Introduction
During mitosis, sister chromatids are evenly
segregated into daughter cells on the basis of the precise action
of spindle checkpoint proteins (SCPs) (1). The SCPs sense the existence of
misaligned sister chromatids during mitosis and meiosis and use
multiple mechanisms to inhibit the ubiquitin ligase activity of the
anaphase-promoting complex or cyclosome (APC/C) protein Cdc20
(1). At the same time, the SCPs
indirectly preserve chromosome cohesion and delay the onset of
sister chromatid separation (1),
thus stabilising the cell and delaying the onset of anaphase
(2). SCPs are vital in
establishing a physical association among sister chromatids in the
S phase and maintaining this cohesion until their separation
(3). The molecular components of
the SCPs include at least two evolutionarily conserved protein
families, Mad and Bub (4). Bub1
not only phosphorylates Cdc20 but also interacts with Mad2 by an
unknown mechanism (5). Therefore,
SCPs are essential for cell proliferation and differentiation.
Recently, the role of SCPs in carcinogenesis has gained increased
attention.
Alterations in cellular signalling pathways that
respond to external stimuli regulating cell mitosis, growth,
differentiation and death commonly contribute to cancer. Viruses
have developed mechanisms for modulating cellular signalling
pathways that reprogram host cells to support their viral life
cycles or modulate the host defence responses (6,7).
One such case is human papillomavirus (HPV). HPV has been
associated with benign and malignant epithelial lesions. In
particular, infection with high-risk (HR) HPV (the most epidemic
types of which are HPV16/18) is key in cervical cancer (CC)
development. In the present study, we investigated whether any
changes involving SCPs occur during the development of CC and we
explored the possible mechanism by which these proteins change
during cancer.
Materials and methods
Tumour specimens and immunohistochemistry
(IHC)
Samples of cervical neoplasms at different stages
were retrieved from files at the Department of Pathology of Tongji
Hospital. The experimental protocols were reviewed and approved by
the Institutional Review Board of the Tongji Hospital Ethics
Committee. Ninety cervical specimens, which were all
HPV16-positive, as determined by HPV DNA gene array (8), were classified as follows: 65 were
cervical intraepithelial neoplasm (CIN) biopsy specimens, and
included 15 CIN I, 23 CIN II and 27 CIN III; 25 were cervical
squamous cell carcinoma tissue samples that had been surgically
removed. The patients were 23–65 years of age, with a mean age of
43.5 years. Ten samples from healthy women were used as
controls.
The sections were deparaffinized in xylene and
rehydrated in graded alcohols as described (9). The slides were incubated with a
monoclonal antibody against Bub1 (1:50) or Mad2 (1:50) (BD
Biosciences, USA).
The slides were analysed by two pathologists who
were blinded to the clinical data using light microscopy. Any
appreciable brown staining was considered positive and graded as
follows: −, no staining; +, barely detectable staining; ++, easily
observed fine granules diffusely present throughout the nucleus or
cytoplasm; +++, staining so strong that nuclear details were
obscured. A corresponding H&E stain was reviewed for
determining the diagnosis and mapping the location of the various
histological patterns (10).
Immunofluorescence, immunoprecipitation
(IP) and western blot analysis
For immunofluorescence, cells were fixed with 4%
paraformaldehyde and incubated with 0.1% Triton X-100 in PBS,
followed by staining the nuclei with propidium iodide (PI).
For the IP, cells were harvested and lysed. The
clarified total cell lysates were pre-cleaned with protein
G-agarose (Upstate Biotechnology, Inc., USA) for 3 h at 4°C, and
then incubated with primary antibodies overnight at 4°C with gentle
rotation. Anti-Bub1, anti-Mad2 (BD Biosciences), anti-GFP or
anti-His (Santa Cruz Biotechnology, Inc., USA) antibody was added
individually to 1 mg of pre-cleared cell lysate in parallel tests.
Immunocomplexes were recovered by incubation with protein
G-agarose for 2 h at 4°C on a rotating platform. The
antibody and protein G-agarose complexes were separated by SDS-PAGE
and analysed by western blot analysis.
For the western blot analysis, proteins were
separated by SDS-PAGE and transferred to a polyvinylidene
difluoride membrane. The membranes were incubated with primary
antibodies [anti-GFP (1:500), anti-His (1:200) (Santa Cruz
Biotechnology, Inc.), anti-p21 (1:200), anti-Bub1 (1:200),
anti-Mad2 (1:100), or β-actin antibody (1:500) (BD Biosciences)]
overnight at 4°C. The membranes were then incubated with
horseradish peroxidase-conjugated secondary antibody (1:
1,500) for 1 h at 37°C prior to detection by ECL (Pierce, USA).
Construction of recombinant HPV-16/18 E5
expression plasmids
HPV-16/18 E5 genes were amplified using PCR from a
plasmid (pBR322-HPV16/18) that contained the complete genome of
HPV16/18 and was kindly provided by Professor Zur Hausen
(Heidelberg University, Germany). The PCR primers are shown in
Table I (11). The reaction conditions were: 30
denaturation cycles at 94°C for 30 sec, annealing at 56°C for 30
sec, and extension at 72°C for 45 sec. The E5 PCR products were
ligated into pEGFP-C1 (Clontech, USA) or pcDNA3.1-V5-His vector
(Invitrogen, USA). Positive colonies were screened by PCR and
sequenced to confirm the identity of the DNA insertions.
 | Table IHPV16/18 E5 PCR primers for pEGFP-C1
and pcDNA3.1/his. |
Table I
HPV16/18 E5 PCR primers for pEGFP-C1
and pcDNA3.1/his.
| Gene | pEGFP-C1 | pcDNA3.1/his |
|---|
| HPV16 E5 |
| F CCCAAGCTTACTGCATCCACAACATTACTGGC | GGGATCCACCATGTACTGCATCCACAACATTACTGGC |
| R GGGATCCATTATGTAATTAAAAAGCGTGCA | AAAGCGGCCGCAATGTAATTAAAAAGCGTGCA |
| HPV18 E5 |
| F GCGAATTCCATGTTATCACTTATTTTTTTATTTTGC | GGGATCCGCCACCATGTTATCACTTATTTTTTTATTTTGC |
| R CCGGATCCCAACCTATACAATTACTGTAAAGACAA | AAAGCGGCCGCAACCTATACAACTGTAAAGACAA |
Preparation of stable cell lines
expressing HPV E5
The following cell lines were used: SiHa (a CC cell
line that contains an integrated HPV16 genome, ATCC HTB-35™), HeLa
(a CC cell line containing HPV-18 sequences, ATCC CCL-2™), C33A (a
CC cell line negative for HPV DNA and RNA, which was used as
control for the potential effects of other HPV components) and the
HaCaT cell line (an immortal human keratinocyte cell line from
Wuhan University Typical Object Preservation Centre, also used as a
control). All cell lines were maintained in DMEM/FCS. Cells were
transfected with E5-containing plasmids (HPV16 E5 was transfected
into SiHa, C33A, HaCaT cells; HPV18 E5 was transfected into HeLa
cells) using Lipofectin™ (Invitrogen) and grown for 3 weeks in
DMEM/FCS containing 600–1,000 mg/l G418 (a cytotoxic dose
for non-transfected cells within 1 week). Individual colonies of
G418-selected cells were isolated and expanded. The expression of
His-E5 or GFP-E5 protein was confirmed using anti-His or anti-GFP
antibody in western blot analyses, as a commercial antibody against
E5 is not available.
Analysis of gene expression by RT-PCR and
real-time RT-PCR
Total-RNA was reverse-transcribed by the RT-PCR
System (Life Technologies, USA) (12). The primer sets used are shown in
Table II (generated by Takara
Biotechnology Co., Ltd., Japan). Real-time RT-PCR was performed on
an ABI PRISM 7700 Sequence Detector. Amplified products were
detected with SYBR Green PCR Master Mix (PE Biosystems, USA). Each
primer set was tested in triplicate for each sample. Real-time
RT-PCR was performed for 35 cycles of 15 sec at 95°C and 1 min at
56°C, preceded by a 2-min incubation at 65°C and a 10-min
incubation at 95°C. The instrument software calculated the number
of cycles required for the accumulated signal to reach a designated
threshold value (cycle threshold, Ct) at least 10 standard
deviations greater than the baseline. Thus, the Ct value was
proportional to the number of starting copies of the target
sequence. The relative quantity of gene expression was calculated
using the ΔΔCt method, ΔΔCt = [Ct gene of interest (stimulated
sample)-Ct GAPDH (stimulated sample)]-[Ct gene of interest (vehicle
control)-Ct GAPDH (vehicle control)] (13).
| Table IIReal-time RT-PCR primer sets. |
Table II
Real-time RT-PCR primer sets.
| Gene | Forward primer | Reverse primer |
|---|
| Bub1 |
GCGACTTTGGATTCTTGTGAGGA |
RGGCTGGCTCAGACGAAGTAAGG |
| Mad2 |
GTGCAGAAATACGGACTCACCTTG |
TTCCAGGACCTCACCACTTTCA |
| P21 |
GCTGCGTTCACAGGTGTTTC |
CATGGGTTCTGACGGACATC |
| HPV16 E5 |
TGACAAATCTTGATACTGCATCCAC |
ATAGGCAGACACACAAAAGCACAC |
| HPV18 E5 |
CCGCTTTTGCCATCTGTCT |
GCAGGGGACGTTATTACCACA |
| GAPDH |
ACCCACTCCTCCACCTTTGA |
TGACAAAGTGGTCGTTGAGGG |
Cell proliferation and cell cycle
analyses
Cells were collected and fixed as usual, and then
incubated in Ki-67 antibody (mouse) (1:50) (Santa Cruz
Biotechnology, Inc.) at 4°C overnight. The cells were then
incubated in the dark with fluorescein isothiocyanate
(FITC)-labeled rabbit anti-mouse immunoglobulin
(1:1,000) (Santa Cruz Biotechnology, Inc.). After an additional
wash in PBS, the cells were resuspended and stained with 1 ml of
PI/RNase solution (50 g PI and 200 g RNase/l in PBS) for 15 min at
room temperature before analysing on a FACSCalibur™ flow cytometer
(Becton-Dickinson, USA). The cells were characterised based on
green (FITC) and red (PI) fluorescence.
Statistical analysis
Statistical analyses and graphical presentations
were performed by SPSS 13.0 and Sigma Plot 10.0, and the results
are reported as the means ± standard error of the mean (SEM).
Statistical analysis of significance was based on analysis of
variance or χ2 analysis. P<0.05 was considered to
indicate statistically significant differences.
Results
As cervical disease progresses from CIN I
to CIN II, CIN III, and finally CC, the expression levels of Bub1
and Mad2 are gradually suppressed, as shown by IHC
We examined Bub1 and Mad2 expression in cervical
neoplasm tissue samples by IHC. As cervical disease progressed from
CIN I to CIN II, CIN III, and eventually CC, the expression of the
two proteins decreased gradually. Indeed, in some cases of CC, Bub1
and Mad2 were undetectable (Table
III and Fig. 1). In 10
healthy samples, the levels of Bub1 and Mad2 were significantly
higher (P<0.01) than in the CIN and CC groups. Furthermore, the
expression levels in the CIN groups were also significantly
different from those in the CC group (P<0.05). We concluded that
the expression levels of Bub1 and Mad2 decrease with the severity
of cervical disease and the decrease possibly starts from the CIN I
stage.
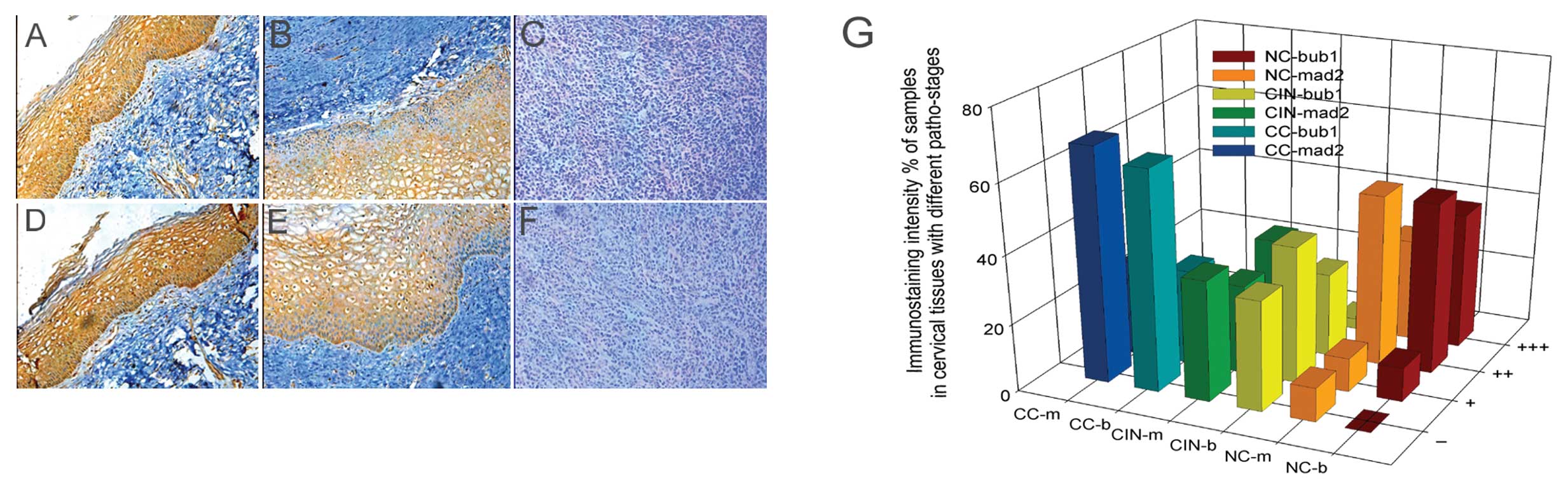 | Figure 1Immunostaining intensity of Bub1 and
Mad2 proteins in cervical tissues at various pathological stages
with different staining classes by IHC. (A-C) Expression of Bub1 in
NC-CIN I, CIN II–III, CC; (D-F) expression of Mad2 in NC-CIN
I, CIN II–III, CC; (G) comparison of the expression of Bub1
and Mad2 proteins in cervical tissues with different pathological
stages and staining classes. NC, normal cervical; CIN, cervical
intraepithelial neoplasm; CC, cervical cancer; b, Bub1; m, Mad2; −,
no staining; +, barely detectable staining; ++, easily seen fine
granules present diffusely throughout the nucleus or cytoplasm;
+++, staining so strong that nuclear detail is obscured. |
| Table IIIImmunostaining intensity of Bub1 and
Mad2 proteins in cervical tissues at various pathological stages
with different staining classes. |
Table III
Immunostaining intensity of Bub1 and
Mad2 proteins in cervical tissues at various pathological stages
with different staining classes.
| | Bub1 (%) | Mad2 (%) |
|---|
| |
|
|
|---|
| Category | N | − | + | ++ | +++ | − | + | ++ | +++ |
|---|
| NC | 10 | 0 (0.00) | 1 (10.00) | 5 (50.00) | 4 (40.00) | 1 (10.00) | 1 (10.00) | 5 (50.00) | 3 (30.00) |
| CIN | 65 | 21 (32.31) | 26 (40.00) | 16 (24.62) | 2 (3.08) | 23 (35.38) | 17 (26.15) | 21 (32.31) | 4 (6.15) |
| CINI | 15 | 1 (6.67) | 6 (40.00) | 7 (46.67) | 1 (6.67) | 0 (0.00) | 7 (46.67) | 6 (40.00) | 2 (13.33) |
| CINII | 23 | 7 (30.43) | 10 (43.48) | 5 (21.74) | 1 (4.35) | 9 (39.13) | 2 (8.7) | 10 (43.48) | 2 (8.70) |
| CINIII | 27 | 13 (48.15) | 10 (37.04) | 4 (14.81) | 0 (0.00) | 14 (51.85) | 8 (29.63) | 5 (18.52) | 0 (0.00) |
| CC | 25 | 16 (64.00) | 7 (28.00) | 2 (8.00) | 0 (0.00) | 17 (68.00) | 7 (28.00) | 1 (4.00) | 0 (0.00) |
| SCC | 15 | 10 (66.67) | 4 (26.67) | 1 (6.67) | 0 (0.00) | 11 (77.33) | 3 (20.00) | 1 (6.67) | 0 (0.00) |
| AC | 10 | 6 (60.00) | 3 (30.00) | 1 (10.00) | 0 (0.00) | 6 (60.00) | 4 (40.00) | 0 (0.00) | 0 (0.00) |
Interaction between HPV16 E5 and SCPs
explored by IP
To explore the mechanism for the downregulation of
SCPs in HPV16-related cervical disease, an
immunoprecipitation/immunoblot (IP/IB) experiment was carried out
on extracts prepared from primary and transfected cells. We tested
several possible target genes, such as HPV16 E6/E7, FLT4, Akt, PI3K
and HPV16 E5 (using the HPV16 E5-GFP plasmid from our lab that had
been checked by an antibody to GFP) (data not shown). Equivalent
amounts of total cell protein from transfected cells were first
immunoprecipitated with Bub1 antibody followed by immunoblotting
for GFP revealed GFP+E5 bands. Conversely, IP with GFP antibody
followed by immunoblotting for Bub1 revealed clear Bub1 bands in
the E5-transfected cells, but not in the mock-transfected (empty
vector) or untransfected cells (Fig.
2).
These data suggest that Bub1 and HPV16 E5 can
interact directly or indirectly in vitro. This result was
verified in a parallel experiment using His+E5, which showed
exactly the same results (data not shown). Control lanes showed no
interaction between GFP and Bub1 (Fig. 2). With the preliminary test, we
conducted a thorough experiment.
Expression of Bub1 and Mad2 is
significantly suppressed by HPV16/18 E5 further supporting an
interaction between HPV16 E5 and Bub1
To analyse the interaction between E5 and Bub1, we
tested the most epidemic types of HPV, HPV16/18. We prepared four
stable expression HPV16/18 E5-expressing cell lines as described in
Materials and methods. As a result, HPV16/18 E5 was expressed in
either a His-labelled native form or as a GFP-fusion protein.
We used RT-PCR to determine the expression of
HPV16/18 E5 prior to and following transfection. HPV16 E5 was
detected in all the transfected cells and at low levels in the
control groups (mock-transfected with empty plasmids and
untransfected HeLa cells) but was not present in other groups
(Fig. 3A). To further
substantiate these results and rule out potential effects resulting
from the GFP tag, we transfected cells with the His-E5 recombinant
plasmids. The protein extracts were blotted with a GFP or His
antibody. Bands with compatible molecular weights (GFP+E5 or
His+E5) were observed (Fig. 3B and
C). When HeLa cells were stably transfected with HPV18 E5, the
subcellular localisation of GFP+E5 fluorescence accumulated at the
perinuclear region of the HeLa cells (Fig. 3D). These results are consistent
with those of Krawczyk et al (14).
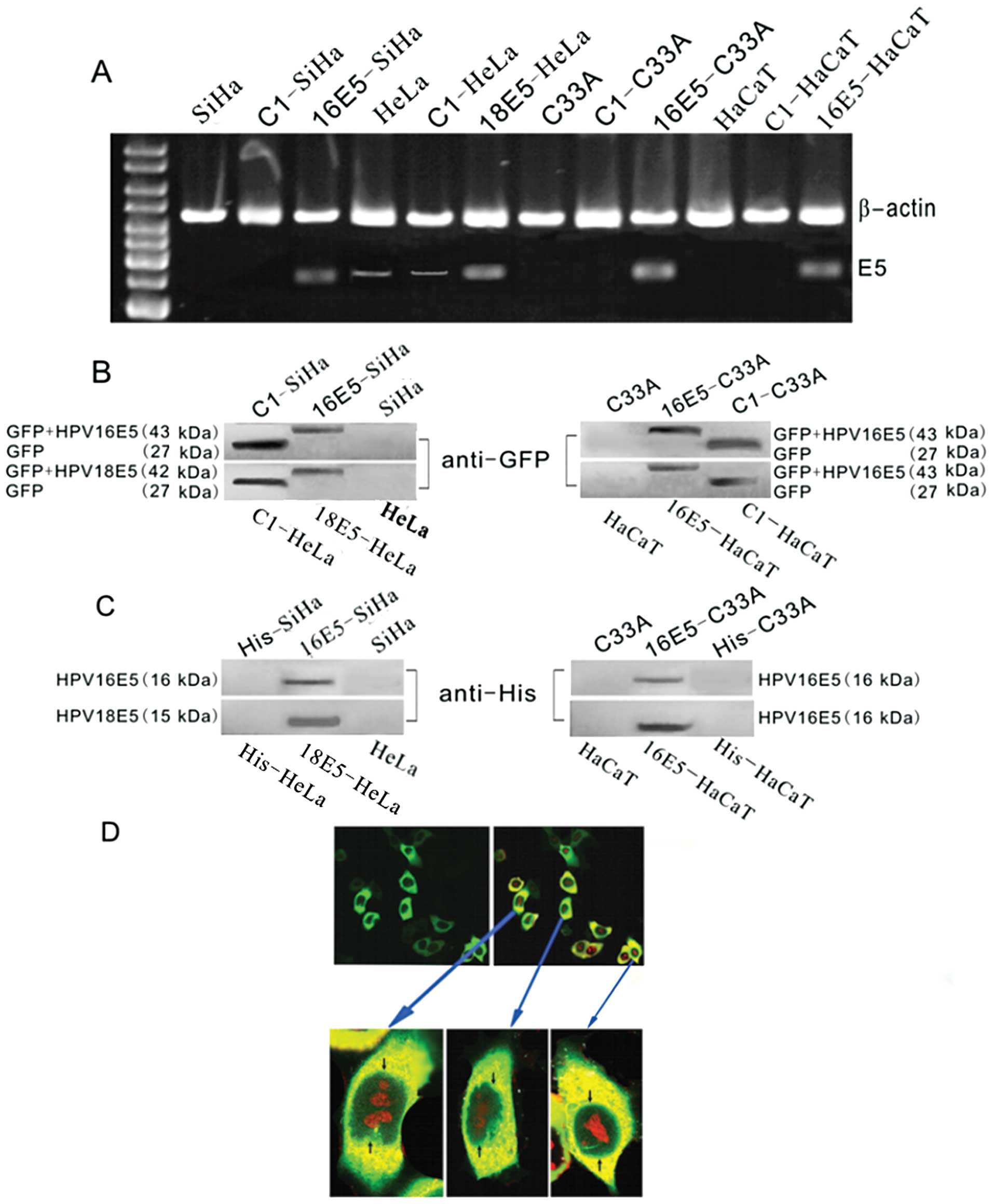 | Figure 3Detection of HPV 16/18 E5 in
recombinant plasmids and protein expression verification. (A)
RT-PCR for HPV16/18 E5 expression in different cell lines. In the
E5-stably transfected cells (16E5-SiHa, 18E5-HeLa, 16E5-C33A and
16E5-HaCaT lanes), E5 was detected. In the empty plasmid pEGFP-C1
transfected groups (C1-SiHa, C1-C33A and C1-HaCaT lanes), E5 was
not observed. In the untreated SiHa, C33A and HaCaT cell lanes, no
E5 band was found; however, E5 expression in the C1-HeLa group and
HeLa cells was scarcely detected. (B and C) Western blot analysis
of the GFP and His fusion recombinant proteins of HPV16/18 E5.
Using the (B) anti-GFP and (C) anti-His antibodies, respectively, a
positive band was observed with a molecular weight corresponding to
(B) GFP+HPV16 E5 (43 kDa)/GFP+HPV18 E5 (42 kDa) or (C) His+HPV16 E5
(16 kDa)/His+HPV18 E5 (15 kDa) in the E5-stably transfected cells
(16E5-SiHa, 18E5-HeLa, 16E5-C33A, 16E5-HaCaT lanes). In the empty
plasmid pEGFP-C1 transfected groups (C1-SiHa, C1-HeLa, C1-C33A and
C1-HaCaT lanes), (B) a GFP band was detected (27 kDa), but (C) as
the His tag has a low molecular weight, no band was tested in the
anti-His group. (B and C) In the untreated SiHa, HeLa, C33A and
HaCaT cell lanes, no bands were found. (D) Immunofluorescence for
detecting the subcellular localization of HPV16 E5, photographed
with a confocal fluorescence microscope and nuclei were stained
with PI. Most of the fluorescence in GFP+E5 transfectants
accumulated at the perinuclear region of the 18E5-HeLa cells that
were transfected with pEGFP-C1-HPV18 E5. |
Additionally, we analysed the relationship between
HPV16/18 E5 and Bub1 in all the HPV 16/18 E5 positive groups by
western blot analysis and real-time RT-PCR; Bub1 and Mad2 were
significantly downregulated (Fig.
4). This result was verified in a parallel experiment using
cells that had been stably transfected with His+E5.
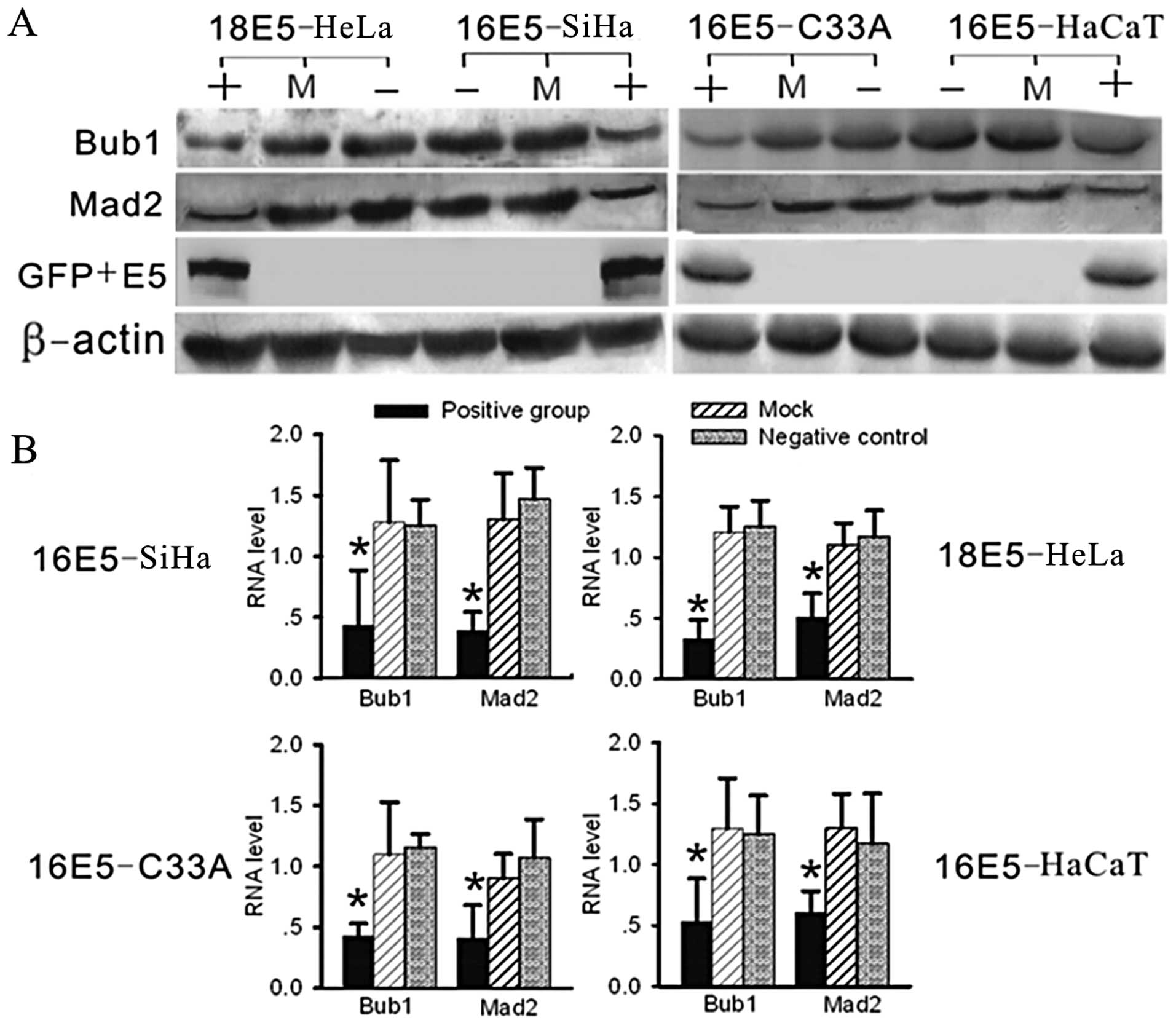 | Figure 4Analysis of RNA and protein levels of
Bub1 and Mad2 by real-time RT-PCR and western blot analysis. (A)
Using western blot analysis, compared with the negative and mock
groups, the expression levels of Bub1 and Mad2 were significantly
suppressed in the E5-expressing groups (in two parallel
experiments, the pcDNA3.1-V5-His and pcDNA3.1-V5-His-E5 transfected
groups showed exactly the same results, data not shown). (B)
Compared with the other groups by real-time RT-PCR, the RNA levels
of Bub1 and Mad2 were significantly decreased in the E5-expressing
groups. (+, positive group, stably transfected with E5-expression
plasmids; −, negative group, untreated cells as negative control;
M, mock group, transfected with empty vectors, as blank control;
*P<0.05). |
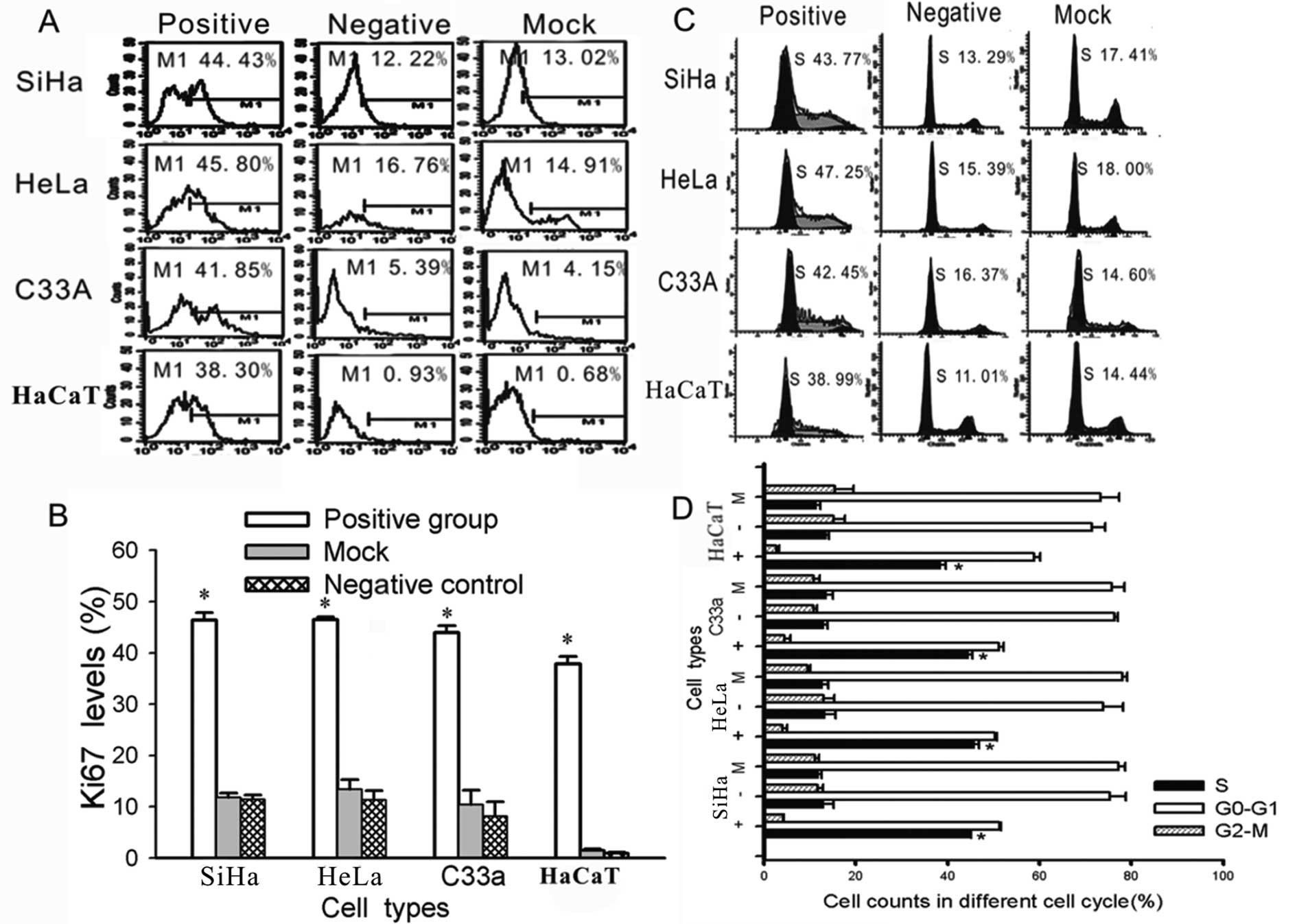
HPV16/18 E5 increases cell proliferation
and induces cells to leave G1 arrest and enter the S phase
To further investigate the effect of HPV16/18 E5 on
the biological characteristics of cervical cells, we performed an
additional test. As previously described (15), the expression of the human Ki-67
protein is closely associated with cell proliferation. The Ki-67
protein has been extensively used as a proliferation marker for
determining the so-called growth fraction of a given cell
population. Therefore, we used FACS to measure the expression of
Ki-67. There were significantly higher levels of Ki-67 in the
HPV16/18 E5-transfected cells than in the untreated (negative
group) and blank vector-transfected cells (mock group)
(Fig. 5). Meanwhile, the cycle
phase distribution of the cells that were transfected with HPV16/18
E5 was significantly different from the control groups (negative
and mock groups) (P<0.01); a greater proportion was in the S
phase as shown by FACS (Fig.
5).
Discussion
In the present study, we tested 100 specimens from
patients with cervical diseases, using IHC, and demonstrated that
from CIN I to CIN II, CIN III, and eventually CC, the expression of
Bub1 and Mad2 gradually decreased (Table III and Fig. 1). This decrease in the SCPs may
influence sister chromatid separation and promote mis-separation
producing a heteroploid cell or polyploidy. Given these results, we
postulated that an essential mechanism involving these proteins
must exist. We examined many possible target genes, such as HPV16
E6/E7, FLT4, Akt and PI3K, and we found that a decrease in the
expression of Bub1 and Mad2 may be induced in some manner by HPV16
E5, as shown by IP (Fig. 2).
HPV16 E5 is a hydrophobic, 83-amino acid polypeptide that
associates with the Golgi apparatus, endoplasmic reticulum, and
perinuclear membrane (Fig. 3)
(14,16,17). E5 is capable of altering the
growth and differentiation of epithelial cells via numerous
pathways, including conferring resistance to apoptosis and inducing
anchorage-independent growth (8,18,19). Currently, HPV16 E5 is considered
an oncogene as it transforms murine fibroblasts and keratinocytes
in tissue culture (20) and
enhances the immortalisation potential of E6 and E7 (17). However, little is known about the
molecular mechanisms by which E5 produces these effects.
To further confirm our results, we prepared four
different cell models to test the function of the HR HPV E5 protein
(Fig. 3). In our experiment, the
expression of Bub1 and Mad2 was significantly decreased in cell
lines that stably expressed HPV16/18 E5 at both the RNA and protein
levels (Fig. 4). We then
investigated the underlying molecular mechanisms and how Bub1, Mad2
and E5 interacted with each other. To do this, we noted that some
cytoplasmic organelles (such as the Golgi apparatus and the
endoplasmic reticulum) disintegrated to form membrane bubbles
during cell prophase when mitotic spindles were emerging (1,3).
In addition, we noted that these disintegrated organelles were
precisely colocalized with the E5 protein (14). Therefore, we hypothesized that at
the beginning of mitosis and at the moment of spindle formation,
organelles that were associated with E5 disintegrated and offered a
right time and location for E5 and its spindle-related binding
partners to associate. This hypothesis would explain the possible
interaction between E5 and SCPs. We are currently exploring more
direct proof of this interaction.
Next, we thoroughly explored whether there were any
changes in cell characteristics as a result of HPV16/18 E5
expression. In this study, we tested the level of Ki-67 by FACS
analysis in different groups since the expression of Ki-67 is
closely associated with cell proliferation. Ki-67 is absent in
resting cells (G0), making it an excellent marker for determining
all active phases of the cell cycle (G1, S, G2, and mitosis
stages). The expression of Ki-67 is also correlated with the
clinical course of the disease (15). We showed that HPV16/18
E5-transfected cells had much higher Ki-67 expression and
faster proliferation than controls (Fig. 5). Nevertheless, we did not find a
significant difference between the HPV-positive and negative
groups.
We showed that E5 affected the cell cycle and
increased the percentage of S phase cells (Fig. 5), which resulted in increased DNA
synthesis. As previously described (21), E6 increases the turnover of p53,
which mediates G1/S arrest in response to DNA-damaging agents
through the abrogation of p21 activity (a cyclin-dependent kinase
tumour suppressor gene product that causes pocket-protein
phosphorylation and releases E2F). Moreover, keratinocytes
expressing E7 alone also fail to undergo a G1/S arrest through the
deregulation of E2F activity (22). Each of the E5, E6 and E7 proteins
can rescue cells from G1 arrest and promote S phase entry in
different ways. They cooperate with and supplement each other,
forming a network that regulates the cell cycle (Fig. 6). At the same time, we showed the
decreasing expression of p21 in cells that stably express E5
(Fig. 7). E5 might promote cell
proliferation in this way (23).
A previous study (24) indicated
that intradermal injection of cottontail rabbit papillomavirus DNA,
a virus with a natural history of infection similar to that of
HPV-16 but with mutations in E7 of critical residues for the
binding of pRb, can still induce papilloma formations in rabbits.
One possible explanation for these observations is that the
suppression of the p21 gene by E5 may facilitate the activation of
CDK4-cyclin D complexes, which are known to phosphorylate
pRb and inactivate Rb-checkpoint control (22). Furthermore, E6 leads to p53
degradation and contributes to malignant transformation (21). Since p53 can upregulate p21
expression (17), we also assume
that a network including E5, E6, E7, Rb, p53 and p21 exists
(Fig. 6).
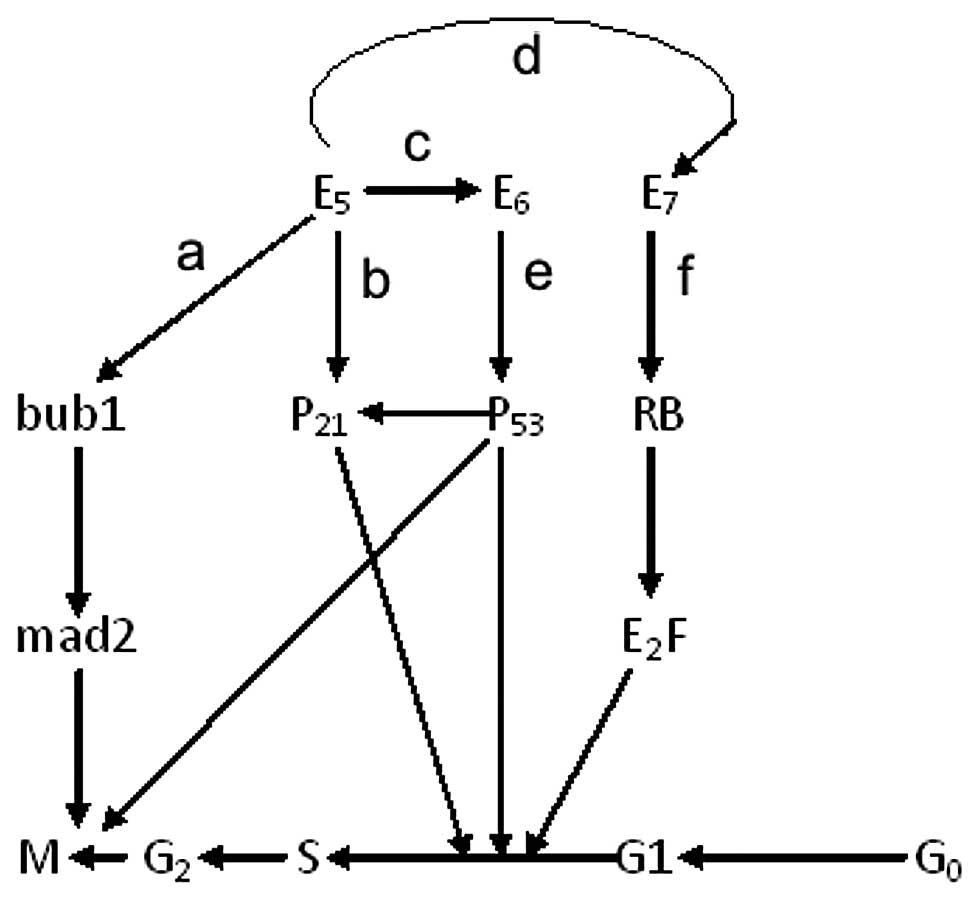 | Figure 6The potential network between E5, E6
and E7 in different cell cycle stages. (a) In our experiment, the
down-regulation of the Bub1 and Mad2 may be induced by HPV16 E5 in
some ways as proven by IP (Fig.
3) and the expression of Bub1 and Mad2 were decreased
significantly in the HPV16/18 E5 stable expression groups on both
of the RNA and protein levels (Fig.
5). (b) We also proved that the expression of p21
(WafI/SdiI/CipI) was reduced in stably expressing E5 cells
(Fig. 7). (c) HPV16 E5 enhances
the immortalization potential of E6 and E7 (17). Furthermore, E6 targets the tumor
suppressor protein p53, leads to p53 degradation and contributes to
malignant transformation (22),
and p53 can upregulate p21 expression (17). (d) The repression of the p21 gene
by the E5 protein may complement the altered pRb binding activity
of mutated E7. (e) E6 proteins increase the turnover of p53, which
leads to abrogation of p21-mediated or P21 unrelated G1/S arrest in
response to DNA-damaging agents (22). (f) The keratinocytes expressing E7
alone also fail to undergo a G1/S arrest, not directly via p21
suppression but through deregulation of E2F activity (23). Notably, all the details about E5,
E6 and E7 can rescue G1 arrest and promote S phase entry in
different ways, forming a potential network. |
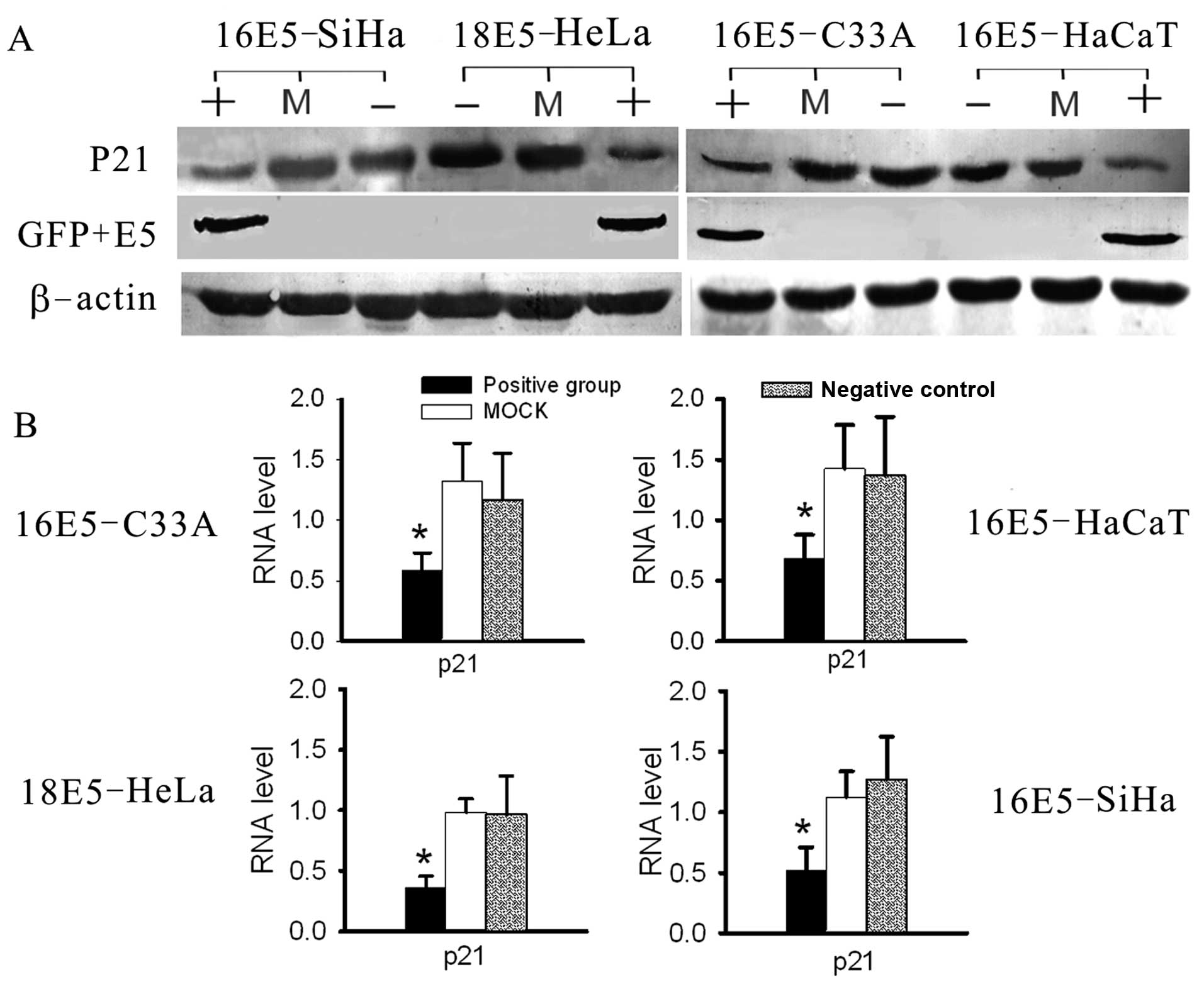 | Figure 7Analysis of the RNA and protein levels
of P21 by real-time RT-PCR and western blot analysis. (A) The
expression levels of P21 were suppressed in E5-expressing cell
lines, compared with the negative and mock groups, as tested by
western blot analysis. (B) The RNA levels of P21 were decreased
significantly in E5-expressing cells, compared with the other
groups, as determined by real-time RT-PCR. (+, positive group,
stably transfected with E5-expression vectors; −, negative group,
untreated cells as negative control; M, mock group, transfected
with empty plasmids, as blank control; *P<0.05). |
In conclusion, initial infection with HR HPV causes
low-grade disease and the viral DNA is located in the cell nucleus.
E5 is present in the major abundant viral transcripts, as are E6
and E7 (21). During the
progression of malignant disease, the HPV DNA integrates into the
host cell genome, and E5 is often deleted (25) suggesting that E5 might play an
important role at the beginning of HPV infection and in the early
stages of HPV-related cervical diseases. The mechanism stimulates
cell growth, leads to an increased tendency to enter S phase,
compensates for the function of E6 and E7 and impairs SCPs. These
processes would result in aggravation of the malignant
transformation potential of HPV-infected cells and acceleration of
the carcinogenic process.
Acknowledgements
This study was supported by grants from the National
Science Foundation of China (30901586; 30973205; 30973148); the
‘973’ Program of China (2009CB521808); the Huibei Province Science
Foundation (2011CDB191) and the Central University Basic Science
Research Foundation (2012QN188).
Abbreviations:
|
HPV
|
human papillomavirus
|
|
SCP
|
spindle checkpoint protein
|
|
CC
|
cervical cancer
|
|
APC/C
|
anaphase-promoting complex or
cyclosome
|
|
IHC
|
immunohistochemistry
|
|
CIN
|
cervical intraepithelial neoplasm
|
|
FACS
|
fluorescence-activated cell
sorting
|
|
IP
|
immunoprecipitation
|
|
PI
|
propidium iodide
|
References
|
1
|
Tang Z, Sun Y, Harley SE, Zou H and Yu H:
Human Bub1 protects centromeric sister-chromatid cohesion through
Shugoshin during mitosis. Proc Natl Acad Sci USA. 101:18012–18017.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yu H: Regulation of APC-Cdc20 by the
spindle checkpoint. Curr Opin Cell Biol. 14:706–714. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nasmyth K: Segregating sister genomes: the
molecular biology of chromosome separation. Science. 297:559–565.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cleveland DW, Mao Y and Sullivan KF:
Centromeres and kinetochores: from epigenetics to mitotic
checkpoint signaling. Cell. 112:407–421. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tang Z, Shu H, Oncel D, et al:
Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for
APC/C inhibition by the spindle checkpoint. Mol Cell. 16:387–397.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Genther Williams SM, Disbrow GL, Schlegel
R, et al: Requirement of epidermal growth factor receptor for
hyperplasia induced by E5, a high-risk human papillomavirus
oncogene. Cancer Res. 65:6534–6542. 2005.PubMed/NCBI
|
|
7
|
Burgert HG, Ruzsics Z, Obermeier S, et al:
A subversion of host defense mechanisms by adenoviruses. Curr Top
Microbiol Immunol. 269:273–318. 2002.PubMed/NCBI
|
|
8
|
Vazquez-Ortiz G, Ciudad CJ, Pina P, et al:
Gene identification by cDNA arrays in HPV-positive cervical cancer.
Arch Med Res. 36:448–458. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Medema RH, Klompmaker R, Smits VA and
Rijksen G: P21waf1 can block cells at two points in the cell cycle,
but does not interfere with processive DNA replication or
stress-activated kinases. Oncogene. 16:431–441. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Freitas S, Moore DH, Michael H and Kelley
MR: Studies of apurinic/apyrimidinic endonuclease/ref21 expression
in epithelial ovarian cancer: correlations with tumor progression
and platinum resistance. Clin Cancer Res. 9:4689–4694.
2003.PubMed/NCBI
|
|
11
|
Kozak M: At least six nucleotides
preceding the AUG initiator codon enhance translation in mammalian
cells. J Mol Biol. 196:947–950. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Biswas C, Kell B, Mant C, et al: Detection
of human papillomavirus type 16 early-gene transcription by reverse
transcription-PCR is associated with abnormal cervical cytology. J
Clin Microbiol. 35:1560–1564. 1997.PubMed/NCBI
|
|
13
|
Winer J, Jung CK, Shackel I and Williams
PM: Development and validation of real-time quantitative reverse
transcriptase-polymerase chain reaction for monitoring gene
expression in cardiac myocytes in vitro. Anal Biochem. 270:41–49.
1999. View Article : Google Scholar
|
|
14
|
Krawczyk E, Suprynowicz FA, Sudarshan SR
and Schlegel R: Membrane orientation of the human papillomavirus
type 16 E5 oncoprotein. J Virol. 84:1696–1703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Scholzen T and Gerdes J: The Ki-67
protein: from the known and the unknown. J Cell Physiol.
182:311–322. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao P and Zheng J: High-risk HPV
E5-induced cell fusion: a critical initiating event in the early
stage of HPV-associated cervical cancer. J Virol. 7:238–240. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Boulenouar S, Weyn C, Van Noppen M, et al:
Effects of HPV-16 E5, E6 and E7 proteins on survival, adhesion,
migration and invasion of trophoblastic cells. Carcinogenesis.
31:473–480. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pedroza-Saavedra A, Lam EW,
Esquivel-Guadarrama F and Gutierrez-Xicotencatl L: The human
papillomavirus type 16 E5 oncoprotein synergizes with EGF-receptor
signaling to enhance cell cycle progression and the down-regulation
of p27(Kip1). Virology. 400:44–52. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kivi N, Greco D, Auvinen P and Auvinen E:
Genes involved in cell adhesion, cell motility and mitogenic
signaling are altered due to HPV 16 E5 protein expression.
Oncogene. 27:2532–2541. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nath R, Mant CA, Kell B, et al: Analyses
of variant human papillomavirus type-16 E5 proteins for their
ability to induce mitogenesis of murine fibroblasts. Cancer Cell
Int. 6:192006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim MK, Kim HS, Kim SH, et al: Human
papillomavirus type 16 E5 oncoprotein as a new target for cervical
cancer treatment. Biochem Pharmacol. 80:1930–1935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ruesch MN and Laimins LA: Initiation of
DNA synthesis by human papillomavirus E7 oncoproteins is resistant
to p21-mediated inhibition of cyclin E-cdk2 activity. J Virol.
71:5570–5578. 1997.PubMed/NCBI
|
|
23
|
Tsao YP, Li LY, Tsai TC and Chen SL: Human
papillomavirus type 11 and 16 E5 represses p21 (WafI/SdiI/CipI)
gene expression in fibroblasts and keratinocytes. J Virol.
70:7535–7539. 1996.PubMed/NCBI
|
|
24
|
Defeo-Jones D, Vuocolo GA, Haskell KM, et
al: Papillomavirus E7 protein binding to the retinoblastoma protein
is not required for viral induction of warts. J Virol. 67:716–725.
1993.PubMed/NCBI
|
|
25
|
Maufort JP, Shai A, Pitot HC and Lambert
PF: A role for HPV16 E5 in cervical carcinogenesis. Cancer Res.
70:2924–2931. 2010. View Article : Google Scholar : PubMed/NCBI
|