;Gly477Arg] | 4 to −2 | Probably
damaging | Affect protein
function | Novel |
| 6 to −2 | Possibly
damaging | Tolerated |
| CACNA1F | 15 | HM358 |
c.[1984_1986delCTC;3001G>A] |
p.[Leu662del;Gly1001Arg] | N/A | N/A | N/A | Novel |
| 25 | 6 to −2 | Probably
damaging | Tolerated |
| GRM6 | 6 | HM660 | c.1267T>C | p.Cys423Arg | 9 to −3 | Probably
damaging | Affect protein
function | Novel |
| GRM6 | 8 | QT499 | c.1537G>A | p.Val513Met | 4 to −2 | Benign | Tolerated | Reported (36) |
| Table III.Clinical information on individuals
with CSNB gene mutations. |
Table III.
Clinical information on individuals
with CSNB gene mutations.
| | | | | | | Refraction
| Visual acuity
| ERG responses
|
|---|
| Patient | Mutation | | Gender | Age | Age at onset | Inheritance | OD | OS | OD | OS | Rod | Cone |
|---|
| HM321 | NYX | c.92G>A | Male | 9 | EC | Sporadic | −5.50D | −5.00D | 0.3 | 0.3 | N/A | N/A |
| QT767 | NYX | c.149G>C | Male | 13 | EC | Unknown | −14.00D | −14.75D | 0.2 | 0.4 | Undetectable | Reduced |
| QT411 | NYX |
c.[272T>A;1429G>C] | Male | 3 | EC | XL | −6.00D | −6.50D | 0.5 | 0.8 | Undetectable | Reduced |
| HM358 | CACNA1F |
c.[1984_1986delCTC;3001G>A] | Male | 1 | EC | XL | N/A | N/A | N/A | N/A | Reduced | Reduced |
| HM660 | GRM6 | c.1267T>C | Female | 12 | EC | Sporadic | −10.00D | −12.50D | 0.2 | 0.2 | N/A | N/A |
| QT499 | GRM6 | c.1537G>A | Female | 2 | EC | Sporadic | −6.50D | −6.00D | N/A | N/A | Undetectable | Reduced |
| | | | | | | | | | | | |
The c.92G>A (p.Cys31Tyr), c.149G>C
(p.Ary50Pro) and c.[272T>A;1429G>C] (p.[Leu91Gln;Gly477Arg])
mutations in NYX were detected in an isolated case and 2 families
with possible X-linked pattern of inheritance (Fig. 1), respectively. These variations
are predicted to affect the function of the encoded protein.
Segregation analysis of the compound c.[272T>A;1429G>C]
(p.[Leu91Gln;Gly477Arg]) mutation in family QT411 confirmed the
hemizygous mutation in other two affected patients (III7 and III9)
and the heterozygous status in the unaffected mother (Fig. 1). Patients with the three NYX
mutations had a complete form of CSNB.
The c.[1984_1986delCTC;3001G>A] (p.[Leu662del;
Gly1001Arg]) mutation in CACNA1F was detected in a patient, who had
incomplete form of CSNB and a family history of the disease showing
X-linked recessive pattern of inheritance (Fig. 1). This mutation is predicted to be
probably damaging by PolyPhen-2.
Two heterozygous mutations in GRM6, c.1267T>C
(p.Cys423Arg) and c.1537G>A (p.Val513Met), were detected in two
isolated male patients with a complete form of CSNB (Fig. 1), respectively. The c.1267T>C
(p.Cys423Arg) mutation is novel and predicted to be probably
damaging (Table II). The
c.1537G>A (p.Val513Met) mutation is predicted to be benign and
has been previously detected in a Chinese patient with high myopia
(36). These two mutations are
located in the extracellular N-terminal domain that is vital in
glutamate binding and the activation or inactivation of mGluR6
(37,38). However, mutations in another
allele of these 2 patients have not been identified.
Discussion
In this study, analysis of the TRPM1, GRM6, NYX and
CACNA1F genes in probands from 24 Chinese families with CSNB
detected 6 mutations in 6 unrelated patients, including five novel
and one known mutations. Three of the 6 mutations in NYX and 1
mutation in CACNA1F are likely to be the cause responsible for CSNB
in those 4 families. However, additional study is needed to reveal
how a heterozygous GRM6 mutation could associate with CSNB as
mutations in GRM6 have been demonstrated to cause autosomal
recessive CSNB.
TRPM1 is identified as the mGluR6-coupled cation
channel in retinal ON-bipolar cells (39). Several studies have reported that
TRPM1 mutations are associated with arCSNB in Caucasian or Japanese
populations (9–11,33). No mutation was detected in the
Chinese patients in this study although mutations in TRPM1 have
been found in about half of the cases with CSNB1 (29).
The c.1267T>C (p.Cys423Arg) mutation in GRM6 is
located in the ligand-binding domains of mGluR6 and probably will
affect the folding of the protein (40). The c.1537G>A (p.Val513Met) in
GRM6 was previously reported in high myopia patient without CSNB
(36). We found this mutation in
a CSNB patient with high myopia. The valine at codon 513 is located
in the second conserved cysteine-rich domain (CRD) of the mGluR6
receptor, which is important in the intermolecular signal
transmission (41). It is unclear
why the same mutation is associated with high myopia alone in one
patient but with CSNB and high myopia in another patient.
The c.92G>A (p.Cys31Try) and c.149G>C
(p.Ary50Pro) mutations in NYX locate in the N-terminal
cysteine-rich LRRs (leucine-rich repeats, LRPs). For the former, it
is worth noting that a different mutation affecting the same codon,
c.92G>C, has been reported before (21). The c.[272T>A;1429G>C]
(p.[Leu91Gln;Gly477Arg]) would affect the second LRRs (total 11
LRRs) and the GPI-anchor region, respectively, and therefore may
impair the structure or function of the encoded protein.
The (c.[1984_1986delCTC;3001G>A] (p.[Leu662del;
Gly1001Arg]) mutation in CACNA1F is present in a patient with
incomplete CSNB who has a family history of this disease showing
X-linked recessive pattern. The deletion in this mutation would
affect the domain II S5 region that is evolutionarily conserved.
The missense change of this mutation involving the domain III S5
region is predicted to be probably damaging, which may disrupt the
channel function (42).
In this study, 3 mutations in NYX, 1 mutation in
CACNA1F, 2 mutations in GRM6 were identified in 6 of 24 Chinese
patients with CSNB. The results expand the mutation spectrum of
these genes. Further analysis of additional genes may enrich our
understanding of the molecular basis of CSNB in those patients
without mutation.
Acknowledgements
We would like to thank all subjects
for their participation. This study was supported in part by the
National Science Found for Distinguished Young Scholars (30725044
to Q.Z.) and the Fundamental Research Funds of State Key
Laboratory.
References
|
1.
|
C ZeitzMolecular genetics and protein
function involved in nocturnal visionExpert Rev
Ophthalmol2467485200710.1586/17469899.2.3.467
|
|
2.
|
Y MiyakeK YagasakiM HoriguchiY KawaseT
KandaCongenital stationary night blindness with negative
electroretinogram. A new classificationArch
Ophthalmol10410131020198610.1001/archopht.1986.010501900710423488053
|
|
3.
|
VR RaoGB CohenDD OprianRhodopsin mutation
G90D and a molecular mechanism for congenital night
blindnessNature367639642199410.1038/367639a08107847
|
|
4.
|
TP DryjaTL McGeeEL BersonNight blindness
and abnormal cone electroretinogram ON responses in patients with
mutations in the GRM6 gene encoding mGluR6Proc Natl Acad Sci
USA10248844889200510.1073/pnas.050123310215781871
|
|
5.
|
N al-JandalGJ FarrarAS KiangA novel
mutation within the rhodopsin gene (Thr-94-Ile) causing autosomal
dominant congenital stationary night blindnessHum
Mutat137581199910.1002/(SICI)1098-1004(1999)13:1%3C75::AID-HUMU9%3E3.0.CO;2-49888392
|
|
6.
|
TP DryjaLB HahnT ReboulB ArnaudMissense
mutation in the gene encoding the alpha subunit of rod transducin
in the Nougaret form of congenital stationary night blindnessNat
Genet13358360199610.1038/ng0796-3588673138
|
|
7.
|
A GalU OrthW BaehrE SchwingerT
RosenbergHeterozygous missense mutation in the rod cGMP
phosphodiesterase beta-subunit gene in autosomal dominant
stationary night blindnessNat Genet7551199410.1038/ng0894-551c
|
|
8.
|
C ZeitzM van GenderenJ NeidhardtMutations
in GRM6 cause autosomal recessive congenital stationary night
blindness with a distinctive scotopic 15-Hz flicker
electroretinogramInvest Ophthalmol Vis
Sci4643284335200510.1167/iovs.05-0526
|
|
9.
|
I AudoS KohlBP LeroyTRPM1 is mutated in
patients with autosomal-recessive complete congenital stationary
night blindnessAm J Hum
Genet85720729200910.1016/j.ajhg.2009.10.01319896113
|
|
10.
|
Z LiPI SergouniotisM MichaelidesRecessive
mutations of the gene TRPM1 abrogate ON bipolar cell function and
cause complete congenital stationary night blindness in humansAm J
Hum Genet85711719200910.1016/j.ajhg.2009.10.00319878917
|
|
11.
|
MM van GenderenMM BijveldYB
ClaassenMutations in TRPM1 are a common cause of complete
congenital stationary night blindnessAm J Hum
Genet85730736200919896109
|
|
12.
|
SA RiazuddinA ShahzadiC ZeitzA mutation in
SLC24A1 implicated in autosomal-recessive congenital stationary
night blindnessAm J Hum
Genet87523531201010.1016/j.ajhg.2010.08.01320850105
|
|
13.
|
C ZeitzB Kloeckener-GruissemU
ForsterMutations in CABP4, the gene encoding the
Ca2+-binding protein 4, cause autosomal recessive night
blindnessAm J Hum Genet79657667200616960802
|
|
14.
|
KA WyciskB BuddeS FeilStructural and
functional abnormalities of retinal ribbon synapses due to Cacna2d4
mutationInvest Ophthalmol Vis
Sci4735233530200610.1167/iovs.06-027116877424
|
|
15.
|
KA WyciskC ZeitzS FeilMutation in the
auxiliary calcium-channel subunit CACNA2D4 causes autosomal
recessive cone dystrophyAm J Hum
Genet79973977200610.1086/50894417033974
|
|
16.
|
S YamamotoKC SippelEL BersonTP
DryjaDefects in the rhodopsin kinase gene in the Oguchi form of
stationary night blindnessNat
Genet15175178199710.1038/ng0297-1759020843
|
|
17.
|
S FuchsM NakazawaM MawM TamaiY OguchiA
GalA homozygous 1-base pair deletion in the arrestin gene is a
frequent cause of Oguchi disease in JapaneseNat
Genet10360362199510.1038/ng0795-3607670478
|
|
18.
|
M NakamuraS YamamotoM OkadaS ItoY TanoY
MiyakeNovel mutations in the arrestin gene and associated clinical
features in Japanese patients with Oguchi’s
diseaseOphthalmology11114101414200415584351
|
|
19.
|
M MawG KumaramanickavelB KarS JohnR
BridgesM DentonTwo Indian siblings with Oguchi disease are
homozygous for an arrestin mutation encoding premature
terminationHum MutatSuppl
1S317S319199810.1002/humu.13801101999452120
|
|
20.
|
Q ZhangF ZulfiqarSA RiazuddinA variant
form of Oguchi disease mapped to 13q34 associated with partial
deletion of GRK1 geneMol Vis11977985200516319817
|
|
21.
|
CM PuschC ZeitzO BrandauThe complete form
of X-linked congenital stationary night blindness is caused by
mutations in a gene encoding a leucine-rich repeat proteinNat
Genet26324327200010.1038/8162711062472
|
|
22.
|
NT Bech-HansenMJ NaylorTA
MaybaumLoss-of-function mutations in a calcium-channel
alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital
stationary night blindnessNat
Genet19264267199810.1038/9479662400
|
|
23.
|
TM StromG NyakaturaE Apfelstedt-SyllaAn
L-type calcium-channel gene mutated in incomplete X-linked
congenital stationary night blindnessNat
Genet19260263199810.1038/9409662399
|
|
24.
|
I ZitoLE AllenRJ PatelMutations in the
CACNA1F and NYX genes in British CSNBX familiesHum
Mutat21169200310.1002/humu.910612552565
|
|
25.
|
KM BoycottTA MaybaumMJ NaylorA summary of
20 CACNA1F mutations identified in 36 families with incomplete
X-linked congenital stationary night blindness, and
characterization of splice variantsHum
Genet1089197200110.1007/s00439010046111281458
|
|
26.
|
C ZeitzR MinottiS FeilNovel mutations in
CACNA1F and NYX in Dutch families with X-linked congenital
stationary night blindnessMol Vis11179183200515761389
|
|
27.
|
X XiaoX JiaX GuoS LiZ YangQ ZhangCSNB1 in
Chinese families associated with novel mutations in NYXJ Hum
Genet51634640200610.1007/s10038-006-0406-516670814
|
|
28.
|
JC HodaF ZaghettoA KoschakJ
StriessnigCongenital stationary night blindness type 2 mutations
S229P, G369D, L1068P, and W1440X alter channel gating or functional
expression of Ca(v)1.4 L-type Ca2+ channelsJ
Neurosci25252259200510.1523/JNEUROSCI.3054-04.200515634789
|
|
29.
|
CW MorgansRL BrownRM DuvoisinTRPM1: the
endpoint of the mGluR6 signal transduction cascade in retinal
ON-bipolar
cellsBioessays32609614201010.1002/bies.20090019820544736
|
|
30.
|
NT Bech-HansenMJ NaylorTA MaybaumMutations
in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause
X-linked complete congenital stationary night blindnessNat
Genet26319323200010.1038/8161911062471
|
|
31.
|
CW MorgansG RenL AkileswaranLocalization
of nyctalopin in the mammalian retinaEur J
Neurosci2311631171200610.1111/j.1460-9568.2006.04647.x16553780
|
|
32.
|
PJ BojangJN PearringRG GreggNyctalopin
Interacts with Transient Receptor Potential Channels in YeastARVO
2009 Annual Meeting, Florida, Abst no. 51762009
|
|
33.
|
M NakamuraR SanukiTR YasumaTRPM1 mutations
are associated with the complete form of congenital stationary
night blindnessMol Vis16425437201020300565
|
|
34.
|
Q WangP WangS LiMitochondrial DNA
haplogroup distribution in Chaoshanese with and without myopiaMol
Vis16303309201020208987
|
|
35.
|
A BrautigamT MullickS SchlieskyAP
WeberCritical assessment of assembly strategies for non-model
species mRNA-Seq data and application of next-generation sequencing
to the comparison of C3 and C4 speciesJ Exp
Bot6230933102201110.1093/jxb/err02921398430
|
|
36.
|
X XuS LiX XiaoP WangX GuoQ ZhangSequence
variations of GRM6 in patients with high myopiaMol
Vis1520942100200919862333
|
|
37.
|
D TsuchiyaN KunishimaN KamiyaH JingamiK
MorikawaStructural views of the ligand-binding cores of a
metabotropic glutamate receptor complexed with an antagonist and
both glutamate and Gd3+Proc Natl Acad Sci
USA9926602665200210.1073/pnas.05270859911867751
|
|
38.
|
N KunishimaY ShimadaY TsujiStructural
basis of glutamate recognition by a dimeric metabotropic glutamate
receptorNature407971977200010.1038/3503956411069170
|
|
39.
|
C KoikeT ObaraY UriuTRPM1 is a component
of the retinal ON bipolar cell transduction channel in the mGluR6
cascadeProc Natl Acad Sci
USA107332337201010.1073/pnas.091273010719966281
|
|
40.
|
C ZeitzU ForsterJ NeidhardtNight
blindness-associated mutations in the ligand-binding,
cysteine-rich, and intracellular domains of the metabotropic
glutamate receptor 6 abolish protein traffickingHum
Mutat28771780200710.1002/humu.20499
|
|
41.
|
P RondardJ LiuS HuangCoupling of agonist
binding to effector domain activation in metabotropic
glutamate-like receptorsJ Biol
Chem2812465324661200610.1074/jbc.M60227720016787923
|
|
42.
|
JB PeloquinR RehakCJ DoeringJE
McRoryFunctional analysis of congenital stationary night blindness
type-2 CACNA1F mutations F742C, G1007R, and
R1049WNeuroscience150335345200710.1016/j.neuroscience.2007.09.02117949918
|