Introduction
Anorectal malformations (ARMs) comprise a broad
spectrum of anomalies including anal atresia, congenital anal
fistula and persistence of the cloaca. The estimated incidence of
ARMs is approximately 1 in 2,500 to 3,000 live births (1–5)
and a male to female ratio of 1.7 has been reported for isolated
(non-syndromic) forms (6).
Isolated forms account for 40–50% of all ARM cases, and these are
sometimes associated with other developmental anomalies, such as
renal and urogenital malformations (5,6).
In the remaining cases, ARMs occur within the context of defined
genetic syndromes or multiple congenital anomalies.
Research suggests that ARMs have a heterogeneous
etiology and include Mendelian and multifactorial forms. The latter
probably arise as a result of a complex interplay between genetic
risk variants and environmental factors. Possible maternal risk
factors include use of multivitamins or medications for severe
chronic dyspepsia and asthma during pregnancy (7,8),
periconceptional injuries or pyrexia, obesity and diabetes.
Research also suggests that smoking and certain occupational
exposures in either parent may be associated with a higher risk of
ARMs (9–12).
Although mutations for some ARM-related syndromes
have been identified, the majority of the underlying genetic
factors for ARMs remain unknown (13,14). Previously reported genetic factors
include the homeobox gene MNX1 in Currarino syndrome
(15), the SALL1 zinc-finger
protein in Townes-Brocks syndrome (16) and the GLI3 gene in
Pallister-Hall syndrome (17).
Since many ARM phenotypes are negatively associated
with reproductive fitness, it is reasonable to assume that a
substantial proportion of ARM patients carry de novo
mutations. Thus candidate gene sequencing to identify rare,
high-penetrance mutations is a rational approach.
The processes of urogenital and anorectal
embryogenesis involves the wingless-type MMTV integration site
family (WNT)/fibroblast growth factor (FGF) signaling pathways.
Mammalian WNT proteins constitute a family of roughly 20 secreted
glycoproteins (18), which act as
short-range paracrine signaling effectors. The FGF family comprises
22 extracellular ligands, whose signals are mediated through a
family of tyrosine kinase receptors. These are termed the five FGF
receptors (FGFR1-5) (19).
Alternative splicing generates multiple isoforms for each FGFR.
Each isoform is characterized by a differing affinity for the
respective ligand (20).
Multiple lines of evidence from mouse models suggest
that genes in these signaling pathways (Fig. 1) are implicated in the etiology of
ARMs. Mice that are homozygous for a hypomorphic Wnt3a
allele display vertebral defects, a short tail due to loss of
caudal vertebrae, deficient cloacal development and incomplete
uro-rectal septation (21).
Moreover, studies involving human pluripotent stem cells have shown
that WNT3A is required for hindgut specification (22). Wnt5a is expressed in the
embryonic colon and rectum and affects the development of the
proximal cloacal plate (23).
Wnt5a-knockout mice display ARMs such as imperforate anus
and the presence of fistulas between the urinary and intestinal
tracts (24). As with Wnt3a and
Wnt5a, Wnt11 has been identified in the developing mouse urogenital
tract (25). Two studies have
reported expression of human WNT11 in the embryonic uro-rectal
septum, the urogenital folds, the labioscrotal swellings and the
epithelium of the esophagus and colon (26,27). Studies in Chinese hamster ovary
cells have shown that Wnt11 signaling leads to downregulation of
the key signaling pathways Wnt/β-catenin, JNK/AP-1 and NF-κB
(28).
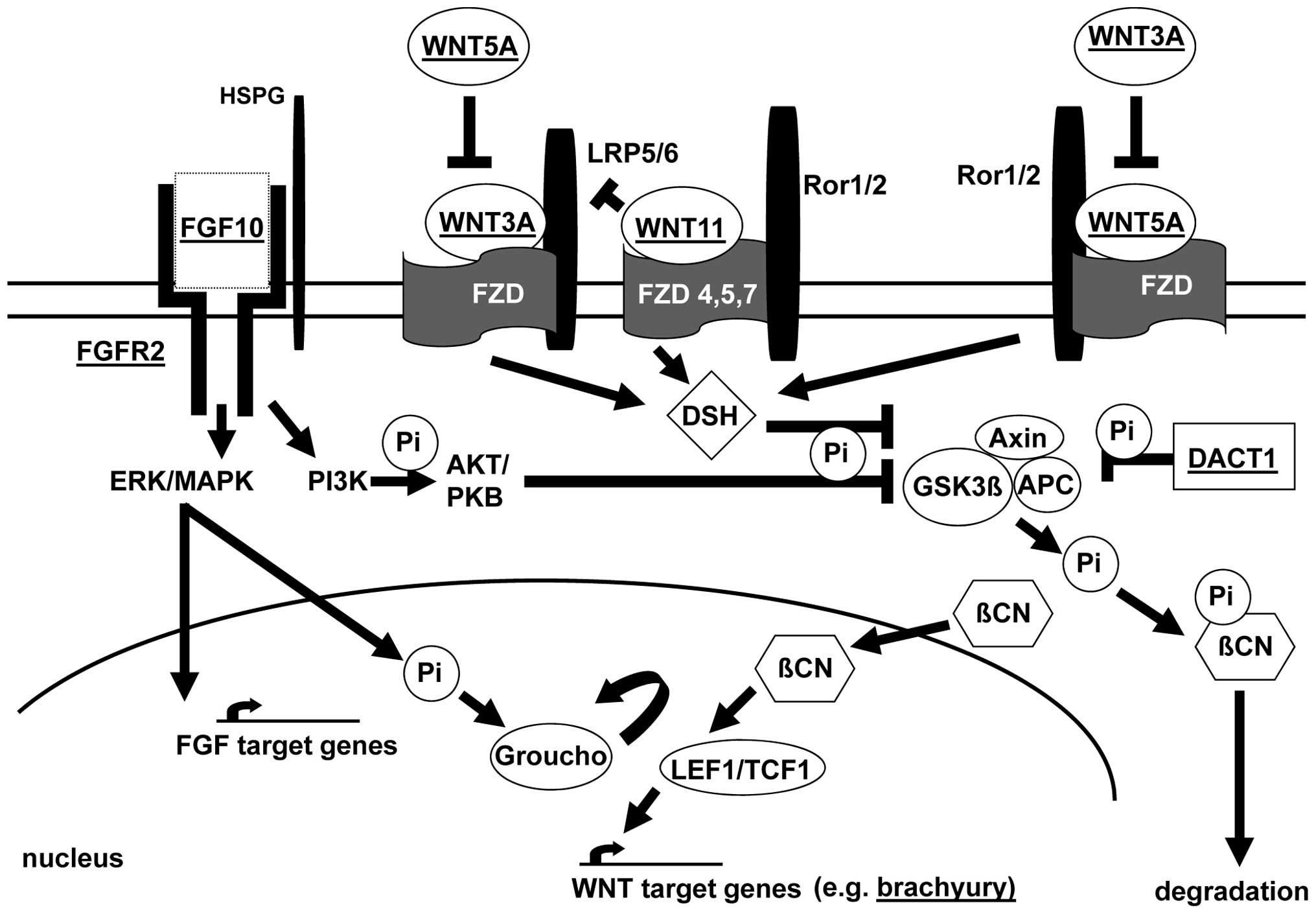 | Figure 1Molecular FGF/WNT signaling factor
pathways as candidates for ARM. Selected proteins and interactions
are shown. FGF signal transduction (binding of FGF10 to its
receptor FGFR2) is shown. Formation of the FGF:FGFR:HSPG (heparan
sulfate proteoglycans) signaling complex activates extra-cellular
signal-regulated kinases (ERK)/mitogen-activated protein kinases
(MAPK) and phosphoinositide-3 kinase (PI3K). PI3K activates protein
kinase AKT or protein kinase B (PKB), with subsequent inhibition of
glycogen synthase kinase 3β (GSK3β) by phosphorylation. MAPK
dependent phosphorylation of transcription factors allows
transcription of FGF target genes. In addition, phosphorylation may
promote the release of transcriptional repressor Groucho from
transcription factor (TCF) 1. This allows interaction between
TCF/lymphoid enhancer-binding factor 1 (LEF1) and β-catenin (βCN)
and stimulation of the transcription of WNT-dependent genes, e.g.
the T-gene (brachyury). In the absence of nuclear βCN,
TCF1/LEF1 act as transcriptional repressors by binding to Groucho.
βCN can also displace Groucho from TCF1/LEF1. Stabilization of βCN
is the major effect of WNT signaling. Absence of this effect leads
to phosphorylation of βCN via a destruction complex including axin,
the APC gene (mutant in adenomatous polyposis) product and GSK3β.
This mechanism primes βCN-Pi for degradation by the ubiquitin
pathway. WNT ligands include the Frizzled (FZD) family of receptors
and these signal through co-receptors such as low-density
lipoprotein receptor related protein 5/6 (LRP5/6) and the orphan
tyrosine kinase receptors ROR1 and ROR2. Binding of WNT3A
(inhibited by WNT5A) to a receptor from the Frizzled (FZD) family
leads to the activation of Dishellved (DSH), a core component of
WNT signaling, thereby enhancing the phosphorylation and subsequent
inhibition of GSK3β. In addition to FZD receptors, WNT5A can also
bind and activate ROR2, resulting in the activation of the
actin-binding protein filamin A and the JNK signaling pathway.
WNT3A and WNT5A exert reciprocal pathway inhibition. WNT11 binds to
several FZD (type 4, 5 and 7) receptors. Inhibition of WNT/βCN
signaling may be mediated by competition for FZD receptors. DACT1
can bind βCN and this complex then inhibits GSK3β. This inhibition
represses the destruction complex and leads to the release of βCN,
thereby increasing its nuclear and cytoplasmic fraction. The figure
is adapted from previous studies (31,60–62). |
Dapper homolog 1 (Dact1) also functions as a
negative regulator of Wnt signaling (29), and its inactivation leads to
perinatal lethality in Dact−/− mice. Wen et
al (30) reported that the
phenotype of Dact null embryos resembled human congenital caudal
regression syndrome, including features such as caudal vertebrae
agenesis, ARMs, renal agenesis/dysplasia, fused kidneys and absence
of the bladder.
The orchestration of embryogenesis via crosstalk of
the WNT and FGF signaling pathways is mediated by several
intracellular cascades (19,31), and integration of WNT/FGF
signaling can also occur at the level of an individual gene
promoter (32). The T gene
(brachyury; T-box containing transcription factor) is a
direct target of WNT3A signaling during paraxial mesoderm
specification (33), and a
T missense mutation (p.Ala338Val) has been observed in four
patients with sacral agenesis/anorectal atresia (34,35). Bagai et al (36) reported that FGF10 plays an
important role in regulating the growth, differentiation and repair
of the urothelium. Other studies found that complete invalidation
of fibroblast growth factor 10 (FGF10) in mice resulted in a
genetically reproducible ARM (37) and failure of ventral fusion in the
urethral plate (38).
FGF10 signaling is mediated by the FGFR2 protein,
which is encoded by the receptor encoding gene FGFR2.
Mutations in this gene cause various forms of autosomal dominant
craniosynostosis syndrome and have been associated with ARMs in
patients with Apert syndrome (acrocephalosyndactyly type I; MIM,
101200), Pfeiffer syndrome types 1 and 2 (acrocephalosyndactyly
type V; MIM, 101600), Crouzon syndrome (craniofacial dysostosis;
MIM, 123500) and Beare-Stevenson cutis gyrata syndrome (MIM,
123790) (39–47).
To explore the possible involvement of the above
genes in the etiology of human ARMs, we performed sequencing
analysis in a sample of 78 patients with ARMs occurring within the
context of multiple congenital anomalies.
Patients and methods
Subjects
Patients were contacted and recruited through the
German self-help organization for patients with anorectal
malformations (SoMA e.V.) as well as the Departments of (i)
Neonatology, Children’s Hospital, University of Bonn, (ii)
Paediatric Surgery and Urology, Centre for Child and Adolescent
Health, Hospital Bremen-Mitte, (iii) Pediatric Surgery and
Pediatric Urology, Children’s Hospital, Cologne, (iv) Pediatric
Surgery, Campus Virchow Clinic, Charité University Hospital Berlin
and (v) Pediatric Surgery, Cnopf’sche Kinderklinik, Nuremberg.
Blood samples were taken from the patients and (if available) their
parents (n=55 trios). In 18 cases only one parent agreed to
participate and in five cases no parental sample could be
obtained.
All 78 ARM patients were of European descent and had
a normal karyotype. The study was approved by the Ethics Committee
of the University of Bonn and written informed consent was obtained
from all subjects prior to inclusion and blood sampling.
Gene analysis
Standard procedures were used for the isolation of
genomic DNA, amplification of DNA via polymerase chain reaction
(PCR) and performance of the automated sequencing analyses. In
brief, primers (sequences available on request) were directed to
all exons of the genes WNT3A, WNT5A, WNT11,
DACT1, FGF10, FGFR2 and T (GenBank acc.
nos.: NM_033131.3, NM_001256105.1, NM_004626.2, NM_016651.5,
NM_004465.1, NM_000141.4, NM_001144913.1-001144919.1, NM_022970.3
and NM_003181.2). The resultant PCR products were subjected to
direct automated sequencing (3130xl Genetic Analyzer; Applied
Biosystems, Foster City, CA, USA). For each patient, both strands
of each amplicon were sequenced. All nucleotide variations were
confirmed via the performance of independent PCR reactions.
Segregation of these variants in family members was investigated by
sequencing the respective PCR products. In the course of direct
sequencing, information was obtained concerning various single
nucleotide polymorphisms (SNPs) in the analyzed genes.
RNA analysis
RNA analyses were performed to determine the effect
of the DNA variation observed in index patient (case B10) and his
mother. The respective blood samples were collected in PAXgene
tubes (PreAnalytiX, Hombrechtikon, Switzerland) in order to
stabilize the intracellular RNA. RNA for reverse transcription (RT)
PCR was prepared with the RNeasy Plus Micro kit (Qiagen, Hilden,
Germany) in accordance with the manufacturer’s instructions.
Primers were directed to FGFR2 exons 10/11 (10/11F-cDNA:
5′-GACAGTTCTGCCAGCGCCTG-3′) and 13 (13R-cDNA:
5′-GCCTCCTTGGGCTTGTCTTTG-3′). This allowed analysis of the effect
of the c.G2012A (p.Thr454=) variant (numbering according to GenBank
acc. no. NM_022970.3 and Swiss-Prot entry P21802) and detection of
alternatively spliced mRNAs.
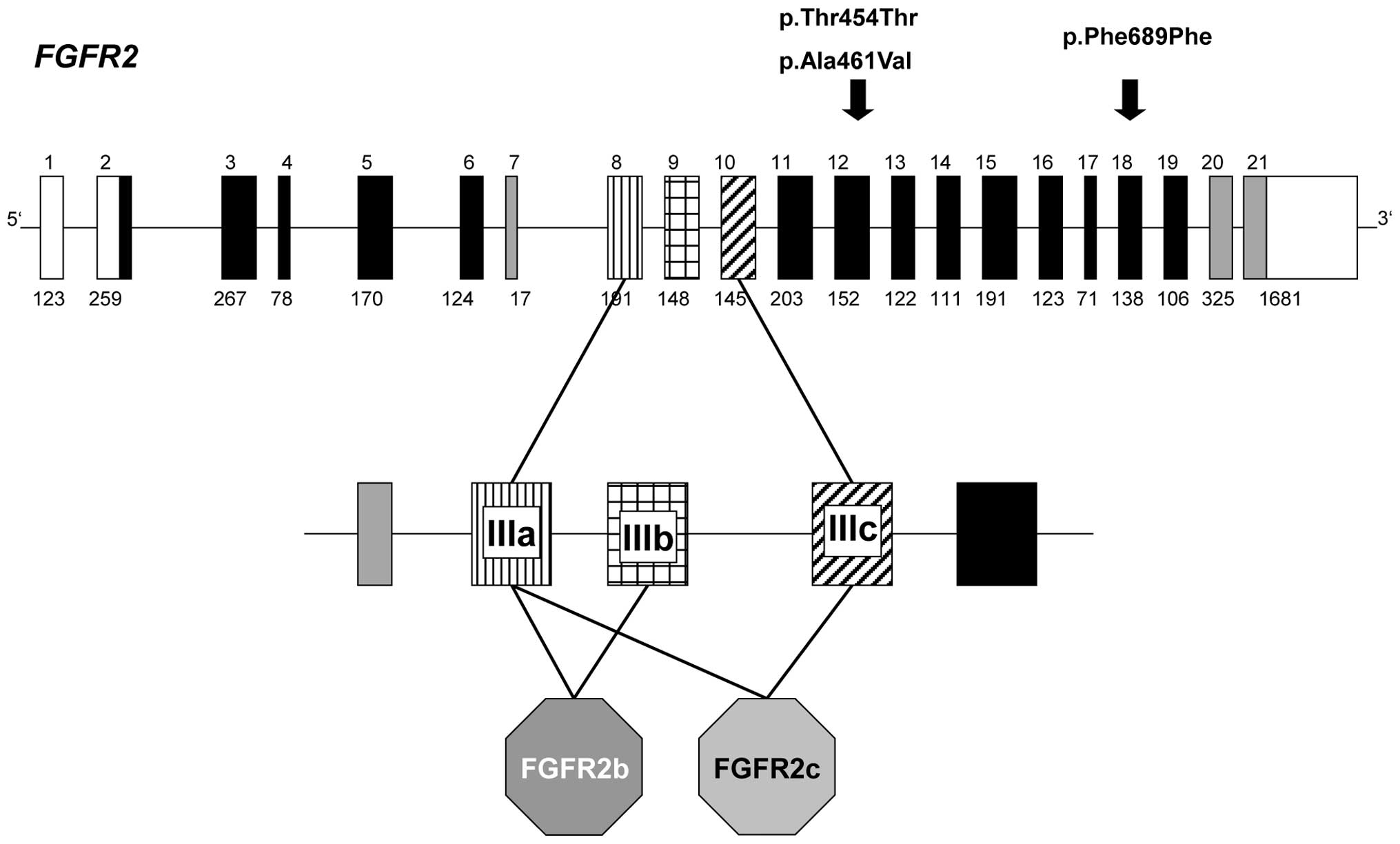
Results
In our candidate gene approach no variant of likely
causal relevance was detected in the genes encoding WNT3A, WNT5A,
WNT11, DACT1, FGF10 and T protein. Sequencing analysis of the
FGFR2 gene revealed the presence of novel heterozygous
variants in three patients (Fig.
2). These variants are listed neither in the current SNP
database (dbSNP136) nor in the 1,000 Genomes catalog.
A synonymous p.Thr454= variant (c.G2012A transition
in exon 12) was identified in patient B10 and his mother. Patient
B10 presented with anal atresia with fistula, hypospadias, left
renal agenesis, rib and vertebral column malformations, lumbar
spina bifida occulta and hexadactylism of the right foot.
Interestingly, his mother also had spina bifida occulta. A closer
look at the sequence surroundings affected by this silent
substitution, revealed the formation of a potential novel 3′
acceptor splice site in exon 12. Calculation of the consensus value
(CV) for splice site recognition (48) revealed a CV of 0.913 for the
wild-type sequence (5′-cattttgtatccag^G; exonic base in capital
letter) and a CV of 0.917 for the novel variant
(5′-cctctcttcaacag^C; substitution underlined). The finding of
nearly identical CVs suggested an alternative usage of these splice
sites with the possible consequence of a deletion of 76 bp
(p.Ala455GlnfsX26). Therefore, RNA analysis was performed.
However, no novel 425-bp fragment (normal, 501 bp) was detected in
the RT-PCR experiments (data not shown). The only detected
variation was due to alternative splicing at the exon 11 donor
splice site, which has been shown to account for the absence of
residues Val428 and Thr429 in several FGFR2 isoforms (see
Swiss-Prot entry P21802).
Another heterozygous exon 12 FGFR2 variant
(c.C2032T) was detected in patient G10. This variant is predicted
to result in a p.Ala461Val amino acid substitution. Patient G10
presented with perineal fistula, hypoplasia of the left thumb,
pre-axial polydactyly of the left hand, wedged vertebra (thoracic
and cervical), rib malformations, dextroversio cordis and double
kidney (left). The mother showed wild-type sequence only, and no
DNA was available from the father. To test if this variant had
arisen de novo on the maternal allele, a search for neutral
heterozygous variants 5 kb upstream and downstream of exon 12 was
performed. Detection of such variants would have allowed
allele-specific PCR, and a distinction as to whether the variant
resided on the maternal or the paternal allele. However, no
heterozygous SNP or private variant was detected in the 10-kb
flanking exon 12. Interestingly, pathogenicity prediction of this
sequence alteration varied depending on the program used. According
to Mutation Taster (www.mutationtaster.org), this variant had a 0.981
probability of being disease-causing. In contrast, three other
programs predicted that it was benign: [PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/,
benign with a score of 0.005); MutPred (http://mutpred.mutdb.org, probability of being
deleterious 0.235); and SNPs&Go (http://snps-and-go.biocomp.unibo.it/cgi-bin/snps-and-go/runpred.cgi,
reliability index 1].
A third variant was identified in patient A02, who
presented with anal atresia with recto-urethral prostatic fistula,
ventricular septal defect, subaortic stenosis and dystopic kidney.
This synonymous FGFR2 exon 18 variant (c.C2717T, p.Phe689=)
was assumed to have no effect. Hence, no further analyses of this
variant were performed.
Direct sequencing also generated information
concerning several SNPs (dbSNP136) in the investigated genes. For
all genes, similar haplotype data were found in the European
Population of the International HapMap Project, the CEPH (Centre
d’Etude du Polymorphisme Human) pedigrees and the PDR90 (The NIH
Polymorphism Discovery Resource; 90 individual screenings)
subset.
Discussion
WNT/FGF signaling pathways (Fig. 1) orchestrate correct patterning,
cell specification and tissue differentiation during embryogenesis
(19,31). Research has shown that disruption
of this coordinated interplay can result in severe malformations in
mice and humans (49,50), including urogenital and anorectal
anomalies. The present study investigated selected candidate genes
from these pathways, chosen on the basis of observations in mice
and human cell lines and/or their involvement in diseases
associated with ARMs. However, no potential causal variant for ARMs
was detected in the genes encoding WNT3A, WNT5A, WNT11, DACT1,
FGF10 and T protein in the present cohort.
In contrast, FGFR2 analysis revealed the
presence of novel heterozygous variations in three patients
(Fig. 2). We initially considered
the two exon 12 variants to be of potential causal relevance. Our
rationale for this hypothesis was that as well as affecting all
FGFR2 isoforms, a mutation in this exon would affect a cytoplasmic
part of the protein which has not yet been implicated in the
various forms of autosomal dominant craniosynostosis syndrome.
Moreover, the program Mutation Taster (51) predicted that the p.Ala461Val amino
acid substitution was disease-causing. However, Thusberg et
al (52) reported on the
suboptimal performance of this program and in line with their
findings of the performance of mutation pathogenicity prediction
methods, several other - more reliable - programs classified it as
benign. Furthermore, the results of our mRNA experiments suggest
that the p.Thr454= variant had no effect on correct splicing.
Despite these negative findings, the possibility
remains that these nucleotide substitutions contribute to the ARM
phenotype through as-yet-unknown mechanisms. As FGF10 is
expressed in the mesenchyme that lies adjacent to the urethral
plate, a plausible hypothesis is that it is important in the
regulation of endoderm and/or mesenchyme growth, and thus in
proliferation-driven urogenital and anorectal development (37,38). Interestingly, ARM patient B10 and
his mother - both of whom showed spina bifida occulta - carried the
silent p.Thr454= variant. Severe spinal dysraphism has been
observed in association with an FGFR2 p.Ser351Cys mutation
(53) and spina bifida occulta
occurs in the mouse mutant Brachyury curtailed
(Tc/+) (54).
However, whereas Tc/+ mice are tailless, several
of the FGFR2 amino acid substitutions observed in patients with
Beare-Stevenson (55), Crouzon
and Pfeiffer syndromes (56) are
associated with sacral appendage. These findings, together with the
repeated observation of ARMs in mice (37,38) and patients with FGFR2 defects
(39–47), imply that this FGF signaling
pathway is of crucial importance in normal caudal development,
rendering the coincidental co-occurrence of these defects
unlikely.
In summary, no significant association between ARMs
and mutations in WNT3A, WNT5A, WNT11,
DACT1, FGF10, FGFR2 or the T gene was
found in the present cohort. However, although our patient cohort
was larger than those used in previous candidate gene studies of
ARMs (34,57–59), the sample size may have been too
small to detect rare causal mutational events. Furthermore, we
cannot exclude the possibility that mutations in the promoter
region, in as yet unknown regulatory sequences or in non-coding
regions that are not detectable with the method applied were
overlooked. Future studies should consider additional proteins of
the WNT/FGF signaling pathways as possible candidates.
Acknowledgements
M.D., A.H., S.S.M., E.S., E.B.,
M.M.N., H.R. and M.L. are members of the ‘Network for the
Systematic Investigation of the Molecular Causes, Clinical
Implications and Psychosocial Outcome of Congenital Uro-Rectal
Malformations (CURE-Net)’, which is supported by a research grant
(01GM08107) from the German Federal Ministry of Education and
Research (Bundesministerium für Bildung und Forschung, BMBF).
S.S.M. is supported by a research grant from the
Richard-Winter-Stiftung. G.D. and E.B. are supported by the BONFOR
program of the University of Bonn, grant nos. O-149.0096 and
O-149.0099, respectively. We thank all patients and family members
for their cooperation, as well as the German Self-Help Organization
for People with Anorectal Malformations (SoMA e.V.). We thank Pia
Uerdingen for her excellent technical assistance and Dr Christine
Schmael for her expert advice regarding the manuscript.
References
|
1.
|
ED SmithIncidence, frequency of types and
aetiology of anorectal malformationsBirth Defects Orig Artic
Ser2423124619883067766
|
|
2.
|
S ChoSP MooreT FangmanOne hundred three
consecutive patients with anorectal malformations and their
associated anomaliesArch Pediatr Adolesc
Med155587591200110.1001/archpedi.155.5.58711343503
|
|
3.
|
A CuschieriEUROCAT Working
GroupDescriptive epidemiology of isolated anal anomalies: a survey
of 4.6 million births in EuropeAm J Med
Genet103207215200110.1002/ajmg.153211745992
|
|
4.
|
MA LevittA PeñaAnorectal
malformationsOrphanet J Rare Dis233200710.1186/1750-1172-2-33
|
|
5.
|
C StollY AlembikB DottMP RothAssociated
malformations in patients with anorectal anomaliesEur J Med
Genet50281290200710.1016/j.ejmg.2007.04.00217572165
|
|
6.
|
A CuschieriEUROCAT Working GroupAnorectal
anomalies associated with or as part of other anomaliesAm J Med
Genet110122130200210.1002/ajmg.1037112116249
|
|
7.
|
N AcsF BánhidyEH PuhóAE CzeizelA possible
association between maternal dyspepsia and congenital rectal/anal
atresia/stenosis in their children: a population-based case-control
studyActa Obstet Gynecol
Scand8810171023200910.1080/00016340903128447
|
|
8.
|
S LinJPW MunsieML Herdt-LosavioCM
DruschelK CampbellML BrownePA RomittiRS OlneyEM BellNational Birth
Defects Prevention StudyMaternal asthma medication use and the risk
of selected birth
defectsPediatrics129e317e324201210.1542/peds.2010-266022250027
|
|
9.
|
IA Van RooijCH WijersPN RieuHS HendriksMM
BrouwersNV KnoersI de BlaauwN RoeleveldMaternal and paternal risk
factors for anorectal malformations: a Dutch case-control
studyBirth Defects Res A Clin Mol Teratol88152158201020073076
|
|
10.
|
A HackshawC RodeckS BonifaceMaternal
smoking in pregnancy and birth defects: a systematic review based
on 173 687 malformed cases and 11.7 million controlsHum Reprod
Update17589604201110.1093/humupd/dmr02221747128
|
|
11.
|
SC TinkerJ ReefhuisAM DellingerDJ
JamiesonMaternal injuries during the periconceptional period and
the risk of birth defects, National Birth Defects Prevention Study,
1997–2005Paediatr Perinat Epidemiol25487496201121819430
|
|
12.
|
N ZwinkE JenetzkyH BrennerParental risk
factors and anorectal malformations: systematic review and
meta-analysisOrphanet J Rare
Dis625201110.1186/1750-1172-6-2521586115
|
|
13.
|
B SolomonVACTERL/VATER AssociationOrphanet
J Rare Dis656201110.1186/1750-1172-6-56
|
|
14.
|
E MundtMD BatesGenetics of Hirschsprung
disease and anorectal malformationsSemin Pediatr
Surg19107117201010.1053/j.sempedsurg.2009.11.01520307847
|
|
15.
|
E BelloniG MartuccielloD VerderioE PontiM
SeriV JasonniM TorreM FerrariLC TsuiSW SchererInvolvement of the
HLXB9 homeobox gene in Currarino syndromeAm J Hum
Genet66312319200010.1086/30272310631160
|
|
16.
|
J KohlhaseA WischermannH ReichenbachU
FrosterW EngelMutations in the SALL1 putative transcription factor
gene cause Townes-Brocks syndromeNat
Genet188183199810.1038/ng0198-819425907
|
|
17.
|
S KangJM Graham JrAH OlneyLG
BieseckerGLI3 frameshift mutations cause autosomal dominant
Pallister-Hall syndromeNat Genet15266268199710.1038/ng0397-266
|
|
18.
|
H CleversWnt/beta-catenin signaling in
development and
diseaseCell127469480200610.1016/j.cell.2006.10.01817081971
|
|
19.
|
K DoreyE AmayaFGF signalling: diverse
roles during early vertebrate
embryogenesisDevelopment13737313742201010.1242/dev.03768920978071
|
|
20.
|
X ZhangOA IbrahimiSK OlsenH UmemoriM
MohammadiDM OrnitzReceptor specificity of the fibroblast growth
factor family. The complete mammalian FGF familyJ Biol
Chem2811569415700200610.1074/jbc.M60125220016597617
|
|
21.
|
C Van de VenM BialeckaR NeijtsT YoungJE
RowlandEJ StringerC van RooijenF MeijlinkA NóvoaJN FreundConcerted
involvement of Cdx/Hox genes and Wnt signaling in
morphogenesis of the caudal neural tube and cloacal derivatives
from the posterior growth zoneDevelopment138345134622011
|
|
22.
|
JR SpenceCN MayhewSA RankinMW KuharJE
VallanceK TolleEE HoskinsVV KalinichenkoSI WellsAM ZornNF ShroyerJM
WellsDirected differentiation of human pluripotent stem cells into
intestinal tissue in
vitroNature470105109201110.1038/nature0969121151107
|
|
23.
|
M NakataY TakadaT HishikiT SaitoK TeruiY
SatoH KosekiH YoshidaInduction of Wnt5a-expressing
mesencymal cells adjacent to the cloacal plate is an essential
process for its proximodistal elongation and subsequent anorectal
developmentPediatr Res661491542009
|
|
24.
|
CC TaiFG SalaHR FordKS WangC LiP MinooTC
GrikscheitS BellusciWnt5a knock-out mouse as a new model of
anorectal malformationJ Surg
Res156278282200910.1016/j.jss.2009.03.08719577771
|
|
25.
|
V MehtaLL AblerKP KeilCT SchmitzPS JoshiCM
VezinaAtlas of Wnt and R-spondin gene expression in
the developing male mouse lower urogenital tractDev
Dyn24025482560201121936019
|
|
26.
|
M LakoT StrachanP BullenDI WilsonSC
RobsonS LindsayIsolation, characterisation and embryonic expression
of WNT11, a gene which maps to 11q13.5 and has possible
roles in the development of skeleton, kidney and
lungGene21910111019989757009
|
|
27.
|
H LickertA KispertS KutschR
KemlerExpression patterns of Wnt genes in mouse gut developmentMech
Dev105181184200110.1016/S0925-4773(01)00390-211429295
|
|
28.
|
A RailoII NagyP KilpeläinenS VainioWnt-11
signaling leads to down-regulation of the Wnt/β-catenin, JNK/AP-1
and NF- B pathways and promotes viability in the CHO-K1 cellsExp
Cell Res31423892399200818572162
|
|
29.
|
BN CheyetteJS WaxmanJR MillerK TakemaruLC
SheldahlN KhlebtsovaEP FoxT EarnestRT MoonDapper, a
Dishevelled-associated antagonist of beta-catenin and JNK
signaling, is required for notochord formationDev
Cell2449461200210.1016/S1534-5807(02)00140-511970895
|
|
30.
|
J WenYJ ChiangC GaoH XueJ XuY NingRJ
HodesX GaoYG ChenLoss of Dact1 disrupts planar cell polarity
signaling by altering dishevelled activity and leads to posterior
malformation in miceJ Biol
Chem2851102311030201010.1074/jbc.M109.08538120145239
|
|
31.
|
ME PownallHV IsaacsFGF signalling in
vertebrate developmentDevelopmental Biology, Book 2DS KesslerMorgan
& Claypool Life SciencesCA14162010
|
|
32.
|
T HaremakiY TanakaI HongoM YugeH
OkamotoIntegration of multiple signal transducing pathways on Fgf
response elements of the Xenopus caudal homologue
Xcad3Development13049074917200310.1242/dev.0071812930781
|
|
33.
|
TP YamaguchiS TakadaY YoshikawaN WuAP
McMahonT (brachyury) is a direct target of Wnt3a during paraxial
mesoderm specificationGenes
Dev1331853190199910.1101/gad.13.24.318510617567
|
|
34.
|
C PapapetrouF DrummondW ReardonR WinterL
SpitzYH EdwardsA genetic study of the human T gene and its
exclusion as a major candidate gene for sacral agenesis with
anorectal atresiaJ Med Genet36208213199910204846
|
|
35.
|
N GhebraniousRD BlankCL RaggioJ StaubliE
McPhersonL IvacicK RasmussenFS JacobsenT FaciszewskiJK BurmesterA
missense T (Brachyury) mutation contributes to vertebral
malformationsJ Bone Miner Res23157615832008
|
|
36.
|
S BagaiE RubioJF ChengR SweetR ThomasE
FuchsR GradyM MitchellJA BassukFibroblast growth factor-10 is a
mitogen for urothelial cellsJ Biol
Chem2772382823837200210.1074/jbc.M20165820011923311
|
|
37.
|
TJ FairbanksS De LangheFG SalaD
WarburtonKD AndersonS BellusciRC BurnsFibroblast growth factor 10
(Fgf10) invalidation results in anorectal malformation in miceJ
Pediatr Surg39360365200410.1016/j.jpedsurg.2003.11.03415017552
|
|
38.
|
S YucelW LiuD CorderoA DonjacourG CunhaLS
BaskinAnatomical studies of the fibroblast growth factor-10 mutant,
Sonic Hedge Hog mutant and androgen receptor mutant mouse genital
tubercleAdv Exp Med
Biol545123148200410.1007/978-1-4419-8995-6_815086024
|
|
39.
|
H OhashiH NishimotoJ NishimuraM SatoS
ImaizumiT AiharaY FukushimaAnorectal anomaly in Pfeiffer
syndromeClin Dysmorphol2283319938298735
|
|
40.
|
WJ ParkGA MeyersX LiC ThedaD DaySJ OrlowMC
JonesEW JabsNovel FGFR2 mutations in Crouzon and Jackson-Weiss
syndromes show allelic heterogeneity and phenotypic variabilityHum
Mol Genet412291233199510.1093/hmg/4.7.12298528214
|
|
41.
|
WJ ParkC ThedaNE MaestriGA MeyersJS
FryburgC DufresneMM Cohen JrEW JabsAnalysis of phenotypic features
and FGFR2 mutations in Apert syndromeAm J Hum
Genet5732132819957668257
|
|
42.
|
RA PfeifferS RinnertR PoppG
RöckeleinAsymmetrical coronal synostosis, cutaneous syndactyly of
the fingers and toes, and jejunal atresia in a male infantAm J Med
Genet63175176199610.1002/(SICI)1096-8628(19960503)63:1%3C175::AID-AJMG30%3E3.0.CO;2-J8723105
|
|
43.
|
KA PrzylepaW PaznekasM ZhangM GolabiW
BiasMJ BamshadJC CareyBD HallR StevensonSJ OrlowFibroblast growth
factor receptor 2 mutations in Beare-Stevenson cutis gyrata
syndromeNat Genet13492494199610.1038/ng0896-4928696350
|
|
44.
|
BP LeHeupJP MasuttiP DroulléJ TisserandThe
Antley-Bixler syndrome: report of two familial cases with severe
renal and anal anomaliesEur J
Pediatr154130133199510.1007/BF019919167720741
|
|
45.
|
F SchaeferC AndersonB CanB SayNovel
mutation in the FGFR2 gene at the same codon as the Crouzon
syndrome mutations in a severe Pfeiffer syndrome type 2 caseAm J
Med
Genet75252255199810.1002/(SICI)1096-8628(19980123)75:3%3C252::AID-AJMG4%3E3.0.CO;2-S9475591
|
|
46.
|
A KřepelováA BaxováP CaldaR PlavkaJ
KaprasFGFR2 gene mutation (Tyr375Cys) in a new case of
Beare-Stevenson syndromeAm J Med Genet7636236419989545103
|
|
47.
|
T KodakaY KanamoriM SugiyamaK HashizumeA
case of acrocephalosyndactyly with low imperforate anusJ Pediatr
Surg39E32E34200410.1016/j.jpedsurg.2003.09.03714694405
|
|
48.
|
MB ShapiroP SenapathyRNA splice junctions
of different classes of eukaryotes: sequence statistics and
functional implications in gene expressionNucleic Acids
Res1571557174198710.1093/nar/15.17.71553658675
|
|
49.
|
AO WilkieBad bones, absent smell, selfish
testes: The pleiotropic consequences of human FGF receptor
mutationsCytokine Growth Factor
Rev16187203200510.1016/j.cytogfr.2005.03.00115863034
|
|
50.
|
ET ShifleySE ColeThe vertebrate
segmentation clock and its role in skeletal birth defectsBirth
Defects Res C Embryo
Today81121133200710.1002/bdrc.2009017600784
|
|
51.
|
JM SchwarzC RödelspergerM SchuelkeD
SeelowMutationTaster evaluates disease-causing potential of
sequence alterationsNat
Methods7575576201010.1038/nmeth0810-57520676075
|
|
52.
|
J ThusbergA OlatubosunM VihinenPerformance
of mutation pathogenicity prediction methods on missense
variantsHum Mutat32358368201110.1002/humu.2144521412949
|
|
53.
|
K ChunJ Siegel-BarteltD ChitayatJ
PhillipsPN RayFGFR2 mutation associated with clinical
manifestations consistent with Antley-Bixler syndromeAm J Med
Genet77219224199810.1002/(SICI)1096-8628(19980518)77:3%3C219::AID-AJMG6%3E3.0.CO;2-K9605588
|
|
54.
|
CHT ParkJH PruittD BennettA mouse model
for neural tube defects: The curtailed (Tc) mutation
produces spina bifida occulta in Tc/+ animals and spina
bifida with meningomyelocele in
Tc/tTeratology3930331219892658196
|
|
55.
|
J McGaughranS SinnottR SusmanMF BuckleyG
ElakisT CoxT RoscioliA case of Beare-Stevenson syndrome with a
broad spectrum of features and a review of the FGFR2 Y375C mutation
phenotypeClin
Dysmorphol158993200610.1097/01.mcd.0000194407.92676.9d16531735
|
|
56.
|
AL ShanskeD StaffenbergJT GoodrichSacral
appendage in a child with an FGFR2 mutation: a report and reviewAm
J Med Genet A146A21722175200810.1002/ajmg.a.3243618629881
|
|
57.
|
M SeriG MartuccielloL PaleariA BolinoM
PrioloG SalemiP ForaboscoF CaroliR CusanoT ToccoExclusion of the
Sonic Hedgehog gene as responsible for Currarino syndrome and
anorectal malformations with sacral hypodevelopmentHum
Genet104108110199910.1007/s00439005091910071202
|
|
58.
|
V KrügerM KhoshvaghtiH ReutterH VogtTM
BoemersM LudwigInvestigation of FGF10 as a candidate gene in
patients with anorectal malformations and exstrophy of the
cloacaPediatr Surg Int248938972008
|
|
59.
|
NB AgochukwuDE Pineda-AlvarezAA KeatonN
Warren-MoraMS RaamA KamatSC ChandrasekharappaBD SolomonAnalysis of
FOXF1 and the FOX gene cluster in patients with VACTERL
associationEur J Med
Genet54323328201110.1016/j.ejmg.2011.01.00721315191
|
|
60.
|
M CatalaGenetic control of caudal
developmentClin
Genet618996200210.1034/j.1399-0004.2002.610202.x11940082
|
|
61.
|
L GrumolatoG LiuP MongR MudbharyR BiswasR
ArroyaveS VijayakumarAN EconomidesSA AaronsonCanonical and
noncanonical Wnts use a common mechanism to activate completely
unrelated coreceptorsGenes
Dev2425172530201010.1101/gad.195771021078818
|
|
62.
|
P Uysal-OnganerRM KyptaWnt11 in 2011 - the
regulation and function of a non-canonical WntActa Physiol
(Oxf)2045264201210.1111/j.1748-1716.2011.02297.x21447091
|