Introduction
Fabry disease (OMIM 301500), also known as
Anderson-Fabry disease, is a rare X-linked inherited lysosomal
storage disorder (LSD) caused by deficiency of α-galactosidase A
(α-gal A; EC 3.2.1.22) (1,2). A
lack of lysosomal α-gal A enzyme causes accumulation of
glycosphingolipids, mainly globotriaosylceramide (also known as
Gb3, GL-3, or ceramide trihexoside) within the vascular endothelium
in the brain, heart, liver, spleen, eyes, skin and kidney, leading
to cerebrovascular, cardiac and renal complications (3). Renal insufficiency is observed in
the final stage of life in a Fabry patient. However, structural
changes in the kidney, including glomerulosclerosis and
interstitial fibrosis, are observed even in young Fabry patients
(4).
The Fabry mouse model has been established by
disruption of the α-gal A-encoding gene (5). This knockout mouse model appears to
be clinically normal, but ultrastructural analysis shows lipid
inclusions in the liver and kidney. These pathophysiological
changes in the α-gal A-knockout mouse are similar to those in
patients with Fabry disease. An in vitro vascular cell model
has been generated by culturing aortic endothelial cells from α-gal
A-null mice (6). In the vascular
endothelium of α-gal A-null mice, the deposition of Gb3 increases
with age. Excessive Gb3 deposition in vascular endothelial cells is
related to increased thrombosis and atherogenesis (7,8).
The pathological mechanisms by which endothelial dysfunction occurs
are poorly characterized. However, the pathogenic context of other
kidney diseases including diabetes can provide clues for
understanding the nephropathy of Fabry disease (9). Intracellular signaling proteins and
growth factors such as insulin-like growth factors, transforming
growth factor-β1 (TGF-β1) and vascular endothelial growth factor
(VEGF) play a role in the development of diabetic nephropathy.
These growth factors might also be related to the renal
complication in patients with Fabry disease (9). TGF-β1 is known to play a key role in
many diseases such as diabetes and renal disease (10,11). The TGF-β1 signaling pathway is
mediated by mitogen-activated protein kinases (MAPKs), and TGF-β1
induces apoptosis in endothelial cells (12). VEGF is important in angiogenesis.
VEGF binds to the VEGF receptors, fms-like tyrosine kinase [Flt-1,
VEGF receptor 1 (VEGFR1)] and fetal liver kinase 1 (Flk-1, KDR,
VEGFR2). VEGF expression is regulated by hypoxia, cytokines and
growth factors (13). VEGF is
expressed in glomerular podocytes and tubular epithelial cells.
Some investigators have proposed that the expression
of TGF-β1 or VEGF is associated with Fabry nephropathy. Proteomic
studies have demonstrated that circulating levels of VEGF and
VEGFR2 are higher in young Fabry patients compared with controls
(14,15). These observations suggest that
TGF-β1 and VEGF expression is associated with Fabry nephropathy. We
explored the roles of TGF-β1 and VEGF in the relationship between
increased levels of Gb3 and endothelial dysfunction in renal
pathogenesis in a mouse model of Fabry disease.
Materials and methods
Animals
Fabry mice were kindly provided by Dr Roscoe O.
Brady (National Institutes of Health, Bethesda, MD, USA) and bred
to produce sufficient numbers of mice for this study. To genotype
each mouse, PCR was performed as described previously (5). Male mice were grouped into wild-type
and hemizygous (Fabry) mice. Each group included a minimum of three
animals. Sixteen-week-old mice were used for all experiments. All
mice were provided with autoclaved water and diet ad
libitum. All mice were treated in accordance with the Animal
Care Guidelines of the School of Medicine, Ewha Womans University
(Seoul, Korea). Fabry mice were treated with an injection of 1 mg
Fabrazyme/kg (Genzyme, Cambridge, MA, USA) in saline through the
tail vein.
Cell culture
For in vitro study, bovine aortic endothelial
cells (BAECs) were cultured in minimum essential medium (MEM)
supplemented with 5% neonatal calf serum, 2 mM l-glutamine and
penicillin/streptomycin (100 U/ml) at 37°C in a humidified 5%
CO2 incubator. BAECs were used up to passages 7–9. All
reagents were purchased from Gibco-BRL (Carlsbad, CA, USA).
Western blot analysis
Proteins obtained from 50 mg of renal tissue were
homogenized using Pro-Prep buffer (iNtRON Biotechnology, Inc.,
Seongnam-si, Korea) supplemented with phosphatase inhibitor
cocktail solution (Dawinbio, Inc., Hanam-si, Korea). Tissue samples
were incubated on ice for 30 min. Gb3-treated BAECs (80% confluent)
were washed with ice-cold PBS and resuspended in PRO-PREP buffer
supplemented with phosphatase inhibitor cocktail solution for 30
min on ice. Insoluble material was removed by centrifugation at
12,000 × g for 10 min at 4°C. Proteins (30–80 μg) were
separated on a 7.5–13.5% SDS-polyacrylamide gel and
electrophoretically transferred to a polyvinylidene fluoride or
nitrocellulose membrane (Millipore, Billerica, MA, USA). The
membranes were blocked with 5% skim milk in Tris-buffered saline
containing 0.1% Tween 20 (TBST) for 2 h at room temperature. The
blots were then incubated with primary antibody overnight at 4°C.
Antibodies used for western blot analysis were anti-thrombospondin
(TSP)-1 monoclonal (NeoMarker, Oviedo, FL, USA); anti-cleaved
cysteine aspatric acid protease (caspase)-6 polyclonal;
anti-cleaved caspase-9 polyclonal; anti-cleaved caspase-12,
phospho-p38 (P-p38) polyclonal; anti-VEGFR2 polyclonal (Cell
Signaling Technology, Danvers, MA, USA); anti-fibroblast growth
factor 2 (FGF-2) polyclonal (Abfrontier, Seoul, Korea); anti-TGF-β1
monoclonal (R&D Systems, Minneapolis, MN, USA); and anti-VEGF
monoclonal (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA)
antibodies. The blots were washed with TBST three times for 5 min
and then incubated with horseradish peroxidase-labeled secondary
antibody for 1 h at room temperature. Goat anti-mouse IgG (1:2,500;
Santa Cruz Biotechnology, Inc.) and goat anti-rabbit IgG (1:2,500;
Santa Cruz Biotechnology, Inc.) were used as the secondary
antibodies. After additional washes, the blots were detected by
enhanced chemiluminescence using an ECL detection kit (GE
Healthcare, Buckinghamshire, UK) according to the manufacturer’s
instructions. The protein signals were visualized by exposing the
membranes in a luminescent image analyzer (LAS-3000; Fujifilm,
Tokyo, Japan). Each protein expression level was normalized to the
expression of β-actin (Sigma-Aldrich, St. Louis, MO, USA). The
results were quantified using Multi Gauge V3.0 software.
Caspase-3/7 assay
Fifty micrograms of protein from kidney lysates (100
μl) and caspase-3/7 substrate-containing solution (100
μl) were mixed and incubated for 30 min to 1 h at 30°C in
96-well tissue culture test plates (SPL Life Sciences, Pocheon-si,
Korea). The activity was measured on a Veritas Microplate
Luminometer instrument (Promega Corporation, Madison, WI, USA).
Caspase-3/7 substrate was purchased from Promega Corporation.
Gb3 treatment
BAECs were seeded in 60 mm tissue culture plates
(SPL Life Sciences) and were either untreated or treated with Gb3
(Matreya, Pleasant Gap, PA, USA) in complete MEM culture medium.
The final Gb3 [dissolved in 100% dimethyl sulfoxide (DMSO;
Sigma-Aldrich)] concentration of the treatment was 15
μM.
Data analysis and statistics
The values are presented as the mean ± SD or ± SE.
Statistical comparisons between groups were performed using the
Student’s t-test. P<0.05 was considered to indicate a
statistically significant result.
Results
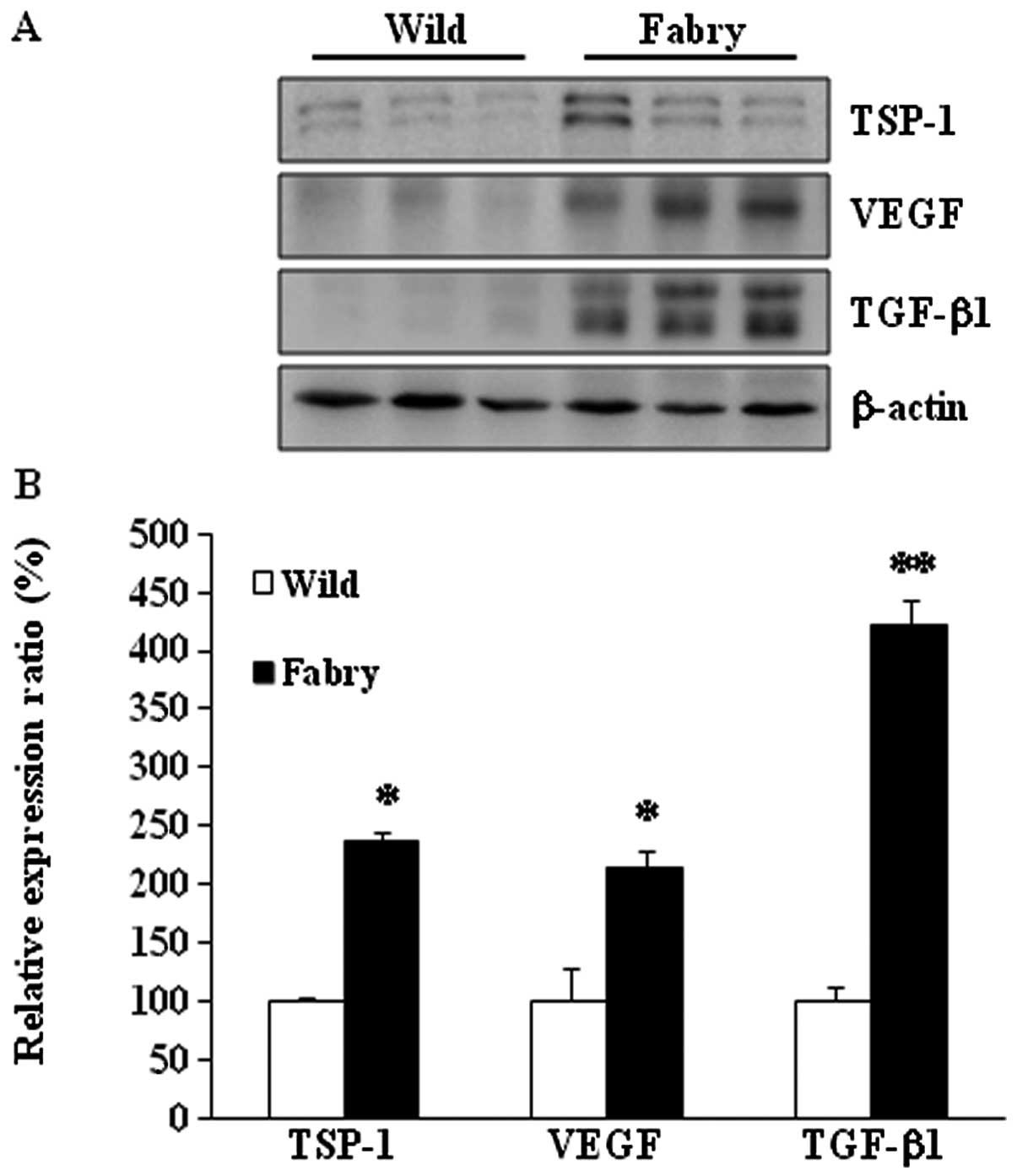
Protein expression of TSP-1, VEGF and
TGF-β1 in kidneys of Fabry mice
The expression patterns of TSP-1, VEGF and TGF-β1
proteins were examined in the kidneys from Fabry and wild-type
mice. As expected, TSP-1 protein expression was 234% higher in
Fabry than in wild-type mice (Fig.
1) (P<0.05). VEGF protein expression was 214% higher in
Fabry mice (P<0.05). TGF-β1 protein expression was 422% higher
in Fabry than in wild-type mice (P<0.01). These results suggest
that VEGF expression is influenced through another pathway
associated with TGF-β1.
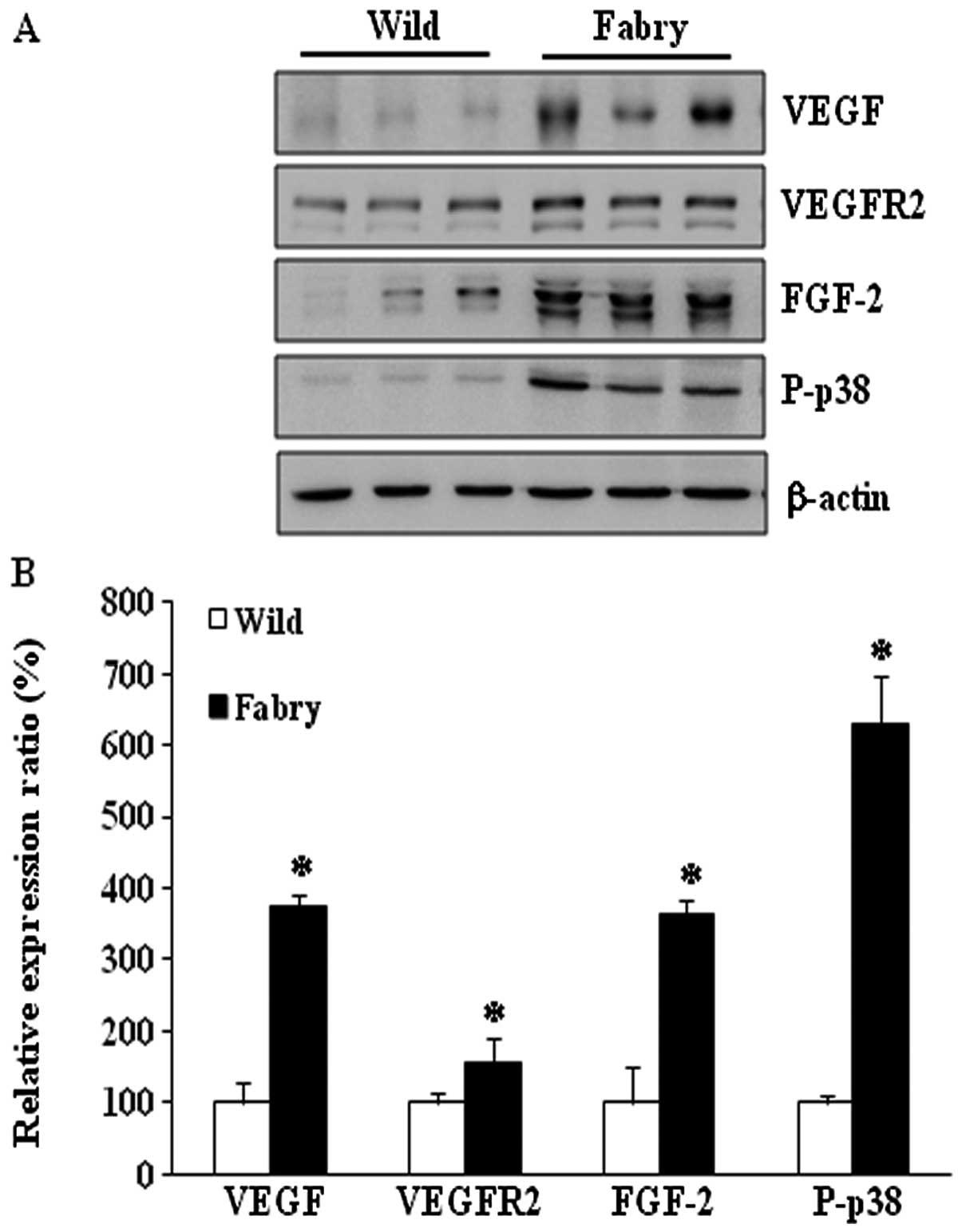
Protein expression of VEGFR2, FGF-2 and
P-p38 in kidneys of Fabry mice
Western blot analysis was performed to test the
hypothesis that the combined overexpression of TGF-β1 and VEGF is
associated with Fabry disease nephropathy. The protein expression
of VEGFR2 was 157% higher in kidneys from Fabry than that in
wild-type mice. FGF-2 expression was 365% higher in Fabry mice and
P-p38 was 631% higher in Fabry than these levels in wild-type mice
(Fig. 2). These results suggest
that apoptosis signaling may be induced by the increased expression
of TGF-β1 and VEGF in the kidney in the Fabry disease mouse
model.
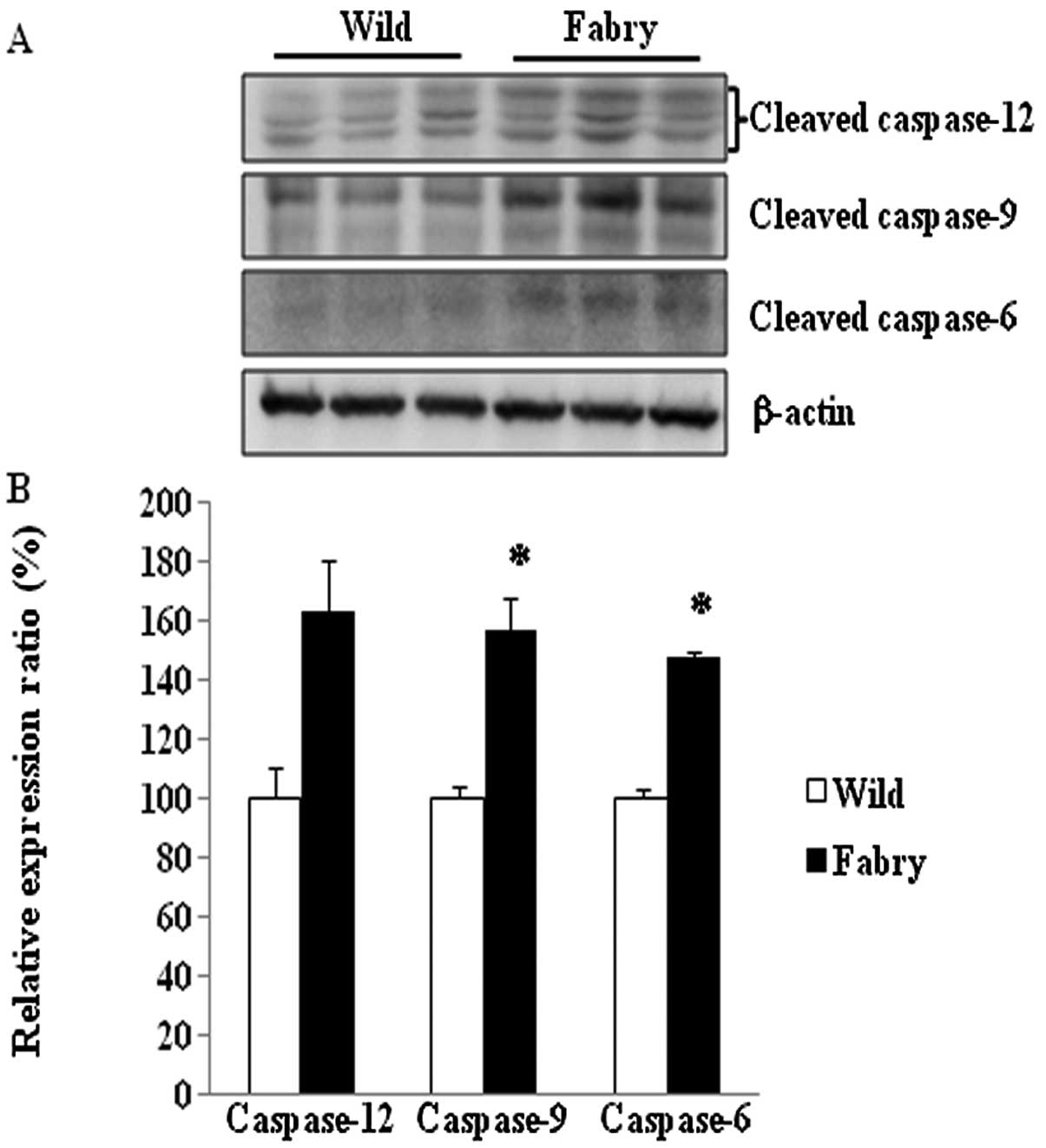
Activation of caspases in the Fabry mouse
kidney
The degree of activation of caspase-6, -9 and -12
was determined by measuring the levels of the cleaved forms of
these caspases. The results shown in Fig. 3 indicate that the levels of
cleaved caspase-6 (148%, P<0.05) and cleaved caspase-9 (157%,
P<0.05) were significantly higher in Fabry mice than in
wild-type mice. Cleaved caspase-12 level increased nonsignificantly
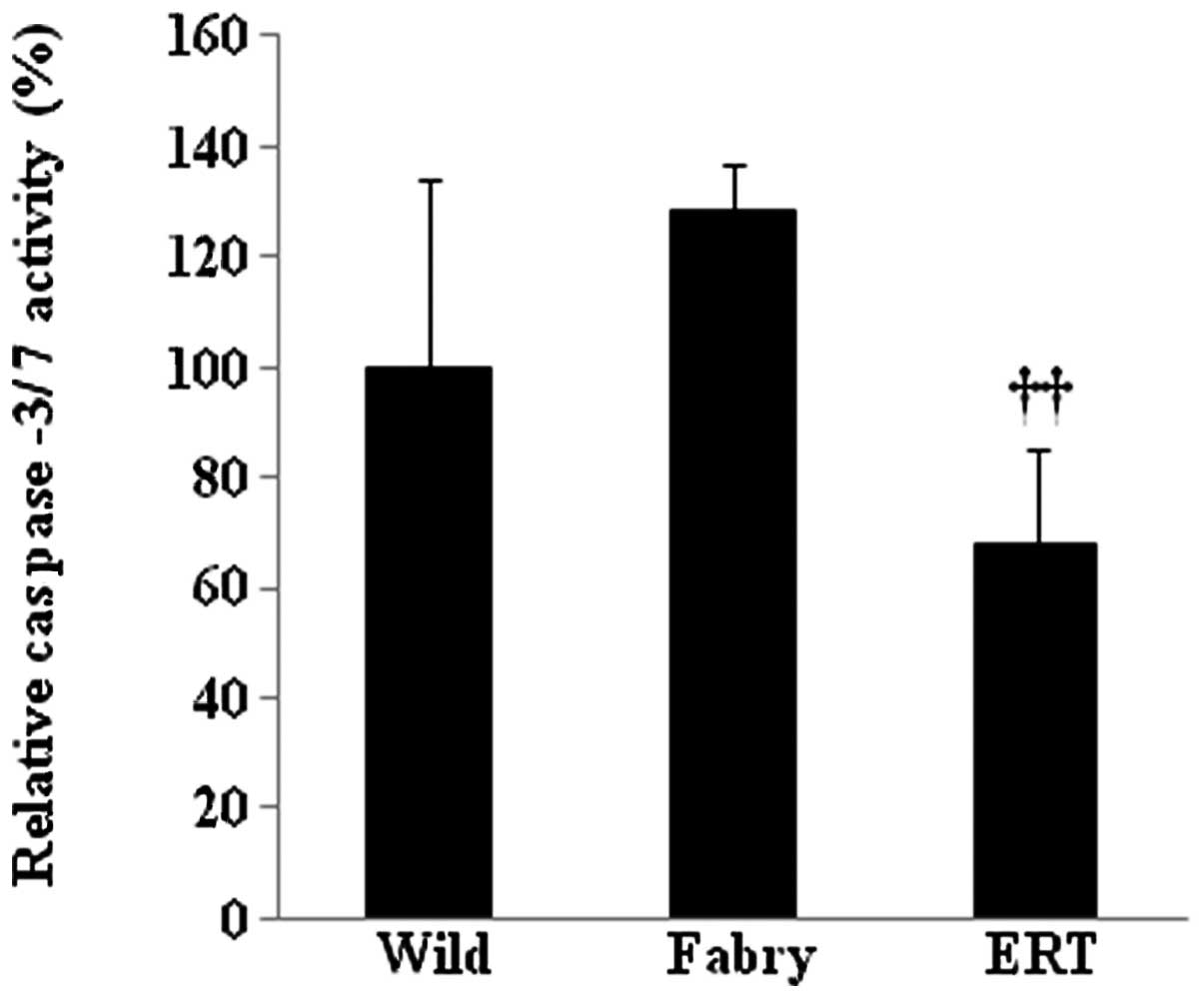
(163%, P=0.06). To confirm that apoptosis was induced in the
kidneys of Fabry mice, caspase-3/7 activity of key apoptotic
proteins was measured using a colorimetric assay. The results shown
in Fig. 4 indicate that
caspase-3/7 activity was 128% higher in the kidney lysates from
Fabry mice than in those from wild-type mice. In enzyme replacement
therapy (ERT)-treated Fabry mice, activity of caspase-3/7 was 68%
lower than that in the untreated Fabry mice (P<0.01).
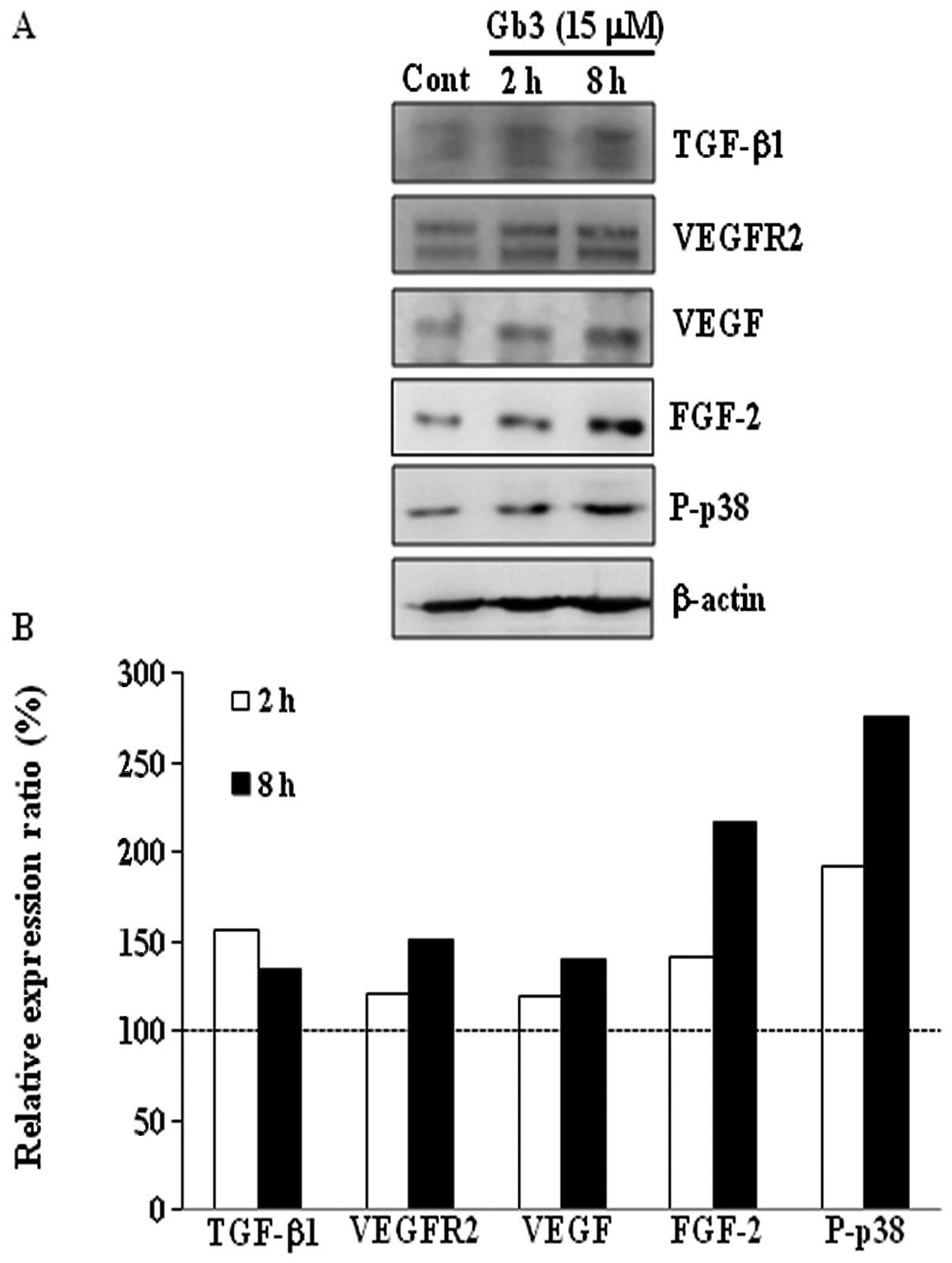
Effect of in vitro Gb3 treatment on BAECs
from Fabry mice
To investigate whether Gb3 accumulation induces the
expression of TGF-β1 and VEGF in vitro in BAECs in a manner
similar to that observed in Fabry mice, BAECs were treated either
with control (vehicle, DMSO alone) or 15 μM Gb3 for 2 or 8 h
(Fig. 5). The protein expression
levels of TGF-β1, VEGFR2, VEGF, FGF-2 and P-p38 were higher in
Gb3-treated BAECs than in control BAECs. Expression of these
proteins was higher in BAECs treated with Gb3 for 8 h compared with
2 h.
Discussion
Endothelial dysfunction related to excess Gb3 leads
to renal complications in Fabry disease (3); however, the pathological mechanism
responsible for the endothelial dysfunction caused by Gb3
accumulation is poorly understood. We hypothesized that growth
factors such as TGF-β1 and VEGF, which play a role in the
development of diabetic nephropathy, are upregulated in
Gb3-accumulated endothelial cells and the Fabry mouse kidney, and
that these factors play a crucial role in the development of the
renal complications of Fabry disease (9).
Previously, we observed upregulation of lipocalin 2
(LCN2) and TSP-1 in the Fabry disease mouse and suggested that
these molecules are candidate biomarker molecules for Fabry disease
(16). To investigate further
whether LCN2 induces TSP-1 expression and inhibits VEGF expression
as reported previously (17), we
examined the expression patterns of TSP-1 and VEGF in kidneys from
Fabry and wild-type mice. We found higher TSP-1, VEGF and TGF-β1
expression levels in the kidneys from Fabry mice when compared with
these levels in the wild-type mice (Fig. 1). These findings suggest that
increased VEGF expression is associated with increased TGF-β1
expression.
TSP-1 is an extracellular matrix-remodeling
glycoprotein and a crucial component of tissue remodeling and is
associated with inhibition of angiogenesis (18). TSP-1 binds to multiple integrins
and their receptors. The regulatory effects of TSP-1 are mediated
by the interactions between TSP-1 and receptors (19). Zhang et al(20) reported that hepatocyte growth
factor/scatter factor induces angiogenesis via TSP-1 downregulation
and VEGF upregulation. In tumor cells, ectopic TSP-1 expression
directly inhibits endothelial cell proliferation and survival,
which promotes endothelial cell apoptosis. Thus, TSP-1 and VEGF can
act as angiogenic regulators. In endothelial cells, TSP-1 induces
apoptosis through activation of the Src-family tyrosine kinase (p59
Fyn), caspase-3-like proteases and p38. Subsequent activation of
activator complex-1 (AP-1) leads to apoptosis (21,22). TSP-1 expression is increased in
progressive renal disease and is associated with renal fibrosis
(23) and TSP-1 stimulates TGF-β1
in diabetes (24). TSP-1 is a
possible activator of TGF-β1 in kidney injury and can induce
apoptosis of endothelial cells in many normal tissues (25,26). Consistent with previous reports,
our data also showed that TSP-1 and TGF-β1 expression levels were
higher in Fabry mice than in wild-type mice (Fig. 1).
VEGF increases vascular permeability, prevents
apoptosis in endothelial cells (27,28) and induces apoptosis in cerebral
endothelial cells after cell injury (29). Ferrari et al(30) suggested that TGF-β1 activates
FGF-2 expression in endothelial cells, which then promotes VEGF
production. They reported that TGF-β1 induces apoptosis via
VEGF/VEGFR2-mediated phosphorylation of MAPK p38. Cross-talk
between TGF-β1 and VEGF expression can alter the response of
endothelial cells to apoptotic signals. Our results, shown in
Figs. 1, 2 and 5, are consistent with these
observations. Other investigators have also supported the
hypothesis that TGF-β1 induces VEGF expression through MAPK (ERK1/2
and p38) activation (31) and
that FGF-2 modulates VEGF expression in endothelial cells (32). Li et al(33) also suggested that VEGF-induced
FGF-2 expression in injured endothelial cells leads to migration
and proliferation of smooth muscle cells. VEGF stimulation results
in TGF-β1-induced fibrosis in proximal tubular (NRK52E) cells
(34). TGF-β1-induced epithelial
cell apoptosis is associated with p38 (35). As discussed above, VEGF expression
may not be downregulated by LCN2, but may be upregulated by
TGF-β1.
We found that TGF-β1 and VEGF expression is
increased in the Fabry mouse kidney and in Gb3-treated BAECs
(Figs. 1 and 5), suggesting that TGF-β1 and VEGF
upregulation may be associated with dysfunction of endothelial
cells. Similarly, Sanchez-Nino et al(9) reported that the expression of
TGF-β1, CD74 and extracellular matrix protein were increased by
adding lyso-Gb3 (deacylated Gb3 form) to human podocytes, showing
that TGF-β1 and CD74 are mediators of podocyte injury. CD74, the
macrophage inhibitory factor receptor, is a potent receptor of
kidney injury in diabetic nephropathy. Increased expression of
TGF-β1 and/or VEGF in podocytes is associated with apoptosis or
nephropathy (36,37). Our results are consistent with
those from a rat nephropathy model in which TGF-β1 and VEGF
expression increases (38).
Therefore, we hypothesized that the combined overexpression of
TGF-β1 and VEGF may be associated with Fabry disease nephropathy
through induction of apoptosis.
We found significantly increased activities of
caspase-6 and -9 in the Fabry mouse kidney and nonsignificant
increases in the relative caspase-3/7 and -12 activities (Figs. 3 and 4). Caspase-9 and -12 are initiator
caspases and caspase-3, -6, and -7 are effector caspases. In the
final stage of the apoptosis pathway, effector caspases are
activated by cleavage of cellular substrates (39). We found that apoptosis was induced
in kidneys from Fabry mice when compared with wild-type mice. This
apoptosis pathway might be related to the intrinsic pathway rather
than to endoplasmic reticulum stress. Activity of caspase-3/7 in
the kidney did not differ significantly between Fabry and wild-type
mice but was lower in ERT-treated Fabry mice compared with Fabry
mice. De Francesco et al(40) suggested that peripheral blood
mononuclear cells (PBMC) from Fabry disease’s patients were in an
apoptotic state, which correlated with the accumulation of Gb3.
This suggests that Fabry nephropathy is associated at least partly
with the intrinsic apoptotic pathway.
To investigate whether Gb3 treatment induces
expression of TGF-β1 and VEGF in the endothelial cells, BAECs were
treated either with control (DMSO alone) or with 15 μM Gb3
for 2 or 8 h (Fig. 5). As
expected, protein expression of TGF-β1 and VEGF increased in
Gb3-treated BAECs. The combined expression of increased TGF-β1 and
VEGF by Gb3 treatment may allow the upregulation of FGF-2, VEGFR2
and P-p38 expression (30). This
finding is consistent with the increased protein expression
observed in kidneys from Fabry mice.
Since apoptotic changes in renal biopsies from Fabry
patients have not been demonstrated, apoptosis caused by the
combined overexpression of TGF-β1 and VEGF cannot be the main
pathogenic mechanism underlying the renal complications of Fabry
disease (41,42). However, a greater apoptotic state
has been observed in PBMC from Fabry patients (40,43), Gb3-induced apoptosis via TGF-β1
and VEGF overexpression may be associated with Fabry
nephropathy.
In conclusion, we found upregulation of TGF-β1,
VEGF, VEGFR2, FGF-2 and P-p38 expression in the Fabry mouse kidney
and in Gb3-treated BAECs. Caspase-6 and -9 activation was also
observed in the Fabry mouse kidney. The combined overexpression of
TGF-β1 and VEGF by Gb3 may allow the upregulation of FGF-2, VEGFR2
and P-p38 expression. These results suggest that increased
expression of these proteins is related to Fabry disease
nephropathy through the induction of apoptosis.
Acknowledgements
This study was supported by grants
from the Life Insurance Philanthropy Foundation and the Korea 21
R&D Project (A010384) of the Ministry of Health and Welfare,
Republic of Korea.
References
|
1
|
Brady RO, Gal AE, Bradley RM, Martensson
E, Warshaw AL and Laster L: Enzymatic defect in Fabry’s disease.
Ceramidetrihexosidase deficiency. New Engl J Med. 276:1163–1167.
1967.
|
|
2
|
Kint JA: The enzyme defect in Fabry’s
disease. Nature. 227:11731970.
|
|
3
|
Desnick RJ, Ioannou YA and Eng CM:
Alpha-galactosidase A deficiency: Fabry disease. The Metabolic and
Molecular Bases of Inherited Disease. Scriver CR, Beaudet AL, Sly
WS and Valle D: 8th edition. McGraw-Hill; New York, NY: pp.
3733–3774. 2001
|
|
4
|
Tondel C, Bostad L, Hirth A and Svarstad
E: Renal biopsy findings in children and adolescents with Fabry
disease and minimal albuminuria. Am J Kidney Dis. 51:767–776. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ohshima T, Murray GJ, Swaim WD, et al:
alpha-Galactosidase A deficient mice: a model of Fabry disease.
Proc Nat Acad Sci USA. 94:2540–2544. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shu L, Murphy HS, Cooling L and Shayman
JA: An in vitro model of Fabry disease. J Am Soc Nephrol.
16:2636–2645. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bodary PF, Shen Y, Vargas FB, et al:
Alpha-galactosidase A deficiency accelerates atherosclerosis in
mice with apolipoprotein E deficiency. Circulation. 111:629–632.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eitzman DT, Bodary PF, Shen Y, et al:
Fabry disease in mice is associated with age-dependent
susceptibility to vascular thrombosis. J Am Soc Nephrol.
14:298–302. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sanchez-Nino MD, Sanz AB, Carrasco S, et
al: Globo-triaosylsphingosine actions on human glomerular
podocytes: implications for Fabry nephropathy. Nephrol Dial
Transplant. 26:1797–1802. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen S, Hong SW, Iglesias-de la Cruz MC,
Isono M, Casaretto A and Ziyadeh FN: The key role of the
transforming growth factor-beta system in the pathogenesis of
diabetic nephropathy. Ren Fail. 23:471–481. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hayashida T, Decaestecker M and Schnaper
HW: Cross-talk between ERK MAP kinase and Smad signaling pathways
enhances TGF-beta-dependent responses in human mesangial cells.
FASEB J. 17:1576–1578. 2003.PubMed/NCBI
|
|
12
|
Pollman MJ, Naumovski L and Gibbons GH:
Vascular cell apoptosis: cell type-specific modulation by
transforming growth factor-beta1 in endothelial cells versus smooth
muscle cells. Circulation. 99:2019–2026. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ferrara N: Vascular endothelial growth
factor: basic science and clinical progress. Endocr Rev.
25:581–611. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Flyvbjerg A: Inhibition and reversibility
of renal changes: lessons from diabetic kidney disease. Acta
Paediatr Suppl. 95:83–92. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moore DF, Krokhin OV, Beavis RC, et al:
Proteomics of specific treatment-related alterations in Fabry
disease: a strategy to identify biological abnormalities. Proc Natl
Acad Sci USA. 104:2873–2878. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park ES, Choi JO, Park JW, Lee MH, Park HY
and Jung SC: Expression of genes and their responses to enzyme
replacement therapy in a Fabry disease mouse model. Int J Mol Med.
24:401–407. 2009.PubMed/NCBI
|
|
17
|
Venkatesha S, Hanai J, Seth P, Karumanchi
SA and Sukhatme VP: Lipocalin 2 antagonizes the proangiogenic
action of ras in transformed cells. Mol Cancer Res. 4:821–829.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dameron KM, Volpert OV, Tainsky MA and
Bouck N: Control of angiogenesis in fibroblasts by p53 regulation
of thrombospondin-1. Science. 265:1582–1584. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen H, Herndon ME and Lawler J: The cell
biology of thrombospondin-1. Matrix Biol. 19:597–614. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang YW, Su Y, Volpert OV and Vande Woude
GF: Hepatocyte growth factor/scatter factor mediates angiogenesis
through positive VEGF and negative thrombospondin 1 regulation.
Proc Natl Acad Sci USA. 100:12718–12723. 2003. View Article : Google Scholar
|
|
21
|
Jimenez B, Volpert OV, Crawford SE,
Febbraio M, Silverstein RL and Bouck N: Signals leading to
apoptosis-dependent inhibition of neovascularization by
thrombospondin-1. Nat Med. 6:41–48. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sargiannidou I, Zhou J and Tuszynski GP:
The role of thrombospondin-1 in tumor progression. Exp Biol Med
(Maywood). 226:726–733. 2001.PubMed/NCBI
|
|
23
|
Kang DH, Anderson S, Kim YG, et al:
Impaired angiogenesis in the aging kidney: vascular endothelial
growth factor and thrombospondin-1 in renal disease. Am J Kidney
Dis. 37:601–611. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yevdokimova N, Wahab NA and Mason RM:
Thrombospondin-1 is the key activator of TGF-beta1 in human
mesangial cells exposed to high glucose. J Am Soc Nephrol.
12:703–712. 2001.PubMed/NCBI
|
|
25
|
Bornstein P: Thrombospondins as
matricellular modulators of cell function. J Clin Invest.
107:929–934. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hugo C and Daniel C: Thrombospondin in
renal disease. Nephron Exp Nephrol. 111:e61–e66. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ferrara N and Gerber HP: The role of
vascular endothelial growth factor in angiogenesis. Acta Haematol.
106:148–156. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Neufeld G, Cohen T, Gengrinovitch S and
Poltorak Z: Vascular endothelial growth factor (VEGF) and its
receptors. FASEB J. 13:9–22. 1999.PubMed/NCBI
|
|
29
|
Narasimhan P, Liu J, Song YS, Massengale
JL and Chan PH: VEGF stimulates the ERK 1/2 signaling pathway and
apoptosis in cerebral endothelial cells after ischemic conditions.
Stroke. 40:1467–1473. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ferrari G, Pintucci G, Seghezzi G, Hyman
K, Galloway AC and Mignatti P: VEGF, a prosurvival factor, acts in
concert with TGF-beta1 to induce endothelial cell apoptosis. Proc
Natl Acad Sci USA. 103:17260–17265. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nakagawa T, Lan HY, Zhu HJ, Kang DH,
Schreiner GF and Johnson RJ: Differential regulation of VEGF by
TGF-beta and hypoxia in rat proximal tubular cells. Am J Physiol
Renal Physiol. 287:F658–F664. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Seghezzi G, Patel S, Ren CJ, et al:
Fibroblast growth factor-2 (FGF-2) induces vascular endothelial
growth factor (VEGF) expression in the endothelial cells of forming
capillaries: an autocrine mechanism contributing to angiogenesis. J
Cell Biol. 141:1659–1673. 1998. View Article : Google Scholar
|
|
33
|
Li D, Zhang C, Song F, Lubenec I, Tian Y
and Song QH: VEGF regulates FGF-2 and TGF-beta1 expression in
injury endothelial cells and mediates smooth muscle cell
proliferation and migration. Microvasc Res. 77:134–142. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakagawa T, Li JH, Garcia G, et al:
TGF-beta induces proangiogenic and antiangiogenic factors via
parallel but distinct Smad pathways. Kidney Int. 66:605–613. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu L, Hebert MC and Zhang YE: TGF-beta
receptor-activated p38 MAP kinase mediates Smad-independent
TGF-beta responses. EMBO J. 21:3749–3759. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Iglesias-de la Cruz MC, Ziyadeh FN, Isono
M, et al: Effects of high glucose and TGF-beta1 on the expression
of collagen IV and vascular endothelial growth factor in mouse
podocytes. Kidney Int. 62:901–913. 2002.PubMed/NCBI
|
|
37
|
Schiffer M, Bitzer M, Roberts IS, et al:
Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin
Invest. 108:807–816. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang W, Wang J, Shi L, et al: Podocyte
injury and overexpression of vascular endothelial growth factor and
transforming growth factor-beta 1 in adriamycin-induced nephropathy
in rats. Cytokine. 59:370–376. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schmitz I, Kirchhoff S and Krammer PH:
Regulation of death receptor-mediated apoptosis pathways. Int J
Biochem Cell Biol. 32:1123–1136. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
De Francesco PN, Mucci JM, Ceci R, Fossati
CA and Rozenfeld PA: Higher apoptotic state in Fabry disease
peripheral blood mononuclear cells: effect of
globotriaosylceramide. Mol Genet Metab. 104:319–324.
2011.PubMed/NCBI
|
|
41
|
Valbuena C, Oliveira JP, Carneiro F, et
al: Kidney histologic alterations in α-galactosidase-deficient
mice. Virchows Arch. 458:477–486. 2011.
|
|
42
|
Noel LH, Laurent B and Grunfeld JP: Renal
biopsies in Fabry disease: a multicenter French study. Nephrol
Ther. May 14–2012.(Epub ahead of print) (In French).
|
|
43
|
Moore DF, Goldin E, Gelderman MP, et al:
Apoptotic abnormalities in differential gene expression in
peripheral blood mononuclear cells from children with Fabry
disease. Acta Paediatr Suppl. 97:48–52. 2008. View Article : Google Scholar : PubMed/NCBI
|