Introduction
Rheumatoid arthritis (RA) is the most common chronic
inflammatory joint disease, affecting ∼0.5–1% of people in the
industrialized world (1).
Clinically, the disorder is characterized by joint pain, stiffness,
and swelling due to synovial inflammation and effusion. The
clinical features of RA are based on several pathological processes
including chronic inflammation, overgrowth of synovial cells, bone
and joint destruction, and fibrosis. Currently, the goal of RA
treatment is the control of underlying inflammatory process to
prevent joint damage using non-steroidal anti-inflammatory drugs,
glucocorticoids, and disease-modifying anti-rheumatic drugs
(DMARD). The most widely used small molecule DMARD is methotrexate,
which shows the highest retention rate compared with other agents
(2). In recent years, biological
agents such as inhibitors of tumor necrosis factor (TNF) signaling
have become available for clinical use; however, this therapy is
prohibitively expensive, and although TNF inhibitors are clinically
as effective as methotrexate, the frequency and extent of response
are more restricted. In fact, many patients can lose the clinical
response to TNF inhibition, highlighting the need for other
treatment modalities to further improve the outcome of RA (3,4).
To address this need, we have been investigating the
mechanism of outgrowth in rheumatoid synovial cells (RSCs). First,
we demonstrated the crucial role of Fas antigen-induced apoptosis
in synovial cell hyperplasia (5).
Then, while studying cellular functions of RSCs, we cloned
synoviolin from these cells (6).
Synoviolin, a mammalian homolog of Hrd1p/Der3p (7–9),
is an endoplasmic reticulum (ER)-resident E3 ubiquitin ligase with
a RING motif that is involved in ER-associated degradation (ERAD)
pathway. Synoviolin is also highly expressed in synoviocytes of
patients with RA (6,10–12). Overexpression of synoviolin in
transgenic mice leads to advanced arthropathy caused by reduced
apoptosis of synoviocytes (6). We
postulated that hyperactivation of the ERAD pathway by
overexpression of synoviolin prevents ER-stress-induced apoptosis,
leading to synovial hyperplasia (13). Synoviolin+/− knockout
mice showed resistance to the development of collagen-induced
arthritis (CIA) due to enhanced apoptosis of synovial cells
(6). Consistent with our
hypothesis, cells from these mice show impaired ERAD due to the
lack of synoviolin. In addition, synoviolin ubiquitinates and
sequesters the tumor suppressor p53 in the cytoplasm, thereby
negatively regulating its biological functions in transcription,
cell cycle regulation, and apoptosis by targeting it instead for
proteasomal degradation (14).
Therefore, synoviolin regulates apoptosis in response to ER stress
(through ERAD) as well as p53-dependent apoptosis.
Together, these studies implicated synoviolin as a
candidate pathogenic factor in arthropathy, and suggested that the
gene dosage of this protein correlates with the onset of
arthropathy. Furthermore, elevated synoviolin levels were
identified in circulating monocytes in association with resistance
to treatment with infliximab (a monoclonal antibody against TNF)
(10). Therefore, blocking the
function of synoviolin could be clinically beneficial in RA
patients. This study attempted to identify an inhibitor of
synoviolin that acts by blocking its enzymatic activity.
Materials and methods
Screening of synoviolin inhibitor
Purified glutathione S-transferase (GST)-synoviolin
Δ transmembrane domain (TM) was mixed with glutathione-SPA beads
(Amersham Pharmacia Biotech) in buffer (50 mM Tris-HCl, pH 7.4,
Protease inhibitor cocktail, 14 mM β-mercaptoethanol, 0.5 μl cell
lysate/well, 0.2 mg SPA bead/well) and incubated for 30 min at room
temperature. Glutathione-SPA beads were washed twice, and then
mixed with the candidate synoviolin inhibitor compounds in buffer
(50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 2 mM NaF, and 10 nM
okadaic acid) in the presence of ATP (2 mM), 33P-labeled
ubiquitin (0.38 μg/well), E1 (25 ng/well) (Affiniti Research), and
E2 (0.3 μg/well) (UbcH5c). After incubation for 90 min at room
temperature, buffer comprising 0.2 M boric acid, pH 8.5, 2 mM
ethylenediaminetetraacetic acid (EDTA), and 2% Triton-X100 was
added to stop the reaction. The beads were allowed to settle and
the amount of 33P-ubiquitin incorporated into the
GST-synoviolin beads was determined using a Microbeta Scintillation
counter.
The primary screen was conducted with multiple
compounds per well (10–20 compounds per well) at an estimated
screening concentration of 2–10 μM. Compound mixtures showing
potential activity in the primary screen were then rescreened at
one compound per well to determine the active compound within the
mixture. Three equivalents of a single compound per well follow-up
screening were evaluated. Reconfirmed active compounds were
resynthesized and tested in a dose-response experiment to determine
potency.
In vitro ubiquitination assay
The in vitro ubiquitination assay used in
this study was described previously (15). Briefly, 40 ng of E1 (Affiniti
Research), 0.3 μg of E2 (UbcH5c), 0.75 μg of 32P-labeled
ubiquitin (a gift from T. Ohta), and 1 μg of recombinant E3
ubiquitin ligases were incubated for 30 min at 37°C. Samples were
analyzed as described above.
Cells
HeLa cells were obtained from ATCC. Synovial cells
were isolated from synovial tissue obtained patients with
rheumatoid arthritis (RA) who met the American College of
Rheumatology criteria for RA at the time of orthopedic surgery.
These cells were cultured in Dulbecco’s modified Eagle’s medium
(Sigma).
Proliferation assay
The proliferation of rheumatoid synovial cells
(RSCs) was evaluated using Alamar blue (BioSource International)
according to the manufacturer’s instructions.
Induction of CIA
CIA was induced as described previously (6). Briefly, bovine type II collagen
(Collagen Research Center) was dissolved overnight in 0.05 M acetic
acid at 4°C, and then emulsified in complete Freund’s adjuvant
(Difco) to a final concentration 1 mg/ml. DBA/1 male mice
(7-week-old) were immunized by subcutaneous injections containing
100 μg of collagen emulsion. After 3 weeks, mice were boosted with
200 μg collagen emulsion in Freund’s complete adjuvant. Then, the
mice were treated daily for 4 weeks with the inhibitor compounds at
1.3, 4.0, and 12.0 mg/kg/day in olive oil, vehicle control
intraperitoneally, or oral administration of 0.25 mg/kg/day
dexamethasone in methylcellulose as a positive control.
The mice were monitored daily for signs of arthritis
using an established scoring system (16): 0, no swelling or redness; 1,
swelling, redness of paw or 1 joint; 2, two joints involved; 3,
more than two joints involved; 4, severe arthritis of entire paws
and joints. All paws were evaluated in each animal and the maximum
score per animal was 16.
Histological studies
The knee and elbow joints were fixed in 4%
paraformaldehyde. After decalcification with EDTA, the joints were
embedded in paraffin, and 4-μm sections were prepared for staining
with hematoxylin and eosin. The extent of arthritis in the joints
was assessed according to the method reported by Tomita et
al(17): 0, normal synovium;
1, synovial membrane hypertrophy and cell infiltration; 2, pannus
and cartilage erosion; 3, major erosion of cartilage and
subchondral bone; 4, loss of joint integrity and ankylosis.
Statistical analysis
All data are expressed as mean ± SEM. Differences
between groups were examined for statistical significance using
Student’s t-test. A P-value <0.05 denoted the presence of a
statistically significant difference.
Ethical considerations
The ethics committee for Animal Experiments of St.
Marianna University School of Medicine approved the mice
experiments described in this study. Furthermore, all the
experimental protocols described in this study were approved by the
Ethics Review Committee of St. Marianna University School of
Medicine (Approval number 01008), and the written informed consent
was obtained from all patients.
Results
High-throughput compound screening for
inhibitors of synoviolin
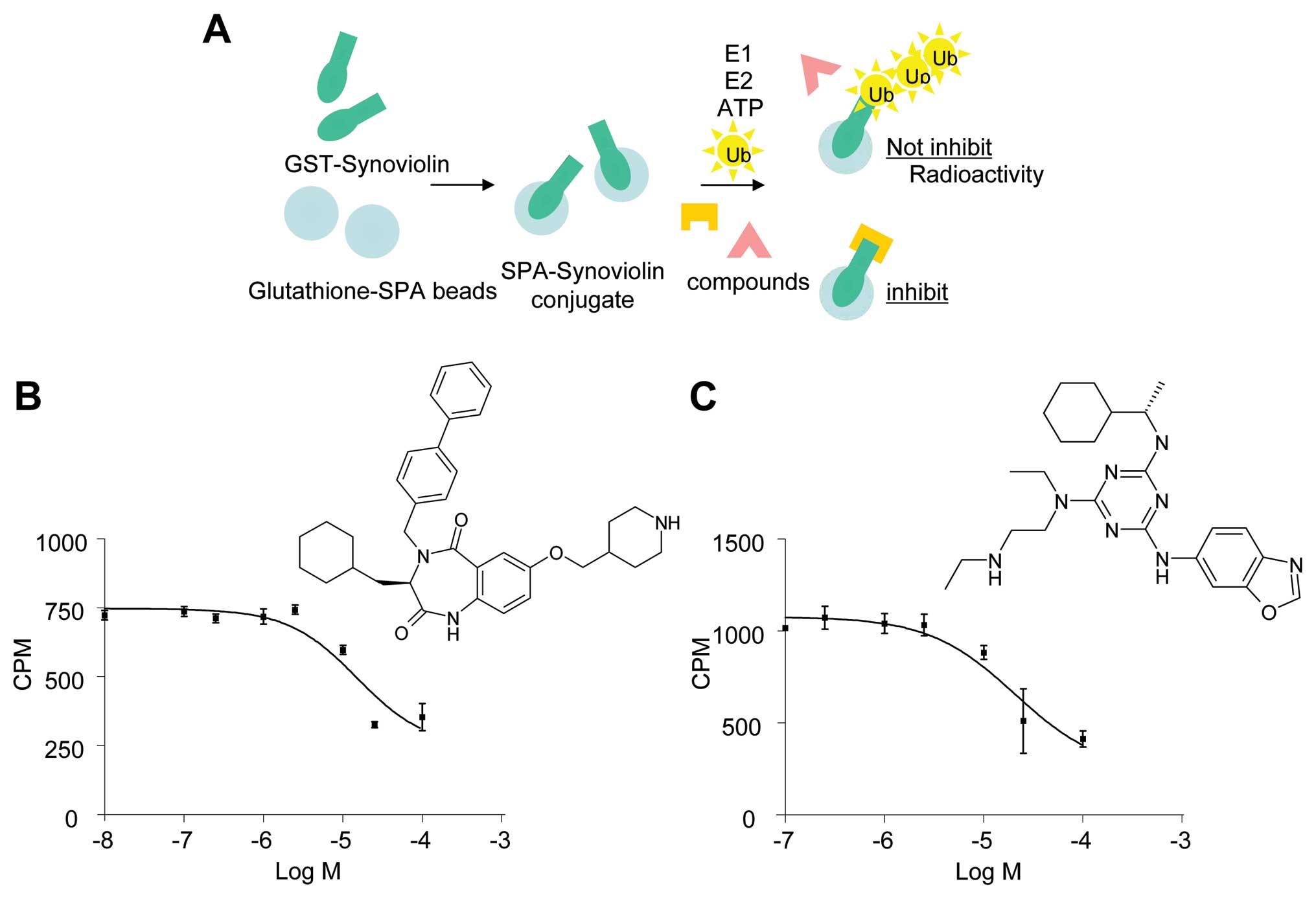
To identify small molecule inhibitors of synoviolin
autoubiquitination, we screened the Lead Discovery Service program
of Pharmacopeia, which includes more than four million compounds
from Pharmacopeia’s Compound Collection (18). Herein we monitored
33P-autoubiquitinated synoviolin in cell lysates
containing GST-synoviolinΔTM in the presence of ATP, E1, E2, and
33P-labeled ubiquitin (Fig. 1A). The primary screen was
conducted with multiple compounds per well (10–20 compounds per
well) at an estimated screening concentration of 2–10 μM. Mixtures
of compounds showing potential activity in the primary screen were
then rescreened individually. Compounds demonstrating activity in
this reconfirmation assay were resynthesized and retested. Two
unique compounds, termed LS-101 and LS-102, inhibited the
autoubiquitination of synoviolin with a 50% inhibitory
concentration value (IC50) of ∼15 μM (Fig. 1B) and 20 μM (Fig. 1C), respectively.
LS-101 and LS-102 inhibit the
autoubiquitination of synoviolin
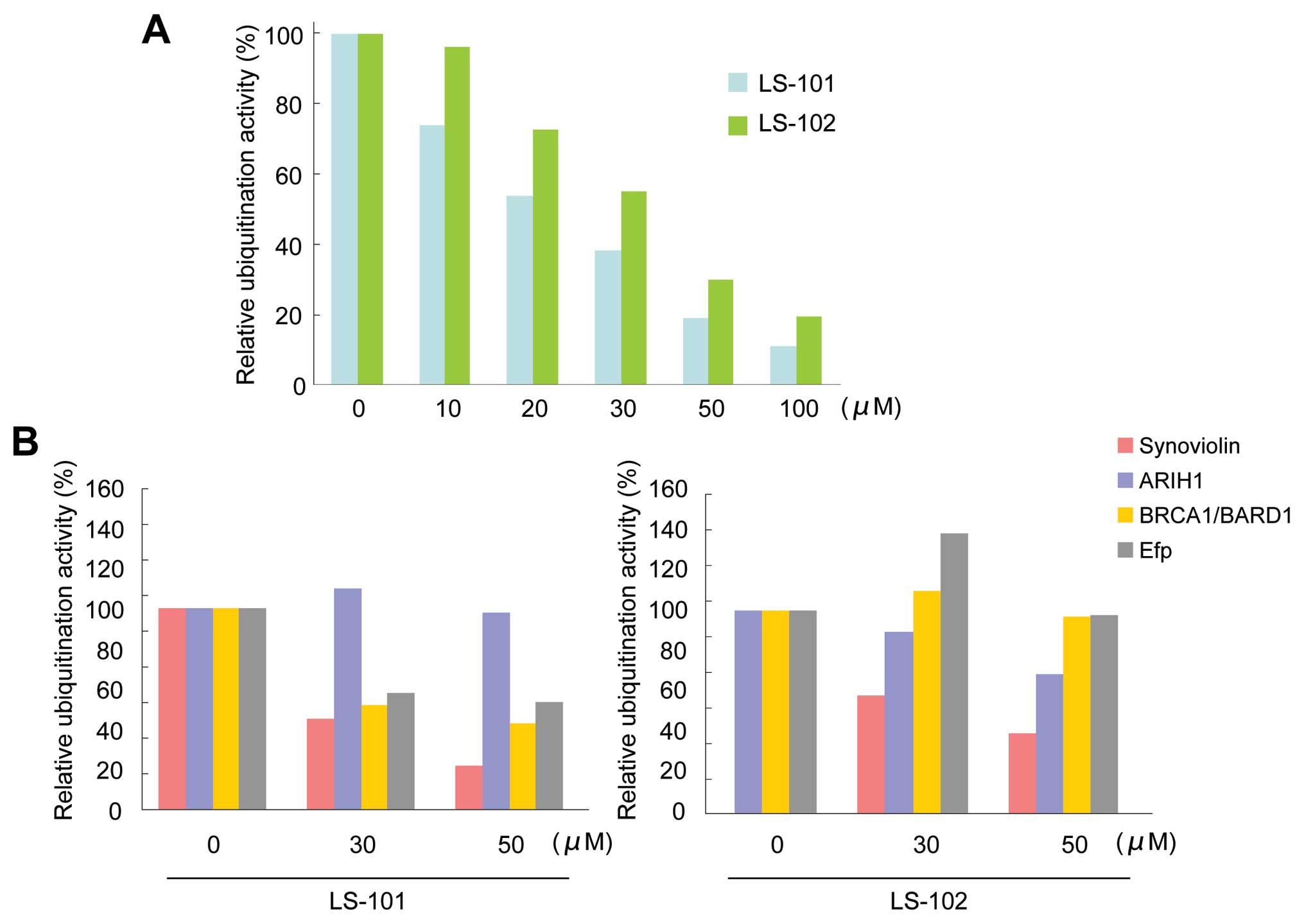
Further evaluation of LS-101 and LS-102 in an in
vitro ubiquitination assay showed that the inhibition of
synoviolin activity by both LS-101 and LS-102 was dose-dependent
(LS-101; IC50=20 μM, LS-102; IC50=35 μM)
(Fig. 2A). To assess the
selectivity of the compounds for other E3 ubiquitin ligases, we
determined the effects of LS-101 and LS-102 on the enzymatic
activity of the following RING-finger type E3 ubiquitin ligases:
ariadne, Drosophila, homolog of, 1 (ARIH1) (19), breast cancer 1 gene
(BRCA1)/BRCA1-associated RING domain 1 (BARD1) (20), and estrogen-responsive RING-finger
protein (Efp) (21). LS-101
inhibited the activity of BRCA1/BARD1 and Efp (Fig. 2B), although this effect was weaker
than that observed with synoviolin (Fig. 2B). Moreover, LS-101 had no effect
against the enzymatic activity of ARIH1 (Fig. 2B). On the other hand, LS-102 did
not inhibit the activity of other E3 ubiquitin ligases, only
affecting synoviolin (Fig. 2B).
These results suggested that LS-102 is a more selective synoviolin
inhibitor than LS-101.
LS-101 and LS-102 inhibit proliferation
of RSCs
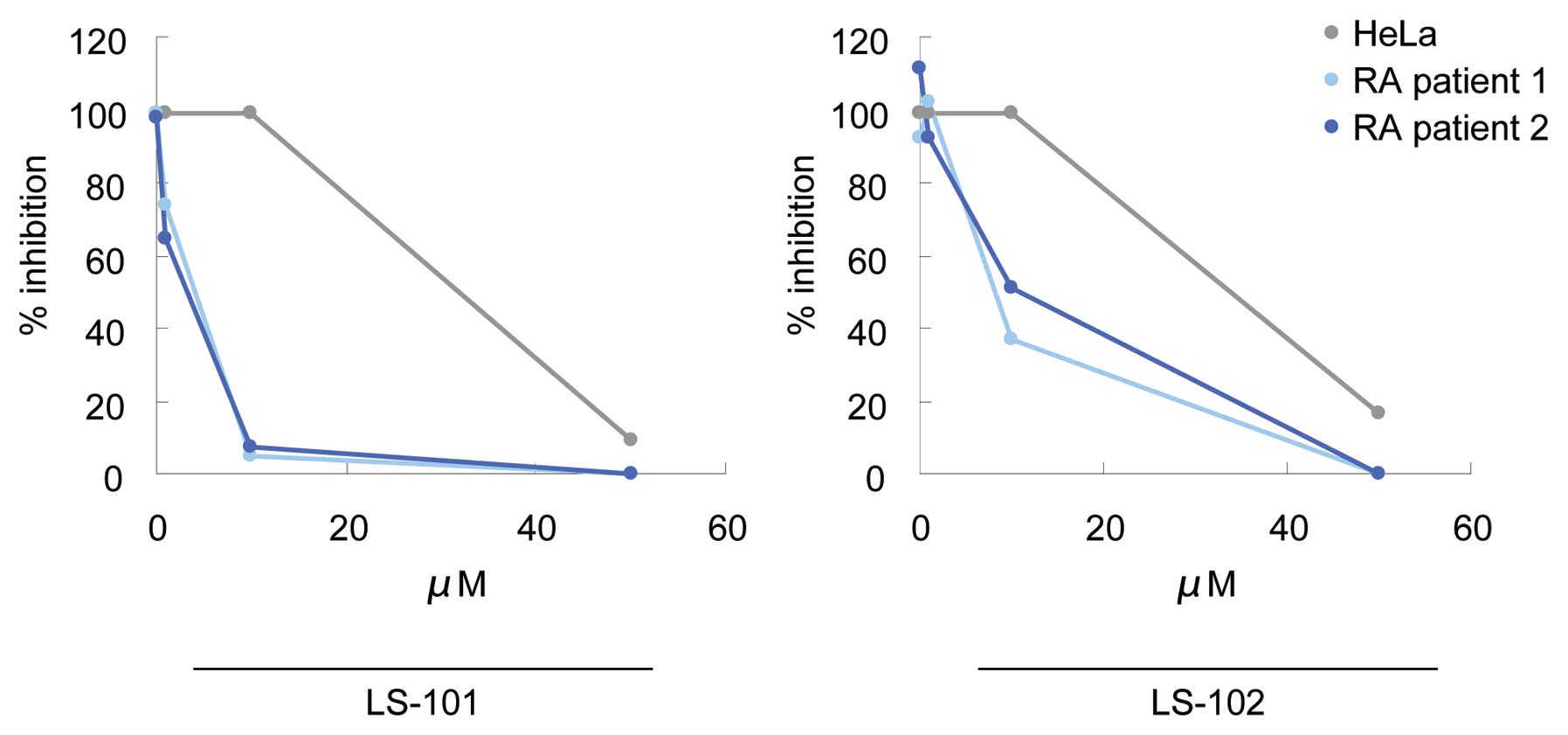
We next tested LS-101 and LS-102 for their effects
on the proliferation of RSCs, using HeLa cells as a control. LS-101
and LS-102 inhibited HeLa cell growth only at very high
concentrations (LS-101; IC50=31.3 μM, LS-102;
IC50=32.7 μM). However, treatment of RSCs with these
compounds suppressed synovial cell growth dose-dependently and with
much greater potency than that observed in HeLa cells (Fig. 3). A similar effect was also
observed in another line of RSCs (Fig. 3). In addition, LS-101 inhibited
synovial cell proliferation more potently than LS-102 (LS-101;
IC50=4.2 μM, LS-102; IC50=5.4 μM). These
results demonstrated that blockade of synoviolin function reduced
the proliferation of RSCs, and that RSCs are more susceptible to
this effect than HeLa cells. Consistent with these findings, higher
expression levels of synoviolin were observed in RSCs than in HeLa
cells (6).
LS-101 and LS-102 reduce clinical
severity scores in a CIA model
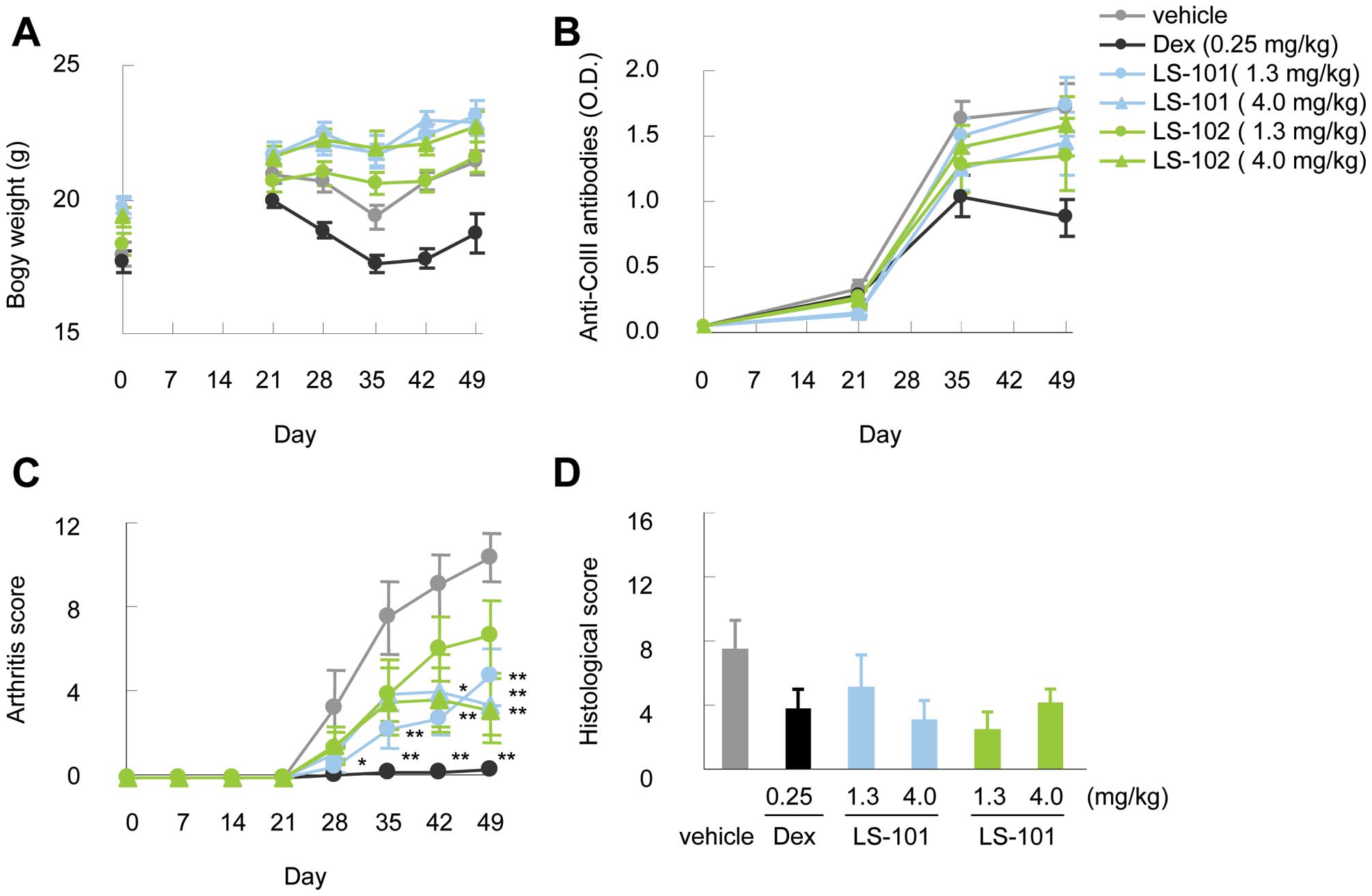
To evaluate the in vivo efficacy of
synoviolin inhibitors, we tested LS-101 and LS-102 in a mouse model
of arthritis over a period of 28 days. No reduction of body weight
was observed during the administration of these compounds (Fig. 4A). Moreover, the production of
anti-type II collagen antibodies resulting from type II collagen
immunization in both the LS-101 and LS-102 group was comparable to
that observed in the vehicle control group (Fig. 4B). Intraperitoneal treatment with
LS-101 or LS-102 starting on day 21 reduced the clinical severity
scores compared to vehicle controls (Fig. 4C). The efficacy was observed at
both 1.3 mg/kg and 4.0 mg/kg doses in this experiment, although the
protective effect of LS-101 at 1.3 mg/kg against CIA was stronger
than the same dose of LS-102. At 4.0 mg/kg, there was no difference
in the effects between LS-101 and LS-102. Finally, histological
analysis showed lower histological arthritis scores in mice treated
with the synoviolin inhibitors compared with wild-type mice
(Fig. 4D).
Discussion
The selective degradation of proteins in eukaryotic
cells is carried out by the ubiquitin proteasome system (UPS),
whereby proteins are targeted for degradation by covalent ligation
to small polypeptide ubiquitin (22,23). This reaction requires the
sequential actions of three enzymes: E1, E2, and E3 ligases
(22,23). E3 ligases are responsible for
conferring selectivity to ubiquitination by recognizing specific
substrates. Bioinformatic analysis has identified over 600 E3
ligases, with RING-type E3 ligases constituting the largest
subfamily within this group (24). Accordingly, RING E3 ligases have
been linked to the control of multiple cellular processes and to
many human diseases such as diabetes mellitus, polyglutamine
disease, and Parkinson’s diseases (24–26). In the UPS, the proteasome
inhibitory agent bortezomib (Velcade) was recently approved for the
treatment of multiple myeloma and mantle cell lymphoma (27). Bortezomib induces apoptosis of a
wide variety of cancer cells, and is the first proteasome inhibitor
to gain FDA approval (28–30).
However, widespread clinical use of bortezomib continues to be
hampered by the appearance of dose-limiting toxicities,
drug-resistance, and interference by some natural compounds
(31). Thus, despite the efficacy
of bortezomib for treating lethal diseases such as cancer, the
associated toxicities prevent its use for the treatment of chronic
diseases such as RA. Thus, it is important to develop inhibitors of
the ubiquitin-proteasome enzymatic cascade upstream from the
proteasome to impact fewer cell processes and reduce toxicity. E3
ligases are attractive such targets given their large number and
substrate specificity. We recently cloned the E3 ubiquitin ligase
synoviolin, which localizes to the ER lumen and has enzymatic
activity. We have also demonstrated that this protein plays crucial
roles in the pathological processes of RA (6), and could therefore be a candidate
novel therapeutic target of RA (32).
In this study, we identified two potent small
compounds as inhibitors of synoviolin enzymatic activity using
high-throughput screening (Fig.
1). Moreover, in vivo studies showed no serious toxicity
associated with these compounds in terms of survival and weight
loss during treatment (Fig. 4A).
Biochemical characterization of the two compounds, LS-101 and
LS-102, demonstrated that they both inhibit the autoubiquitination
activity of synoviolin in vitro (Fig. 2), with LS-101 showing stronger
efficacy (IC50=20 μM) than LS-102 (IC50=35
μM), but less selectivity (Fig.
2). It was unclear from this study why LS-101 showed a weak
inhibitory effect on BRCA1/BARD1 and Efp activity, and further
study is needed to understand the molecular basis for this
observation. LS-101 and LS102 inhibited the proliferation of RSCs
and to a much lesser extent, HeLa cells (Fig. 3). The difference in cell
sensitivities to these compounds could be, at least in part, due to
the expression level of synoviolin, namely, high levels of
synoviolin in RSCs would contribute to the cell overgrowth and
therefore, inhibition of synoviolin in these cells would in turn
suppress proliferation. These cells may also have different
requirements for synoviolin, such that repressing synoviolin
activity in RSCs would lead to growth suppression. Prophylactic
administration of either LS-101 or LS-102 also significantly
reduced the severity of murine CIA (Fig. 4C). Since LS-101, a nonselective
inhibitor, reduced clinical severity scores in CIA similarly to
LS-102, blocking synoviolin enzymatic activity seems crucial in the
pathological process of CIA. These findings suggest that the
suppression level of synovial cell growth and incidence of
arthritis reflect the efficacy of these compounds rather than their
selectivity, and that in RA, synoviolin might have an indispensable
role among E3 ligases.
RA comprises multiple processes such as chronic
inflammation, overgrowth of synovial cells, joint destruction, and
fibrosis. During the course of inflammation, synovial cells,
macrophages, T cells, and B cells all contribute to the production
of cytokines such as interleukin (IL)-1, IL-6, IL-10, TNF, and
transforming growth factor β (TGF-β) (33,34). These cytokines, in turn, stimulate
the overgrowth of synovial cells to form a mass of synovial tissue,
called pannus, which invades and destroys the bone and cartilage
through osteoclast activation and protease production (33–37). This chronic inflammation state
ultimately leads to fibrosis. Our study proved that synoviolin is,
at least in part, involved in the overgrowth of synovial cells
(6) and fibrosis (38) among these processes. The IL-17
induction of synoviolin may also contribute to RA chronicity
(39), and synoviolin has been
shown to target misfolded MHC class I heavy chains (40). In this study, antibody titers were
elevated in synoviolin inhibitor-treated mice to levels comparable
to those in vehicle controls (Fig.
4B). Thus, as with the study of synoviolin+/−
knockout mice in CIA, it is difficult to clarify the function of
synoviolin with respect to the chronicity of inflammation, because
suppressing synoviolin blocks synovial cell outgrowth directly due
to sequential events following immunization of type II collagen
(6). Our results confirm that
further studies of the association between chronic inflammation and
synoviolin are clearly warranted.
Eight biological agents are currently approved for
clinical use in treatment of RA, and these drugs have dramatically
changed the outcome of RA during the past decade (3,4).
However, some patients still fail to respond to the biological
treatment or develop adverse effects such as an increased risk of
infection. Moreover, these agents are associated with high costs
and discomfort arising from the subcutaneous or intravenous
administration. Thus, there is a clear need for the development of
cheaper, orally administered therapies with fewer side effects. In
this regard, spleen tyrosine kinase (Syk) inhibitor, an orally
administered drug, has been developed for the treatment of RA
(41,42). Dual blockade of TNF and IL-17 was
also reported recently as a strategy for halting RA disease from
progression to the extent seen when only one cytokine is blocked
(43). The involvement of
synoviolin in both the TNF and IL-17 pathways further implicates
inhibitors of this enzyme as potential candidate drugs for
treatment of RA.
In conclusion, we identified two strong synoviolin
inhibitors, and confirmed that synoviolin is an ideal molecular
target for RA for disease modification and treatment. We are now
proceeding with the optimization of LS-101 and LS-102, and hope our
research will lead to the development of a new therapy for RA.
Acknowledgements
We thank A. Imai and F. Nagumo for the
technical assistance. We also thank all members of Dr Nakajima’s
laboratory. This work was funded in part by grants from the Naito
Foundation, Natural Science Scholarship Daiichi-Sankyo Foundation
of Life Science, Bureau of Social Welfare and Public Health,
Ministry of Health Labour and Welfare Japan Society for the
Promotion of Science, Takeda Science Foundation.
References
|
1
|
Gabriel SE: The epidemiology of rheumatoid
arthritis. Rheum Dis Clin North Am. 27:269–281. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aletaha D and Smolen JS: Effectiveness
profiles and dose dependent retention of traditional disease
modifying anti-rheumatic drugs for rheumatoid arthritis. An
observational study. J Rheumatol. 29:1631–1638. 2002.PubMed/NCBI
|
|
3
|
Smolen JS, Aletaha D, Koeller M, Weisman
MH and Emery P: New therapies for treatment of rheumatoid
arthritis. Lancet. 370:1861–1874. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nurmohamed MT: Newer biological agents in
the treatment of rheumatoid arthritis: do the benefits outweigh the
risks? Drugs. 69:2035–2043. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nakajima T, Aono H, Hasunuma T, Yamamoto
K, Shirai T, Hirohata K and Nishioka K: Apoptosis and functional
Fas antigen in rheumatoid arthritis synoviocytes. Arthritis Rheum.
38:485–491. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amano T, Yamasaki S, Yagishita N, et al:
Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel pathogenic
factor for arthropathy. Genes Dev. 17:2436–2449. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bordallo J, Plemper RK, Finger A and Wolf
DH: Der3p/Hrd1p is required for endoplasmic reticulum-associated
degradation of misfolded lumenal and integral membrane proteins.
Mol Biol Cell. 9:209–222. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shearer AG and Hampton RY: Structural
control of endoplasmic reticulum-associated degradation: effect of
chemical chaperones on 3-hydroxy-3-methylglutaryl-CoA reductase. J
Biol Chem. 279:188–196. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shearer AG and Hampton RY: Lipid-mediated,
reversible misfolding of a sterol-sensing domain protein. EMBO J.
24:149–159. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Toh ML, Marotte H, Blond JL, Jhumka U,
Eljaafari A, Mougin B and Miossec P: Overexpression of synoviolin
in peripheral blood and synoviocytes from rheumatoid arthritis
patients and continued elevation in nonresponders to infliximab
treatment. Arthritis Rheum. 54:2109–2118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao B, Calhoun K and Fang D: The
proinflammatory cytokines IL-1beta and TNF-alpha induce the
expression of Synoviolin, an E3 ubiquitin ligase, in mouse synovial
fibroblasts via the Erk1/2-ETS1 pathway. Arthritis Res Ther.
8:R1722006. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gao B, Lee SM, Chen A, et al: Synoviolin
promotes IRE1 ubiquitination and degradation in synovial
fibroblasts from mice with collagen-induced arthritis. EMBO Rep.
9:480–485. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yagishita N, Yamasaki S, Nishioka K and
Nakajima T: Synoviolin, protein folding and the maintenance of
joint homeostasis. Nat Clin Pract Rheumatol. 4:91–97. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamasaki S, Yagishita N, Sasaki T, et al:
Cytoplasmic destruction of p53 by the endoplasmic
reticulum-resident ubiquitin ligase ‘Synoviolin’. EMBO J.
26:113–122. 2007.PubMed/NCBI
|
|
15
|
Ohta T, Michel JJ, Schottelius AJ and
Xiong Y: ROC1, a homolog of APC11, represents a family of cullin
partners with an associated ubiquitin ligase activity. Mol Cell.
3:535–541. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hughes C, Wolos JA, Giannini EH and Hirsch
R: Induction of T helper cell hyporesponsiveness in an experimental
model of autoimmunity by using nonmitogenic anti-CD3 monoclonal
antibody. J Immunol. 153:3319–3325. 1994.PubMed/NCBI
|
|
17
|
Tomita T, Takeuchi E, Tomita N, et al:
Suppressed severity of collagen-induced arthritis by in vivo
transfection of nuclear factor kappaB decoy oligodeoxynucleotides
as a gene therapy. Arthritis Rheum. 42:2532–2542. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dunn DA and Feygin I: Challenges and
solutions to ultra-high-throughput screening assay miniaturization:
submicroliter fluid handling. Drug Discov Today. 5:84–91. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moynihan TP, Ardley HC, Nuber U, et al:
The ubiquitin-conjugating enzymes UbcH7 and UbcH8 interact with
RING-finger/IBR motif-containing domains of HHARI and H7-AP1. J
Biol Chem. 274:30963–30968. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hashizume R, Fukuda M, Maeda I, et al: The
RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a
breast cancer-derived mutation. J Biol Chem. 276:14537–14540. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Urano T, Saito T, Tsukui T, et al: Efp
targets 14-3-3 sigma for proteolysis and promotes breast tumour
growth. Nature. 417:871–875. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hershko A and Ciechanover A: The ubiquitin
system. Annu Rev Biochem. 67:425–479. 1998. View Article : Google Scholar
|
|
23
|
Pickart CM: Mechanisms underlying
ubiquitination. Annu Rev Biochem. 70:503–533. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Deshaies RJ and Joazeiro CA: RING domain
E3 ubiquitin ligases. Annu Rev Biochem. 78:399–434. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaufman RJ: Orchestrating the unfolded
protein response in health and disease. J Clin Invest.
110:1389–1398. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Araki E, Oyadomari S and Mori M:
Endoplasmic reticulum stress and diabetes mellitus. Intern Med.
42:7–14. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cvek B and Dvorak Z: The
ubiquitin-proteasome system (UPS) and the mechanism of action of
bortezomib. Curr Pharm Des. 17:1483–1499. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Adams J: Development of the proteasome
inhibitor PS-341. Oncologist. 7:9–16. 2002. View Article : Google Scholar
|
|
29
|
Mitchell BS: The proteasome - an emerging
therapeutic target in cancer. N Engl J Med. 348:2597–2598. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Burger AM and Seth AK: The
ubiquitin-mediated protein degradation pathway in cancer:
therapeutic implications. Eur J Cancer. 40:2217–2229. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen D, Frezza M, Schmitt S, Kanwar J and
Q PD: Bortezomib as the first proteasome inhibitor anticancer drug:
current status and future perspectives. Curr Cancer Drug Targets.
11:239–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hopkins AL and Groom CR: The druggable
genome. Nat Rev Drug Discov. 1:727–730. 2002. View Article : Google Scholar
|
|
33
|
Arend WP: Physiology of cytokine pathways
in rheumatoid arthritis. Arthritis Rheum. 45:101–106. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
McInnes IB and Schett G: Cytokines in the
pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 7:429–442.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stanczyk J, Ospelt C, Gay RE and Gay S:
Synovial cell activation. Curr Opin Rheumatol. 18:262–267. 2006.
View Article : Google Scholar
|
|
36
|
Huber LC, Distler O, Tarner I, Gay RE, Gay
S and Pap T: Synovial fibroblasts: key players in rheumatoid
arthritis. Rheumatology (Oxford). 45669–675. 2006.PubMed/NCBI
|
|
37
|
Knedla A, Neumann E and Muller-Ladner U:
Developments in the synovial biology field 2006. Arthritis Res
Ther. 9:2092007. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hasegawa D, Fujii R, Yagishita N, et al:
E3 ubiquitin ligase synoviolin is involved in liver fibrogenesis.
PLoS One. 5:e135902010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Toh ML, Gonzales G, Koenders MI, et al:
Role of interleukin 17 in arthritis chronicity through survival of
synoviocytes via regulation of synoviolin expression. PLoS One.
5:e134162010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Burr ML, Cano F, Svobodova S, Boyle LH,
Boname JM and Lehner PJ: HRD1 and UBE2J1 target misfolded MHC class
I heavy chains for endoplasmic reticulum-associated degradation.
Proc Natl Acad Sci USA. 108:2034–2039. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gomez-Puerta JA and Bosch X: Therapy:
Spleen tyrosine kinase inhibitors - novel therapies for RA? Nat Rev
Rheumatol. 7:134–136. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Weinblatt ME, Kavanaugh A, Genovese MC,
Musser TK, Grossbard EB and Magilavy DB: An oral spleen tyrosine
kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med.
363:1303–1312. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Koenders MI, Marijnissen RJ, Devesa I, et
al: Tumor necrosis factor-interleukin-17 interplay induces S100A8,
interleukin-1beta, and matrix metalloproteinases, and drives
irreversible cartilage destruction in murine arthritis: rationale
for combination treatment during arthritis. Arthritis Rheum.
63:2329–2339. 2011. View Article : Google Scholar
|