Introduction
Non-alcoholic fatty liver disease (NAFLD) has
emerged as a substantial public health concern. This syndrome is
characterized by steatosis in hepatocytes and elevated serum levels
of free fatty acids (FFAs) (1).
Although the mechanisms responsible for fatty liver are not fully
elucidated, an increased delivery and transport of FFAs into the
liver, and augmented hepatic fatty acid synthesis are likely to
play a significant role in the pathogenesis of NAFLD. Liver cell
apoptosis is a prominent feature of NAFLD and correlates with
disease severity (2). The
toxicity of lipids, or lipotoxicity, and specifically,
lipid-induced apoptosis, or lipoapoptosis, is a potential mechanism
relating apoptosis to NAFLD. Hepatocellular lipid accumulation is
thought to simultaneously stimulate mitochondrial fatty acid
oxidation and the production of ROS, thereby promoting lipid
peroxidation and damage of mitochondrial DNA (mtDNA) (3). Abnormal morphological changes in
liver mitochondria have been observed in patients and animal models
with non-alcoholic steatohepatitis (NASH) (4). There is also growing evidence that
FFA-mediated oxidative stress contributes significantly to
mitochondrial dysfunction in the liver. Therefore, designing
therapies that prevent mitochondrial dysfunction stands to be one
of the most important strategies in for treating of NAFLD and its
complications.
Despite the high prevalence of NAFLD and its
potential to cause serious liver injury, the current therapies for
NAFLD are limited. Hence, developing new therapeutic intervention
is a prerequisite in the treatment of NAFLD. Curcumin has recently
received attention as a promising dietary supplement for liver
protection (5). These recent
studies showed that curcumin inhibited HSCs activation, and
suppressed hepatic fibrogenesis in vitro and in vivo
(6,7). In addition, curcumin eliminated the
effects of leptin on the activation of HSCs in vitro by
reducing the phosphorylation level of the leptin receptor (Ob-R),
leading to the suppression of Ob-R gene expression and the
interruption of leptin signaling (8). Curcumin also exerts potential
anti-inflammatory effects in diverse cell types, including
pancreatic cells, chondrocytes, and hepatic cells (9,10).
Wang et al (11) have
illustrated curcumin’s ability to reduce pro-inflammatory cytokines
in 3T3-L1 adipocytes with FFA-induced insulin resistance. Although
these suppressive effects of pro-inflammatory signaling pathways
might be involved in the pathogenesis of lipotoxicity, there is no
evidence that curcumin is an anti-lipoapoptosis agent against
FFA-induced mitochondrial dysfunction in hepatocytes.
The overall objectives of this study were to examine
whether curcumin has a protective effect on HFFA-induced
lipoapoptosis in hepatocytes and to explore the possibility of a
mechanistic link between oxidative stress and mitochondrial
dysfunction. We showed that HFFA-induced obvious lipoapoptosis and
mitochondrial dysfunction in primary hepatocytes. Following
curcumin treatment, mitochondrial morphology and biogenesis, as
well as the mtDNA copy number, were altered. Decreased levels of
intracellular ROS and attenuated loss of the mitochondrial membrane
potential (MMP) may have contributed to the hepatoprotection.
Materials and methods
Cell culture
Hepatocyte isolation was performed as described
previously (12). Hepatocytes
were seeded at 5×106/dish in 10-cm culture dishes, and
used at 70–80% confluence. The cells were maintained at 37°C with
humidified air in a 5% CO2/95% air atmosphere.
Hepatocytes were incubated with HFFA, which were prepared according
to a slightly modified method described by Kohli et al
(13). Curcumin (purity >94%)
was purchased from Sigma (St. Louis, MO). Confluent cells were
incubated in HFFA medium and with or without curcumin with the
appropriate experimental conditions for the indicated time
points.
Cellular triglyceride assay and Oil-Red O
staining
Cellular lipids were extracted according to the
method described by Folch et al (14). The intracellular triglyceride
content was measured with a diagnostic triglyceride reagent kit
according to the manufacturer’s instructions (Antrim, UK). After
treatment, cells were washed with ice-cold PBS twice and fixed in
4% paraformaldehyde for 15 min. The cells were rinsed with 50%
isopropanol once and stained with Oil-Red O for 15 min. Finally,
the coverslips were washed and analyzed using light microscopy.
Hematoxylin was used as a counterstain.
RNA isolation and real-time PCR
analyses
Total-RNA was extracted from the hepatocytes using
the guanidinium-phenol-chloroform method. Total-RNA (5 μg) was
reverse-transcribed using the RevertAid™ First Strand cDNA
Synthesis kit according to the manufacturer’s protocol. The cDNA
was amplified using the TaqDNA polymerase kit (Vilnius, Lithuania).
RT-PCR products were separated by electrophoresis on a 3% agarose
gel and were quantified by the ImageQuant 5.2 software (Healthcare
Bio-Sciences, Philadelphia, PA). Real-time PCR was performed with a
LightCycler 1.5 apparatus (Roche Diagnostics, Mannheim, Germany)
using the LightCycler FastStart DNA MasterPLUS SYBR-Green I kit
according to the manufacturer’s protocol. Mitochondrial DNA copy
number was determined by real-time PCR as previously described
(15).
Western blotting
Cell lysates were separated by SDS-PAGE and
transferred onto a PVDF membrane. The membrane was blocked with
blocking buffer containing 5% non-fat milk, 50 mM Tris (pH 7.6),
150 mM NaCl and 0.1% Tween-20 (TBS-T) for 1 h at room temperature.
The membrane was then incubated with various primary antibodies for
8 h at 4°C and subsequently with HRP-linked secondary antibodies
for 1 h at room temperature. The signals were detected using an ECL
kit and were quantified by the ImageQuant 5.2 software.
Mitochondria mass and DNA assay and
mitochondrial respiratory complexes and ATP measurement
Mitochondria mass were detected according to the
method of Tedesco et al (16). Hepatocytes seeded onto glass
coverslips were incubated with 100 nM MitoTracker Red 580
(Invitrogen, Carlsbad, CA). After incubation for 30 min at 37°C,
coverslips were rinsed and washed. DAPI was used for nuclear
staining. Finally, the cells were fixed in 4% paraformaldehyde for
15 min and visualized using a microscope. The activity of the
mitochondrial respiratory complex was analyzed as described
(17). The ATP content of
hepatocytes was measured with the ATP bioluminescence assay kit HS
II according to the manufacturer’s protocol (Mannheim, Germany).
The luminescence value was normalized by protein concentration and
the luminescence ratio was compared with the HFFA-free group.
Flow cytometry for ROS and mitochondrial
membrane potential assay
Hepatocytes were incubated with 10 M DCFH-DA
(Molecular Probes, Inc., Eugene, OR) at 37°C for 40 min before the
termination of treatment. Cells were washed and scraped gently with
ice-cold PBS. DCF fluorescence was then detected according to the
manufacturer’s instructions. The mean fluorescence intensity of
JC-1 (BD™ MitoScreen kit) was measured to determine the MMP.
Treated cells were collected and resuspended at a concentration of
1×105/ml in PBS containing 1 g/ml JC-1, and were
incubated at 37°C for 30 min. Samples were analyzed by flow
cytometry using a FACSCalibur (BD Biosciences). The data are
presented in terms of relative fluorescence percentage.
Immunofluoresence staining
Hepatocytes (1×103 cells) were cultured
on glass coverslips. After incubation with HFFA and with or without
curcumin, the cells were fixed in 4% paraformaldehyde for 15 min,
washed with ice-cold PBS and blocked with 7.5% normal goat serum
for 30 min at room temperature. After washing with ice-cold PBS
twice, the cells were incubated with anti-PEPCK,
anti-glucose-6-phosphatase (G6Pase), anti-NF-κB antibodies for 1 h
at room temperature. After washes with ice-cold PBS twice, the
cells were incubated with diluted FITC-conjugated secondary
antibody for 1 h at room temperature. In addition, DAPI was used
for nuclear staining. The slides were mounted in mounting medium
and visualized using a microscope (Olympus, Japan).
Statistical analysis
Data are presented as the mean ± SEM. The
statistical analyses were performed using a one-way analysis of
variance followed by the Student Newman-Keuls multiple-range test.
P<0.05 denote statically significant differences.
Results
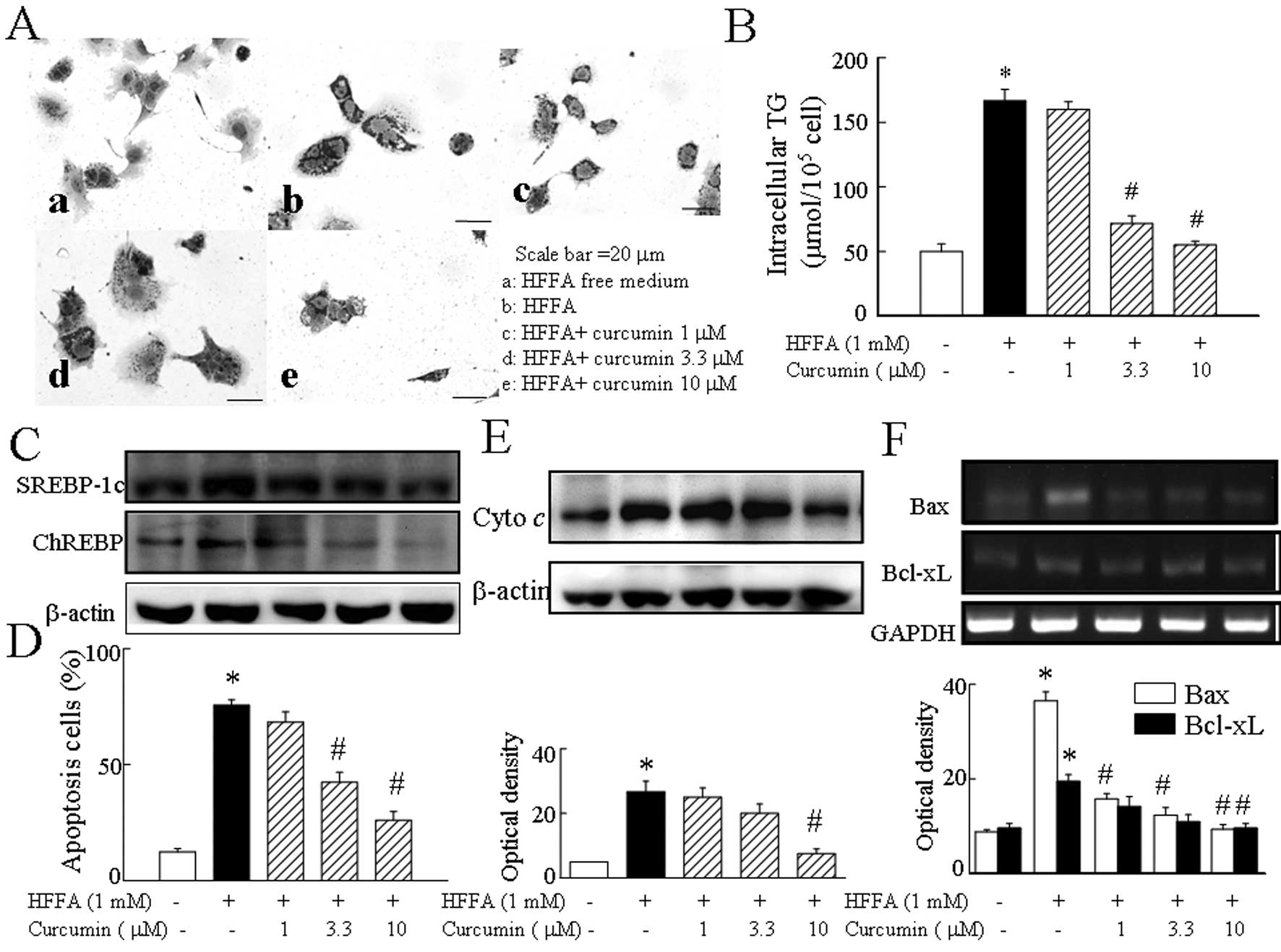
The results from the Oil-Red O staining and from the
intracellular triglyceride assay indicate that fat accumulation was
reduced by curcumin compared with 1 mM HFFA alone (Fig. 1A and B). This effect was
especially visible at 10 μM curcumin. SREBP-1c and ChREBP protein
levels were clearly suppressed by treatment with 10 μM curcumin
(Fig. 1C). The results indicated
that suppression of the HFFA-mediated increase of cellular
triglycerides by curcumin may be associated with downregulation of
the expression of lipogenic factors. HFFA treatment significantly
increased the percentage of positively stained (i.e. apoptotic)
cells to 80% in cultured hepatocytes, whereas curcumin co-treatment
reduced the proportion of apoptotic cells to 30% in hepatocytes
(Fig. 1D). Release of cytochrome
c (Fig. 1E) and Bax (Fig. 1F) into the cytosol with HFFA was
confirmed by immunoblot or RT-PCR analysis of cytosolic fraction.
Curcumin significantly counteracted the HFFA-induced upregulation
of apoptosis-inducing factors.
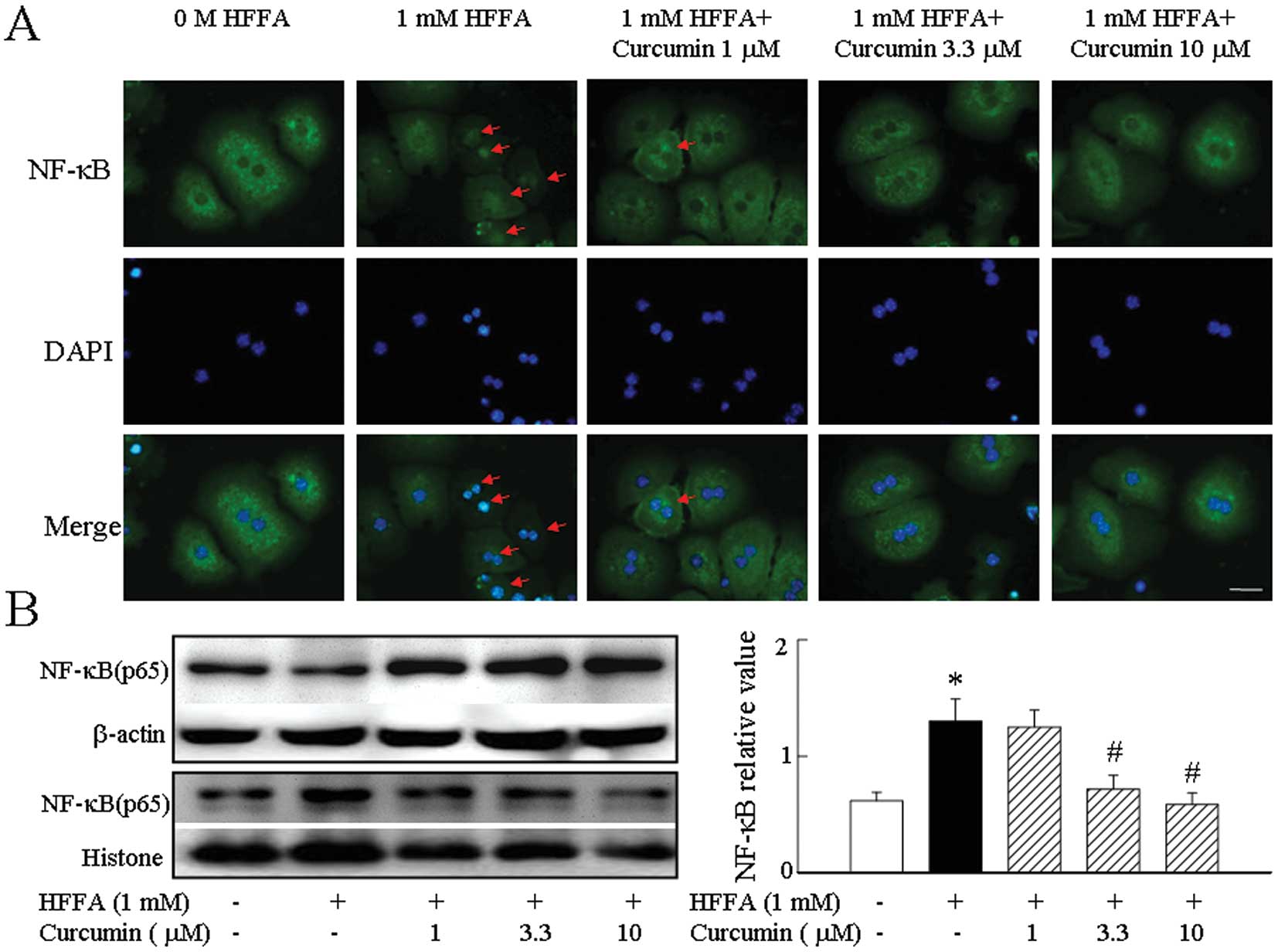
We found that the p65 subunit of NF-κB was
distributed in the cytoplasm in all cells before HFFA stimulation.
Treatment with HFFA resulted in a marked accumulation of p65 in
nuclei after 2 h. HFFA activated NF-κB in hepatocytes and curcumin
was a potent inhibitor of NF-κB activity (Fig. 2A). We further confirmed these
results by western blot analysis to probe nuclear and cytoplasmic
extracts using an antibody specific for the p65 subunit of NF-κB.
Treatment of hepatocytes with curcumin followed by exposure to HFFA
significantly inhibited NF-κB translocation to the nucleus and
returned the p65 subunit of NF-κB into the cytoplasm (Fig. 2B).
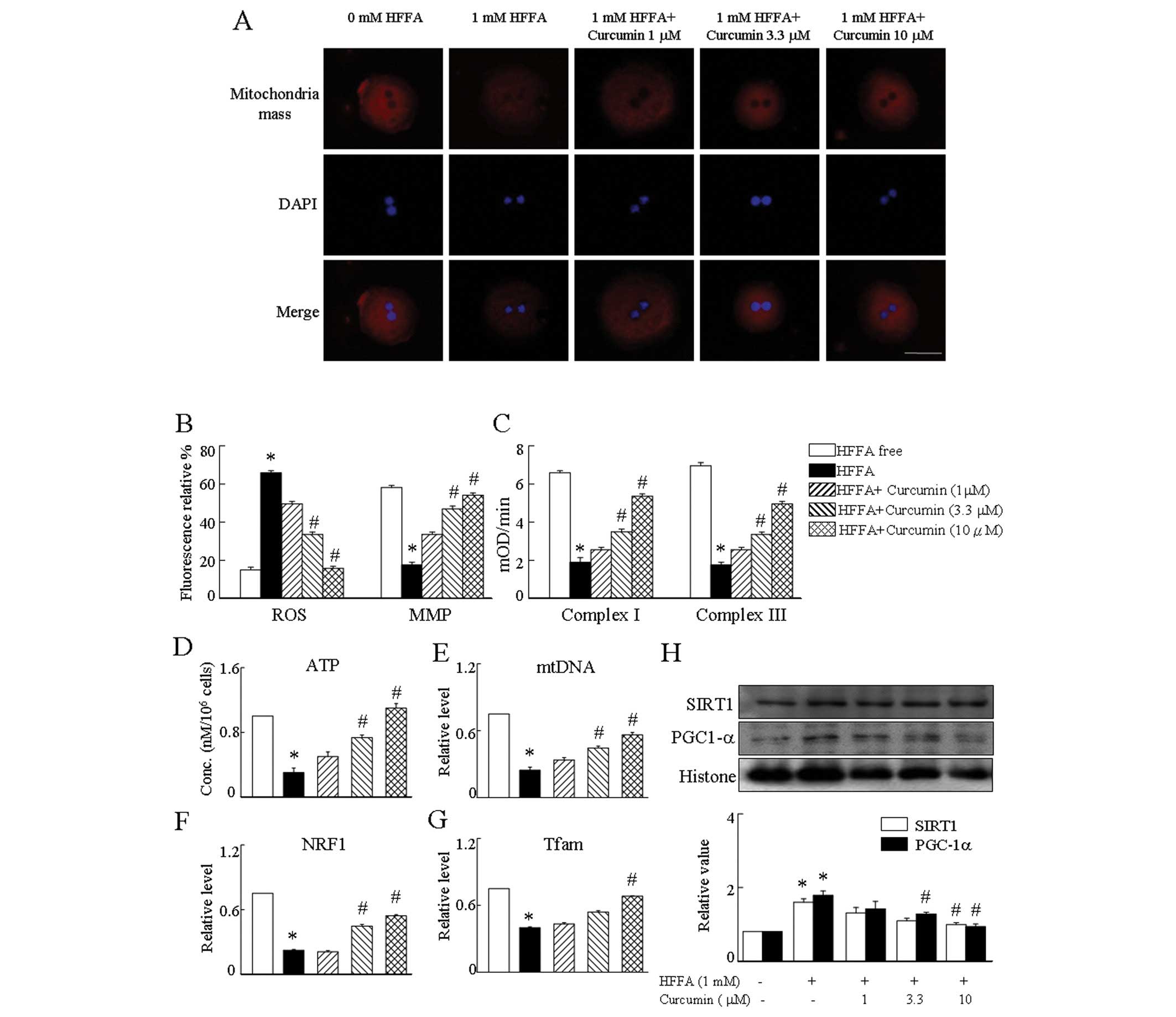
Hepatocytes treated with HFFA showed a significant
decrease of the MitoTracker Red signal, indicating the decrease of
mitochondrial function compared to HFFA-free cells (Fig. 3A). To verify that the inhibition
of mitochondria mass signal by HFFA is a relevant molecular
mechanism by which mitochondrial biogenesis is affected,
hepatocytes were treated with 1 mM HFFA in the presence of curcumin
corcumin prevented the effect of HFFA on mitochondrial biogenesis
with a full restoration of ATP levels (Fig. 3D), NRF1, Tfam mRNA expression
(Fig. 3F and G), PGC-1α and SIRT1
protein levels (Fig. 3H), and the
mtDNA amount in hepatocytes (Fig.
3E). As mitochondrial biogenesis was affected by treatment with
curcumin, we investigated whether these treatments affected
mitochondrial respiratory complexes (MRC). We found that complex I
and III activities were altered after incubation with curcumin
prevented the reduction of MRC in HFFA-treated cells (Fig. 3C). We next sought to determine
whether HFFA affects ROS content and MMP. ROS levels in hepatocytes
treated with HFFA were much higher than that of control cells.
Conversely, HFFA led to a large reduction in MMP. As expected, the
results demonstrated that the presence of curcumin suppressed the
ROS generation and upregulation of MMP expression in HFFA-treated
hepatocytes. These observations suggest that curcumin can attenuate
HFFA-induced aspects of mitochondrial dysfunction, including
depression of the MMP and production of ROS (Fig. 3B).
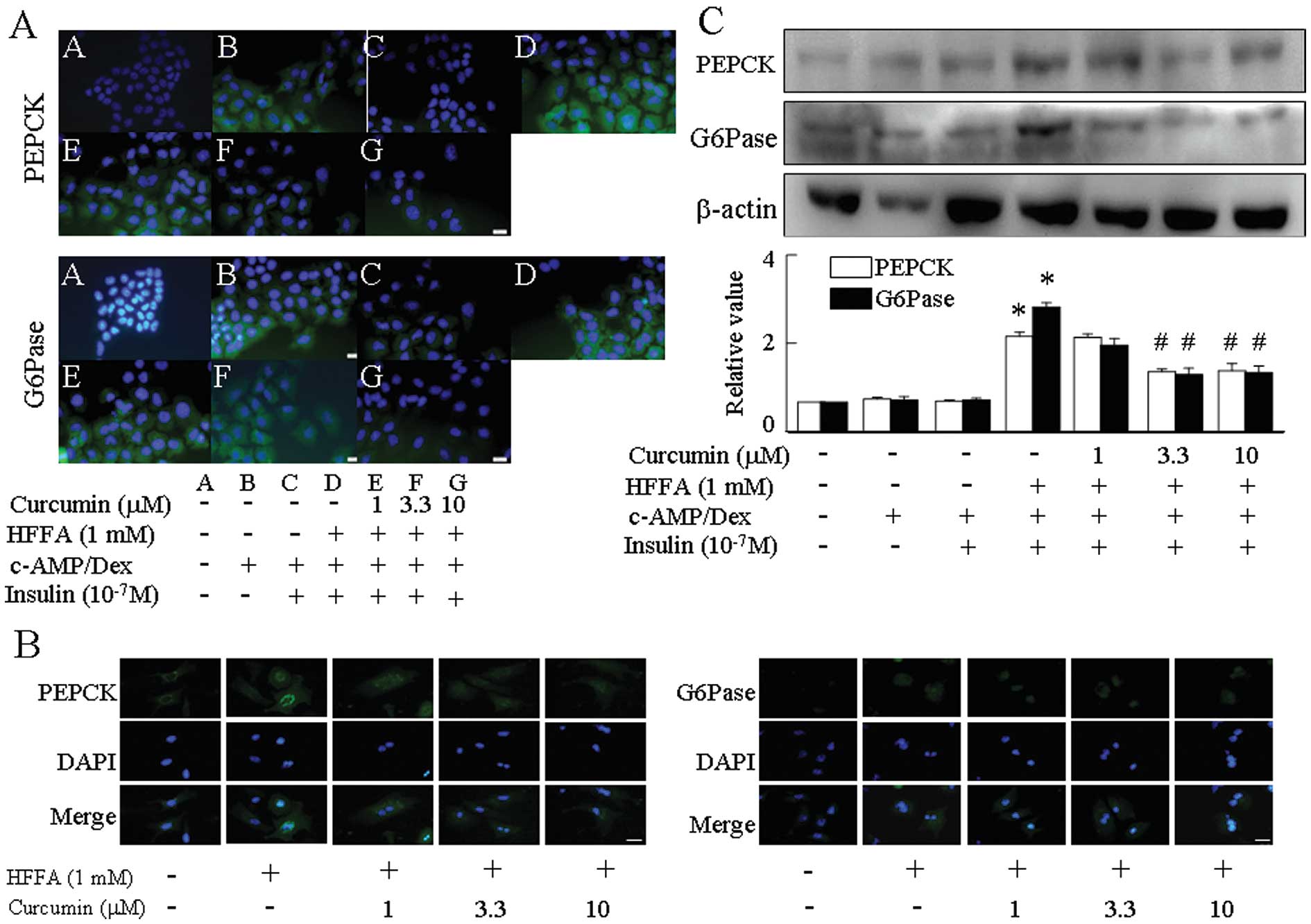
The expression of two key hepatic gluconeogenic
genes, PEPCK and G6Pase, increased in response to
cAMP/dexamethasone (Dex) and was reduced in response to insulin. By
immunofluorescence, treatment with 1 mM HFFA abrogated the insulin
suppression of cAMP/Dex-mediated PEPCK and G6Pase levels when
compared with HFFA-free medium (Fig.
4A and B). Curcumin repressed the expression of PEPCK and
G6Pase in a dose-dependent manner in HFFA-induced aspects of
gluconeogenesis (Fig. 4C).
Discussion
NAFLD is characterized by an elevated serum
concentration of FFAs, liver steatosis and hepatocyte apoptosis
(1,2). Excess free fatty acids may impair
normal cell signaling, causing cellular dysfunction or induced
lipoapoptosis (18). Others have
suggested that mitochondrial dysfunction plays a central role in
FFA-induced apoptosis (19). In
this study, we demonstrated that curcumin could protect hepatocytes
from FFA-induced oxidative damage and could reverse FFA-stimulated
lipoapoptosis. The mechanism of action may be related to
improvements in mitochondrial function and biogenesis. Our results
suggest that curcumin or a similar compound could be used to treat
NAFLD or NASH.
Previous study has shown that apoptotic cells
undergo mitochondrial perturbations including loss of the MMP and
generation of ROS (20). In
addition, the redox-sensitive nuclear transcriptional factor,
NF-κB, acts as a proapoptotic factor in the pathogenesis of NAFLD
and NF-κB activation plays a critical role in the pathophysiology
of cytokine-mediated hypoglycaemia (21,22). It is therefore reasonable to
suppose that the interactions between genes of apoptosis and
gluconeogenesis are regulated by NF-κB. In this study, curcumin has
been shown to have profound effects on mitochondrial function.
Curcumin effectively prevented the loss of MMP in hepatocytes in
response to HFFA challenge, suggesting that curcumin either
increases the expression of a protein that acts to decrease the
activity of a caspase that lies upstream of the mitochondria in the
apoptotic cascade or decreases activation of the NF-κB cascade
(either directly or indirectly). In fact, curcumin has been shown
to act directly on some NF-κB-dependent genes (23). We demonstrate that suppression of
NF-κB enhances the ability of curcumin to rescue hepatocytes from
HFFA-induced lipoapoptosis, strongly suggesting that a decrease in
NF-κB activity by curcumin is at least partially responsible for
this rescue of mitochondrial function. Another attractive idea is
that curcumin might inhibit the decline in ATP production by
affecting energy metabolism in the mitochondria. In the current
study, we observed that curcumin inhibited triglyceride
accumulation in HFFA-treated hepatocytes. The expression of
lipogenic transcription factors (SREBP-1 and ChREBP) was also
reduced. Given that ATP is required for the expression and function
of the lipogenic factors, restoring the ATP levels provides a
potential mechanism to counteract the inhibition of lipogenesis by
curcumin.
Abnormalities in lipid metabolism within hepatocytes
are still poorly understood. Accumulating evidence indicates that
mitochondrial dysfunction plays a central role in the pathogenesis
of NAFLD and that NAFLD is a mitochondrial disease (24). A number of mechanisms may explain
the mitochondrial dysfunction observed in NAFLD patients. One of
these mechanisms is oxidative stress. ROS-induced depletion of
mtDNA can affect mitochondrial function and induces hepatic
steatosis (25). The HFFA-induced
decrease in the mitochondrial respiratory chain (MRC) activity in
hepatocytes is partly due to oxidative stress, as treating cells
with a potent antioxidant, curcumin, improved the activity of
complexes I and III of the MRC. Previous studies showed that the
activity of MRC complexes is decreased in liver tissue from
patients with NASH (26). In the
current study, we confirm that the activity of MRC complexes I and
III are reduced by 30 or 70%, respectively. These results were
similar to those found in NASH patients and indicate that this
in vitro model can be used to investigate the mechanisms of
mitochondrial dysfunction.
Insulin resistance and oxidative stress both
contribute to the pathogenesis of NAFLD and the progression from
steatosis to steatohepatitis (27). High levels of FFA in the plasma
increase hepatic FFA uptake, whereas high insulin levels may
increase FFA synthesis concomitant with hepatic insulin resistance
in some obese individuals (28).
In our current results, the promotion of PEPCK and G6Pase
expression is associated with increased expression of SREBP-1 and
ChREBP in HFFA-treated hepatocytes. These processes demonstrate the
close interrelation between gluconeogenesis and hepatic lipid
metabolism and indicate that NAFLD involves dysregulation in
lipogenesis and gluconeogenesis-mediated hepatic insulin signaling.
It is known that PGC-1α is a coactivator with pleiotropic functions
such as controlling mitochondrial biogenesis and function; both
functions are vital links in the regulatory network for metabolic
homeostasis (29). In our study,
curcumin upregulated mitochondrial biogenesis through reversal of
HFFA-induced upregulation of PGC-1α levels, thereby supporting the
potential utility of curcumin to improve the metabolic status in
patients with fatty liver. For the first time, this study
demonstrated a curcumin-mediated increase in the expression of
genes regulating hepatic mitochondrial biogenesis, including,
PGC-1α, NRF-1 and Tfam, and an increase in the expression of genes
that commonly regulate mtDNA content. Additionally, curcumin may be
involved directly in the modulation of gluconeogenesis. This
observation is supported by previous studies showing that curcumin
suppressed gluconeogenesis and glycemic control (30,31). Thus, it is reasonable to
hypothesize that a curcumin-induced blockade of lipotoxicity may
directly prevent mitochondrial dysfunction and gluconeogenesis.
Taken together, our data suggest that curcumin may serve as a
potential therapeutic approach to ameliorate the cytotoxicity of
HFFA by acting through multiple pathways involved in mitochondrial
function.
Acknowledgements
This study was supported by grant no.
CCMP100-RD-016 from the Committee on Chinese Medicine and Pharmacy,
Department of Health; grant no. NSC98-2320-B-182-021-MY3 from the
National Science Council, Taiwan; and grant no. CMRPD180242 from
the Chang Gung Memorial Hospital, Linkuo, Taiwan.
References
|
1.
|
AE FeldsteinA CanbayME GuicciardiH
HiguchiSF BronkGJ GoresDiet associated hepatic steatosis sensitizes
to Fas mediated liver injury in miceJ
Hepatol39978983200310.1016/S0168-8278(03)00460-414642615
|
|
2.
|
AE FeldsteinA CanbayP AnguloM TaniaiLJ
BurgartKD LindorGJ GoresHepatocyte apoptosis and fas expression are
prominent features of human nonalcoholic
steatohepatitisGastroenterology125437443200310.1016/S0016-5085(03)00907-712891546
|
|
3.
|
SK MantenaDP VaughnKK AndringaHigh fat
diet induces dysregulation of hepatic oxygen gradients and
mitochondrial function in vivoBiochem
J417183193200910.1042/BJ2008086818752470
|
|
4.
|
MS RaoJK ReddyPPARalpha in the
pathogenesis of fatty liver
diseaseHepatology40783786200410.1002/hep.2045315382158
|
|
5.
|
MA O’ConnellSA RushworthCurcumin:
potential for hepatic fibrosis therapyBr J
Pharmacol1534034052008
|
|
6.
|
Y FuS ZhengJ LinJ RyerseA ChenCurcumin
protects the rat liver from CCl4-caused injury and
fibrogenesis by attenuating oxidative stress and suppressing
inflammationMol Pharmacol73399409200818006644
|
|
7.
|
S ZhengA ChenCurcumin suppresses the
expression of extracellular matrix genes in activated hepatic
stellate cells by inhibiting gene expression of connective tissue
growth factorAm J Physiol Gastrointest Liver
Physiol290G883G893200610.1152/ajpgi.00450.2005
|
|
8.
|
Y TangS ZhengA ChenCurcumin eliminates
leptin’s effects on hepatic stellate cell activation via
interrupting leptin signalingEndocrinology150301130202009
|
|
9.
|
AC BhartiY TakadaBB AggarwalCurcumin
(diferuloylmethane) inhibits receptor activator of NF-kappa B
ligand-induced NF-kappa B activation in osteoclast precursors and
suppresses osteoclastogenesisJ
Immunol17259405947200410.4049/jimmunol.172.10.5940
|
|
10.
|
BE BachmeierIV MohrenzV MirisolaCurcumin
downregulates the inflammatory cytokines CXCL1 and -2 in breast
cancer cells via
NFkappaBCarcinogenesis29779789200810.1093/carcin/bgm24817999991
|
|
11.
|
SL WangY LiY WenYF ChenLX NaST LiCH
SunCurcumin, a potential inhibitor of up-regulation of TNF-alpha
and IL-6 induced by palmitate in 3T3-L1 adipocytes through
NF-kappaB and JNK pathwayBiomed Environ
Sci223239200910.1016/S0895-3988(09)60019-219462685
|
|
12.
|
TY LeeFY ChenHH ChangHC LinThe effect of
capillarisin on glycochenodeoxycholic acid-induced apoptosis and
heme oxygenase-1 in rat primary hepatocytesMol Cell
Biochem3255359200910.1007/s11010-008-0019-819132499
|
|
13.
|
R KohliX PanP MalladiMS WainwrightPF
WhitingtonMitochondrial reactive oxygen species signal hepatocyte
steatosis by regulating the phosphatidylinositol 3-kinase cell
survival pathwayJ Biol
Chem2822132721336200710.1074/jbc.M70175920017540768
|
|
14.
|
J FolchM LeesGH Sloane StanleyA simple
method for the isolation and purification of total lipides from
animal tissuesJ Biol Chem226497509195713428781
|
|
15.
|
L TedescoA ValerioC CervinoCannabinoid
type 1 receptor blockade promotes mitochondrial biogenesis through
endothelial nitric oxide synthase expression in white
adipocytesDiabetes5720282036200810.2337/db07-1623
|
|
16.
|
L TedescoA ValerioM DossenaCannabinoid
receptor stimulation impairs mitochondrial biogenesis in mouse
white adipose tissue, muscle, and liver: the role of eNOS, p38
MAPK, and AMPK pathwaysDiabetes5928262836201010.2337/db09-1881
|
|
17.
|
R GonzálezG FerrínAB
HidalgoN-acetylcysteine, coenzyme Q10 and superoxide dismutase
mimetic prevent mitochondrial cell dysfunction and cell death
induced by d-galactosamine in primary culture of human
hepatocytesChem Biol Interact18195106200919523936
|
|
18.
|
JE SchafferLipotoxicity: when tissues
overeatCurr Opin
Lipidol14281287200310.1097/00041433-200306000-0000812840659
|
|
19.
|
Y WeiRS RectorJP ThyfaultJA
IbdahNonalcoholic fatty liver disease and mitochondrial
dysfunctionWorld J
Gastroenterol14193199200810.3748/wjg.14.19318186554
|
|
20.
|
T HirschI MarzoG KroemerRole of the
mitochondrial permeability transition pore in apoptosisBiosci
Rep176776199710.1023/A:10273394186839171922
|
|
21.
|
L LiL ChenL HuNuclear factor high-mobility
group box1 mediating the activation of Toll-like receptor 4
signaling in hepatocytes in the early stage of nonalcoholic fatty
liver disease in miceHepatology5416201630201110.1002/hep.24552
|
|
22.
|
SF LiuAB MalikNF-kappa B activation as a
pathological mechanism of septic shock and inflammationAm J Physiol
Lung Cell Mol
Physiol290L622L645200610.1152/ajplung.00477.200516531564
|
|
23.
|
P RafieeDG BinionM WellnerModulatory
effect of curcumin on survival of irradiated human intestinal
microvascular endothelial cells: role of Akt/mTOR and NF-kappaBAm J
Physiol Gastrointest Liver
Physiol298G865G877201010.1152/ajpgi.00339.200920299603
|
|
24.
|
D PessayreB FromentyNASH: a mitochondrial
diseaseJ Hepatol42928940200510.1016/j.jhep.2005.03.00415885365
|
|
25.
|
C DemeilliersC MaisonneuveA GrodetImpaired
adaptive resynthesis and prolonged depletion of hepatic
mitochondrial DNA after repeated alcohol binges in
miceGastroenterology12312781290200210.1053/gast.2002.3595212360488
|
|
26.
|
M Pérez-CarrerasP Del HoyoMA
MartínDefective hepatic mitochondrial respiratory chain in patients
with nonalcoholic
steatohepatitisHepatology389991007200314512887
|
|
27.
|
LA VidelaR RodrigoJ ArayaJ
PoniachikInsulin resistance and oxidative stress interdependency in
non-alcoholic fatty liver diseaseTrends Mol
Med12555558200610.1016/j.molmed.2006.10.00117049925
|
|
28.
|
EJ GallagherD LeroithE KarnieliInsulin
resistance in obesity as the underlying cause for the metabolic
syndromeMt Sinai J Med77511523201010.1002/msj.2021220960553
|
|
29.
|
J LinH WuPT TarrTranscriptional
co-activator PGC-1 alpha drives the formation of slow-twitch muscle
fibresNature15797801200210.1038/nature0090412181572
|
|
30.
|
SP WeisbergR LeibelDV TortorielloDietary
curcumin significantly improves obesity-associated inflammation and
diabetes in mouse models of
diabesityEndocrinology14935493558200810.1210/en.2008-026218403477
|
|
31.
|
T KimJ DavisAJ ZhangX HeST MathewsCurcumin
activates AMPK and suppresses gluconeogenic gene expression in
hepatoma cellsBiochem Biophys Res
Commun388377382200910.1016/j.bbrc.2009.08.01819665995
|