Introduction
Platelet-derived growth factor (PDGF) is a potent
mitogen and chemotactic factor for numerous cell types of
mesenchymal origin, including glomerular mesangial cells (1,2).
To date, four PDGF family members have been identified: PDGF-A, -B,
-C and -D (3–6). The PDGF family members exert their
biological activities through the cell membrane PDGF receptor
(PDGFR), which comprises the α (PDGFR-α) and β (PDGFR-β) chains,
with different binding specificities and affinities (2,7).
Physiologically, the PDGF system is important in embryonal
development, wound healing, and adult maintenance (4,8,9).
However, excessive signaling of the system has been implicated in
various diseases, including cancer, vasculopathies, fibrosis, and
renal pathologies (10–14). Among components of the PDGF
system, PDGF-D signaling is suggested to be a key factor in the
development of a variety of renal pathologies, including mesangial
proliferative glomerulopathy, renal fibrosis, and
mesangioproliferative glomerulonephritis (15,16).
Previous studies have suggested the involvement of
prostaglandins (PGs) in renal physiology and/or pathology, such as
vasodilatation, renin secretion, and sodium and water excretion,
and/or renal failure (17–20).
Cyclooxygenase (COX) is the rate-limiting enzyme involved in the
biosynthesis of PGs and related eicosanoids from arachidonic acid.
There are two isoforms of COX: COX-1 and COX-2. COX-1 is
constitutively expressed in most cell types and the COX-1-derived
PGs are involved in normal inflammatory responses, bone
development, and wound healing (21–23). By contrast, COX-2 is an inducible
enzyme and its expression is highly increased in cells following
the exposure of extracellular stimuli, including interleukin-1β,
lipopolysaccharide, manganese or growth factor (24–27). Clinical evidence indicates
overexpression of COX-2 and its role in several inflammatory and
neoplastic diseases (28).
Previous studies have demonstrated an upregulation of COX-2
expression in proliferative glomerulonephritis (29) and an increased renal expression of
COX-2 in nephropathies (19,30). It has been shown that inhibition
of COX-2 by specific COX-2 inhibitors ameliorates renal
ablation-induced changes in the kidney function (31) and reduces expression of several
mediators of renal injury in a model of diabetes and hypertension
(32). These previous findings
suggest that exaggerated COX-2 expression and activity may be
involved in various renal pathologies. PDGF-D regulation of COX-2
expression in renal cells, however, remains unknown.
In this study, we investigated the effect of PDGF-D
on COX-2 expression in rat mesangial cells (RMCs) and determined
possible molecular and signaling mechanisms involved.
Materials and methods
Materials
RPMI-1640 medium, fetal bovine serum (FBS),
penicillin, and streptomycin were purchased from WelGENE (Daegu,
Korea). Enzyme-linked chemiluminescence (ECL) western detection
reagents were bought from Thermo Scientific (Waltham, MA, USA).
Nitrocellulose membrane was bought from Millipore (Rockford, IL,
USA). Bradford reagent was purchased from Bio-Rad (Hercules, CA,
USA). PDGF-D was purchased from R&D (Minneapolis, MN, USA).
Protease inhibitor cocktail (100X) and PP1 were purchased from
Calbiochem (Madison, WI, USA). PD98059, LY294002 and GF109203X were
purchased from Biomol (Plymouth Meeting, PA, USA). Antibody of
COX-2 was purchased from Cayman Chemical (Ann Arbor, MI, USA).
Antibodies of phospho-extracellular signal regulated kinase-1/2
(p-ERK-1/2), total-ERK (T-ERK-1/2), phospho-c-Jun N-terminal
kinase-1/2 (p-JNK-1/2), T-JNK-1/2, p-p38 MAPK, T-p38 MAPK, p-PKB
and T-PKB were purchased from Epitomics (Burlingame, CA, USA).
Other reagents, including anti-actin mouse monoclonal antibody,
were purchased from Sigma (St. Louis, MO, USA).
Cell culture
RMCs were grown at 37°C in a humidified condition of
95% air and 5% CO2 in RPMI-1640 supplemented with 10%
heat-inactivated FBS, 100 U/ml penicillin, and 100 μg/ml
streptomycin.
MTS assay
To measure the effect of PDGF-D and/or
pharmacological inhibitors or agents on the viability of RMCs,
cells were grown in 96-well plates at a density of 1×104
cells/well in 100 μl volume and were serum-starved for 24 h.
Cells were then treated without or with PDGF-D in the absence or
presence of LY294002, PD98059, GF109203X or PP1 for 24 h, at which
time point cells were incubated with MTS (20 μl/well) for
1.5 h at 37°C. The absorbance was measured at 595 nm using a
microplate reader. The MTS assay was performed in triplicates. Data
are the means ± standard error (SE) of three independent
experiments.
Preparation of whole cell lysates
To measure the effect of PDGF-D on the expression of
COX-2 protein in RMCs, cells were plated in 6-well plates at a
density of 1×106 cells/well in 2 ml volume and were
serum-starved for 24 h. Cells were then treated without or with
different concentrations of PDGF-D for 24 h. Control or the
PDGF-D-treated cells were washed with PBS and exposed to cell lysis
buffer [50 mM Tris-Cl (pH 7.4), 150 mM NaCl, 0.1% sodium dodecyl
sulfate, 0.25% sodium deoxycholate, 1% Triton X-100, 1% Nonidet
P-40, 1 mM EDTA, 1 mM EGTA, proteinase inhibitor cocktail (1X)].
The cell lysates were collected in a 1.5 ml tube and centrifuged
for 20 min at 4°C at 12,000 rpm. The supernatant was saved and
protein concentrations were determined with the Bradford
reagent.
Western blotting
Proteins (50 μg) were separated by SDS-PAGE
(10%) and transferred onto nitrocellulose membranes (Millipore).
The membranes were washed with TBS (10 mM Tris, 150 mM NaCl)
supplemented with 0.05% (vol/vol) Tween-20 (TBST) followed by
blocking with TBST containing 5% (wt/vol) non-fat dried milk. The
membranes were incubated overnight with antibodies specific for the
protein of interest at 4°C. The membranes were exposed to secondary
antibodies coupled to horseradish peroxidase at room temperature
for 2 h. The membranes were washed, and immunoreactivities were
detected by ECL reagents.
Reverse transcription-polymerase chain
reaction (RT-PCR)
To measure the expression levels of PDGFR-α and
PDGFR-β in RMCs, cells were plated in 6-well plates at a density of
1×106 cells/well in 2 ml volume overnight. To measure
the effect of PDGF-D on the expression of COX-2 mRNA in RMCs, cells
were plated in 6-well plates at a density of 1×106
cells/well in 2 ml volume and serum-starved for 24 h. Cells were
then treated without or with different concentrations of PDGF-D for
24 h. Total RNA from the conditioned cells above was isolated using
the RNAzol-B (Tel-Test, Inc.). Three micrograms of total RNA were
reverse transcribed using a random hexadeoxynucleotide primer and
reverse transcriptase. Single stranded cDNA was amplified by PCR
with the following primers: PDGFR-α, forward, 5′-GGC TTC AAC GGA
ACC TTC AG-3′ and reverse, 5′-CGC TGT CTT CTT CCT TAG CC-3′;
PDGFR-β, forward, 5′-GAG TGC CCT CCC GCA TTG-3′ and reverse, 5′-GGT
AGA CCA GGT GAC ATT TG-3′; COX-2, forward, 5′-CTG TAC TAC GCC GAG
ATT CCT GA-3′ and reverse, 5′-GTC CTC GCT TCT GAT CTG TCT TG-3′;
GAPDH, forward, 5′-GGT GAA GGT CGG TGT GAA CG-3′ and reverse,
5′-GGT AGG AAC ACG GAA GGC CA-3′. The PCR conditions were: PDGFR-α,
30 cycles of denaturation at 94°C for 30 sec, annealing at 50°C for
30 sec, and extension at 72°C for 1 min; PDGFR-β, 35 cycles of
denaturation at 94°C for 30 sec, annealing at 50°C for 30 sec, and
extension at 72°C for 1 min; COX-2, 35 cycles of denaturation at
94°C for 30 sec, annealing at 56°C for 30 sec, and extension at
72°C for 30 sec; GAPDH, 27 cycles of denaturation at 94°C for 30
sec, annealing at 57°C for 30 sec, and extension at 72°C for 30
sec, respectively. GAPDH was used as an internal control to
evaluate the relative expression of COX-2.
Statistical analysis
Cell count analysis was performed in triplicates and
repeated three times. Data are expressed as the means ± standard
error (SE). Significance (P<0.05) was determined by one-way
ANOVA.
Results
PDGF-D induces expression of COX-2 at
both the protein and mRNA levels in RMCs
There are two types of PDGF receptor (PDGFR);
PDGFR-α and PDGFR-β. PDGF-D binds predominantly to PDGFR-β. We thus
initially measured the expression levels of PDGFR in RMCs using
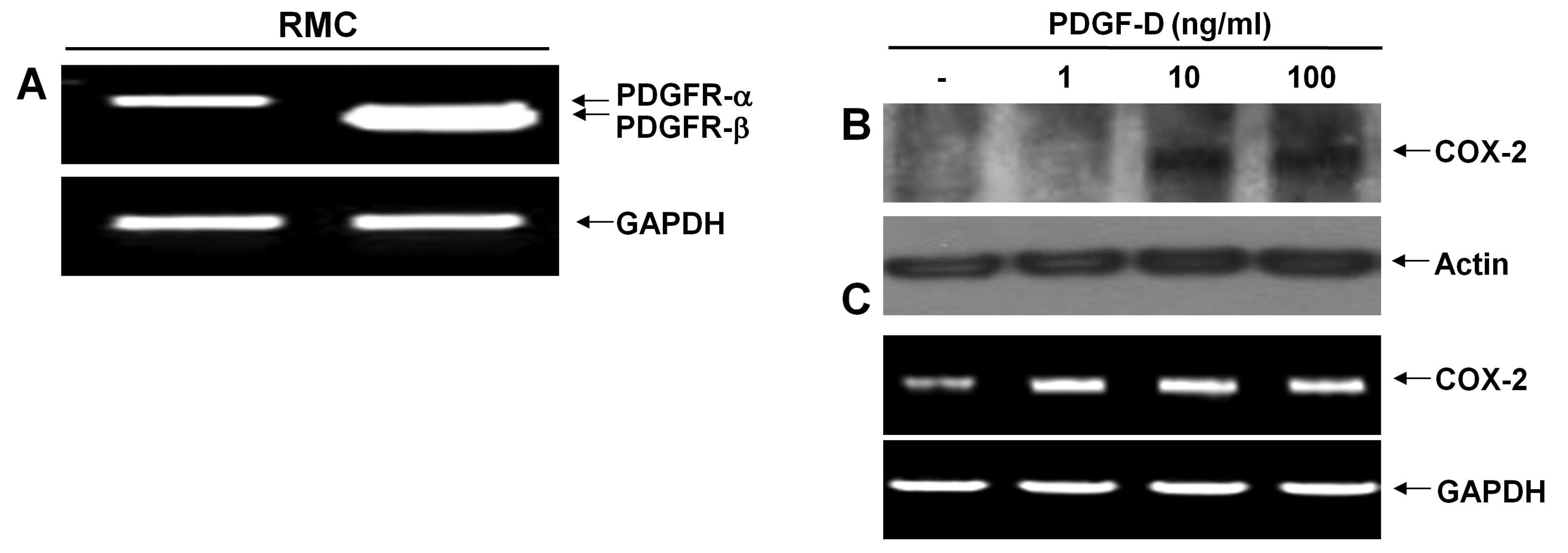
RT-PCR analysis. RMCs expressed mRNA expressions of both PDGFR-α
and PDGFR-β (Fig. 1A). However,
there were much higher expression levels of PDGFR-β than PDGFR-α.
Expression levels of GAPDH mRNA were used as a loading control.
After determining the PDGFRs expressed in RMCs, we next analyzed
the effect of PDGF-D on the induction of COX-2 protein and mRNA
expressions in RMCs using western blotting and RT-PCR analysis,
respectively. COX-2 protein was not detected in control RMCs or
RMCs exposed to PDGF-D at 1 ng/ml for 24 h (Fig. 1B). However, there was an
upregulation of COX-2 protein in RMCs treated with PDGF-D at 10 or
100 ng/ml for 24 h. Notably, there were low levels of COX-2 mRNA in
control RMCs (Fig. 1C). Treatment
with PDGF-D even at 1 ng/ml further enhanced the COX-2 mRNA levels.
However, there was no further enhancement by PDGF-D treatment at 10
or 100 ng/ml. Expression levels of control actin protein and GAPDH
mRNA remained constant in RMCs treated without or with PDGF-D.
PDGF-D leads to a rapid but transient
activation of PKB and ERK-1/2 in RMCs
Due to the strongest COX-2 inducing effect, the 10
ng/ml concentration of PDGF-D was chosen for further studies. Time
course experiments were next used to examine the effect of PDGF-D
on the activation of intracellular signaling proteins, herein PKB
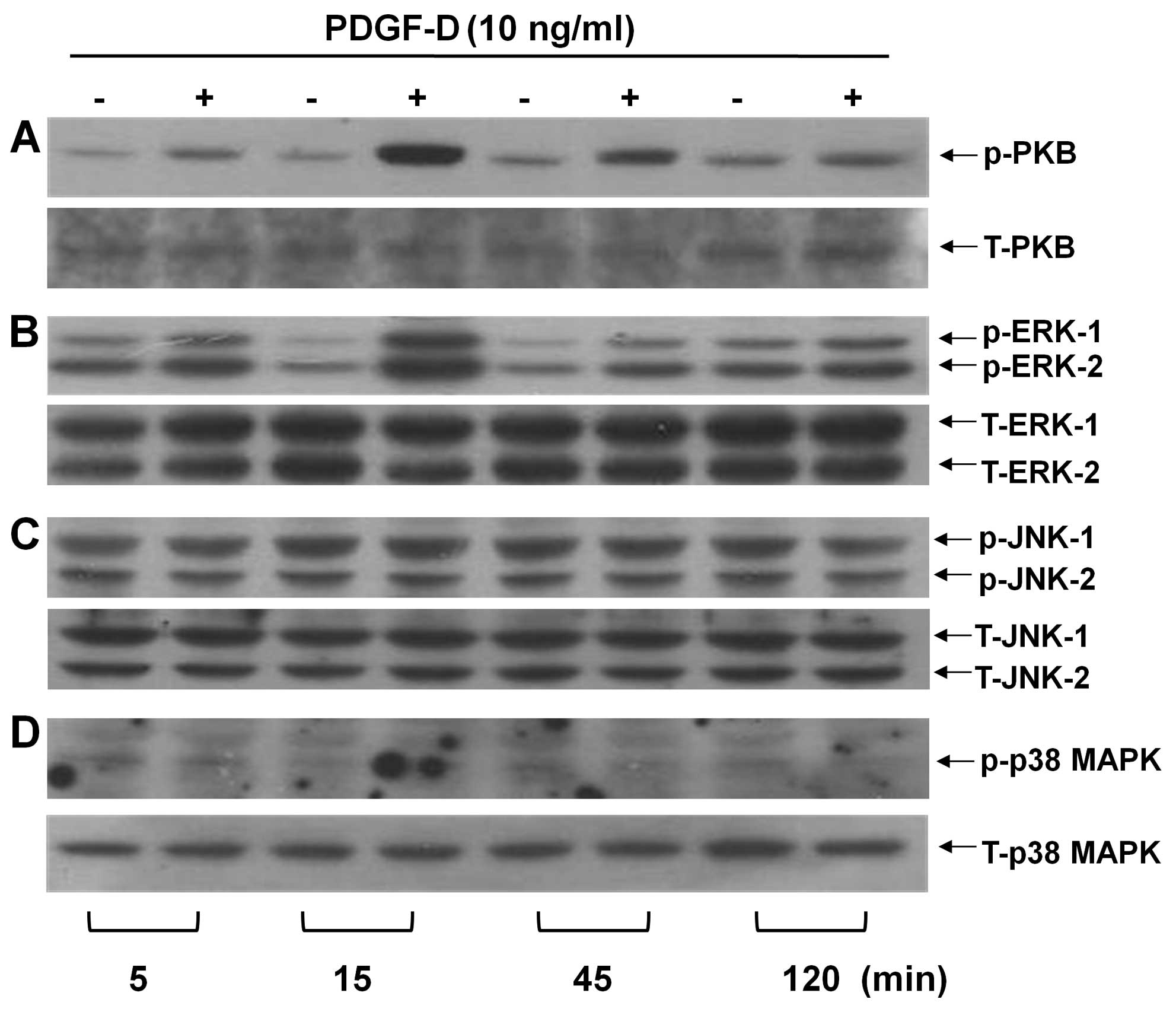
and the family of MAPKs, in RMCs. Treatment with PDGF-D led to a
time-dependent increase in PKB phosphorylation in RMCs (Fig. 2A). PDGF-D treatment even at 5 min
slightly enhanced phosphorylation of PKB. Maximal PKB
phosphorylation occurred at 15 min, followed by a gradual decline
at 45 or 120 min. PDGF-D treatment also resulted in an enhancement
of ERK-1/2 phosphorylation in a time-dependent manner (Fig. 2B). In particular, strong ERK-1/2
phosphorylation was induced by 15 min treatment with PDGF-D,
followed by a sharp decline thereafter. However, treatment with
PDGF-D at the times tested did not change the phosphorylation
levels of JNK-1/2 (Fig. 2C) and
p38 MAPK (Fig. 2D) in RMCs. Total
expression levels of PKB and each member of the MAPK family were
not changed by treatment with PDGF-D at the times tested (Fig. 2, low panels). These results
suggest that PDGF-D treatment specifically increases the
phosphorylation levels of the pre-existing PKB and ERK-1/2 without
de novo synthesis of PKB and ERK-1/2.
Activities of PI3K/PKB and PKCs are
critical for the PDGF-D-induced COX-2 expression in RMCs
Pharmacological inhibition studies were next
performed to investigate the role of PKB and/or ERK-1/2 activation
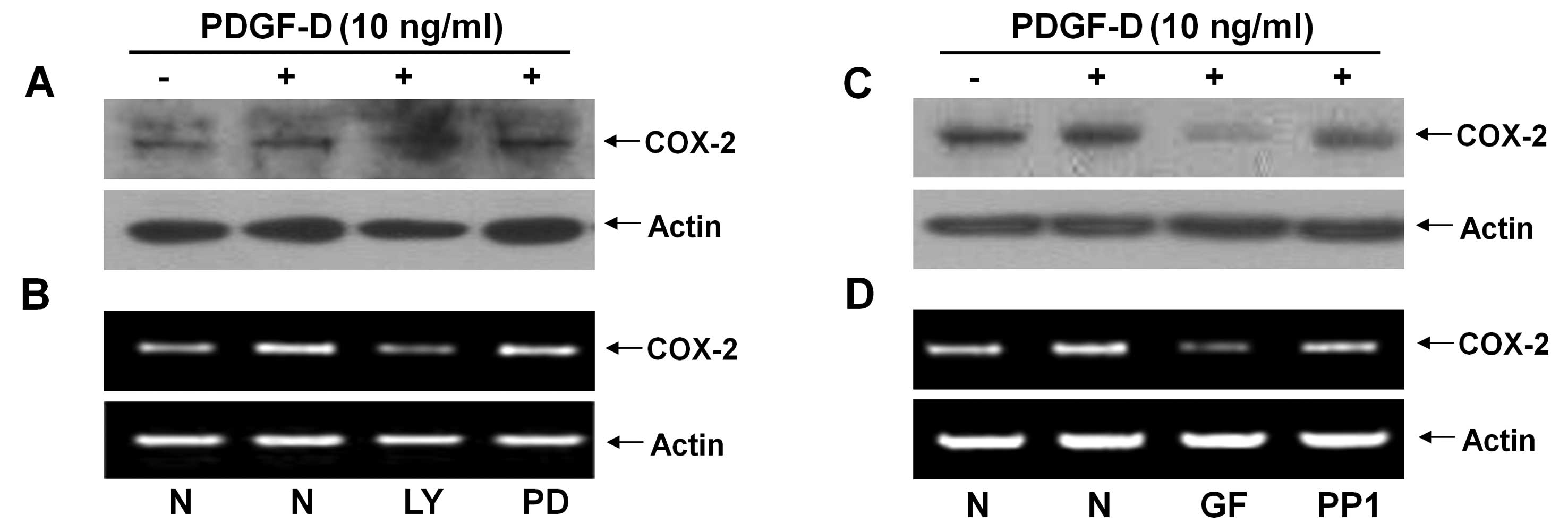
in the PDGF-D-induced COX-2 expression in RMCs. Pretreatment with
LY294002 (LY, a PI3K/PKB inhibitor) strongly suppressed the
PDGF-D-induced expression of COX-2 protein and mRNA, but
pretreatment with PD98059 (PD, an ERK-1/2 inhibitor) had little
effect on the PDGF-D-induced expression of COX-2 protein and mRNA
in RMCs (Fig. 3A and B). Using
additional pharmacological inhibitors, such as GF109203X (GF, a
pan-PKC inhibitor) and PP1 (an Src inhibitor), we also determined
the role of PKCs or Src in the induction of COX-2 expression by
PDGF-D in RMCs. Pretreatment with GF largely inhibited the PDGF-D
effect on the induction of COX-2 protein and mRNA, while
pretreatment with PP1 did not (Fig.
3C and D).
Activities of PI3K/PKB, ERK-1/2 and PKC
(but not COX-2) are necessary for the growth of RMCs
The effect of PDGF-D on the growth of RMCs was next
investigated by MTS assay. Treatment with PDGF-D slightly enhanced
the viability of RMCs (Fig. 4).
The role of COX-2, PKB, ERK-1/2 and/or PKCs induced or activated by
PDGF-D in the growth of RMCs was then evaluated using
pharmacological inhibitors, including NS-398 (NS, a COX-2
inhibitor). Pretreatment with LY, PD or GF reduced the viability of
RMCs treated with PDGF-D. Notably, there was a similar suppressive
effect by LY, PD or GF on the viability of RMCs that were grown in
the absence of PDGF-D. However, NS pretreatment did not affect the
viability of RMCs treated without or with PDGF-D.
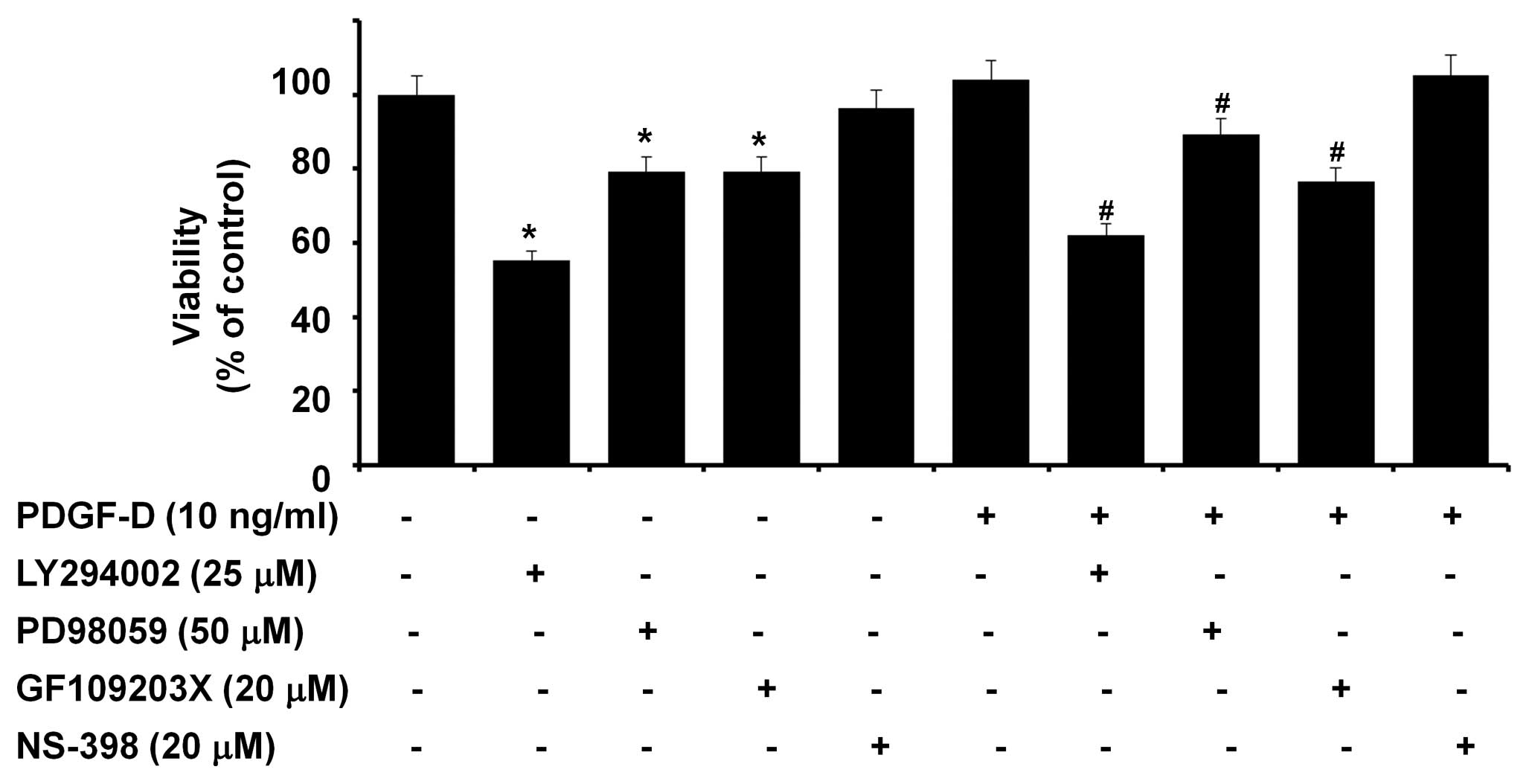 | Figure 4.Effect of LY294002, PD98059,
GF109203X or NS-398 on the viability of RMCs treated without or
with PDGF-D. RMCs seeded in 1×104 cells/100
μl/well/96-well plate the day before treatments were
serum-starved for 24 h. Cells were pretreated without or with
LY294002 (LY, a specific inhibitor of PI3K/PKB), PD98059 (PD, a
specific inhibitor of MEK-1/2, an upstream activator of ERK-1/2),
GF109203X (GF, a pan-PKC inhibitor ) or NS-398 (NS, a specific
COX-2 enzymatic inhibitor) for 1 h and then treated without or with
PDGF-D (10 ng/ml) in the absence or presence of each inhibitor for
an additional 24 h, followed by measurement of the cell viability
by MTS assay. Data are the means ± SE of three independent
experiments. *P<0.05 compared to the value in the
absence of LY, PD or GF. #P<0.05 compared to the
value of PDGF-D treatment in the absence of LY, PD or GF. |
Discussion
In the present study, we demonstrated for the first
time that PDGF-D induces the expression of COX-2 by transcriptional
upregulation in RMCs. Moreover, our data suggest that the
PDGF-D-induced COX-2 expression in RMCs is at least mediated
through modulation of the PI3K/PKB and PKC activities.
Previous studies have shown that PDGF-B, a member of
the PDGF family, induces expression of COX-2 in RMCs (33) or rat smooth muscle cells (34). However, PDGF-D regulation of COX-2
expression in renal cells remains unclear. In initial experiments,
we have shown that RMCs express high levels of PDGFR-β, which binds
predominantly to PDGF-D (Fig. 1A)
and the exposure of PDGF-D into RMCs leads to induction of COX-2
protein expression (Fig. 1B). It
is thus evident that the PDGF-D and PDGFR-β system is functional in
inducing COX-2 in RMCs.
Expression of the COX-2 gene is controlled at
multiple levels, including transcription, post-transcription, and
translation. It has recently been shown that the PDGF-B-induced
COX-2 expression in rat smooth muscle cells is related to both
COX-2 transcriptional and post-transcriptional upregulation
(34). However, there is evidence
demonstrating that PDGF-B fails to induce expression of COX-2 mRNA
in human gingival fibroblasts pretreated with interleukin-1β
(35). The present study shows an
upregulation of COX-2 transcripts in the PDGF-D-treated RMCs
(Fig. 1C), indicating that the
PDGF-D-induced expression of COX-2 protein in RMCs is primarily due
to COX-2 transcriptional upregulation.
There are numerous reports suggesting the
involvement of the activities of PKB and MAPKs in COX-2
transcriptional upregulation (26,36,37). Earlier investigations have shown
activation of PKB, ERK1/2, JNK-1/2 or p38 MAPK and their role in
many cellular changes in response to PDGF-D signal. For instance,
it has been shown that treatment with PDGF-D (100 ng/ml, 10 min)
activates ERK-1/2, which may enhance cell proliferation, in human
schwannoma cells (38). In
malignant mesothelioma cells, it has been demonstrated that
treatment with PDGF-D (40 ng/ml, 10 min) leads to activation of Akt
and ERK, which may facilitate cell chemotaxis (39). In cultured hepatic stellate cells
and myofibroblasts, treatment with PDGF-D (100 ng/ml, 15 min) has
been shown to induce phosphorylation of ERK1/2, JNK-1/2, p38 MAPK
and PKB, which may contribute to matrix accumulation (40). Herein, we demonstrated that
treatment with PDGF-D (10 ng/ml) rapidly but transiently induced
phosphorylation of PKB and ERK-1/2 but did not influence that of
JNK-1/2 and p38 MAPK in RMCs (Fig.
2). It is assumed that PDGF-D may differentially induce
activation of the family of MAPKs in a cell type-dependent manner
and/or that the differential effect of PDGF-D on activation of the
family of MAPKs in previous studies and herein may be due to
different experimental conditions used (such as, serum absence vs.
presence, 100 ng/ml vs. 10 ng/ml of PDGF-D). Notably, in this
study, we showed that activation of PI3K/PKB, but not ERK-1/2, is
critical for the PDGF-D-induced COX-2 transcriptional upregulation
in RMCs (Fig. 3A and B). Previous
studies have suggested a role of PKC-δ in COX-2 expression induced
by epidermal growth factor in gliomas (27) or of Src in COX-2 transcriptional
and post-transcriptional upregulation induced by PDGD-B in rat
smooth muscle cells (34).
Although activities of PKCs and Src in response to PDGF-D treatment
in RMCs are not measured herein, the present findings further
indicate that activities of PKCs and Src are also necessary for the
PDGF-D-induced COX-2 transcriptional upregulation in RMCs (Fig. 3C and D).
Evidence suggests that MC proliferation is an early
event in various renal pathologies and PDGF-D is involved in MC
proliferation in vitro (15,16). In support of the latter, in this
study, we demonstrated that PDGF-D treatment leads to a slight
increase of the viability of RMCs (Fig. 4). The COX-2-dependent and
independent MC proliferation have previously been proposed. This
notion is based on the fact that some COX-2 metabolites have
antiproliferative effects on RMCs (41,42), while COX-2 exerts its
antiproliferative effects on mesangial cells or 293 human embryonic
kidney cells independently of COX activity (43,44). The present findings that treatment
with NS-398, a selective COX-2 inhibitor, does not influence the
viability of both control and PDGF-D-treated RMCs (Fig. 4) support no role of the basal or
agonist-induced COX-2 (and their metabolites) in the viability of
RMCs. There is a large body of evidence suggesting the importance
of the activity of intracellular signaling proteins in MC
proliferation. For instance, it has been shown that the activity of
ERK-1/2 is necessary for MC proliferation induced by insulin-like
growth factor-1, a major mitogenic growth factor for MC (45). It has also been shown that
PI3K/PKB activity is linked to PDGF-B-induced MC proliferation
(46) and PKC activity is
critical for MC proliferation induced by lysophosphatidylcholine, a
major component of oxidized-low density lipoproteins (47). The present study suggests that the
basal (but not the PDGF-D-induced) activities of PI3K/PKB, ERK-1/2
or PKC are important for the growth of RMCs (Fig. 4). Given that MC proliferation has
been largely attributed to the activity of PI3K/PKB, ERK-1/2 and/or
PKC described above, it is suggested that single and/or combined
treatments with pharmacological inhibitor of PI3K/PKB, ERK-1/2 or
PKC may be useful against renal pathologies in which excess MC
proliferation is problematic. Accordingly, a recent study suggested
the potential utility of PKC inhibitor as a therapeutic strategy in
glomerular disease, which is evidenced by the fact that PKC-β
inhibition leads to the amelioration of the pathological findings
of experimental mesangial proliferative glomerulonephritis
(48).
In conclusion, we demonstrated that: i) PDGF-D
induces expression of COX-2 through transcriptional upregulation in
RMCs, ii) the PDGF-D-induced COX-2 expression in RMCs is at least
mediated through the regulation of PI3K/PKB and PKCs activities,
iii) the activities of PI3K/PKB, ERK-1/2 and PKCs, but not COX-2,
are necessary for the growth of RMCs.
Acknowledgements
This study was supported by the
research promoting grant from the Keimyung University Dongsan
Medical Center in 2009.
References
|
1.
|
Heldin CH, Eriksson U and Ostman A: New
members of the platelet-derived growth factor family of mitogens.
Arch Biochem Biophys. 398:284–290. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Fredriksson L, Li H and Eriksson U: The
PDGF family: four gene products form five dimeric isoforms.
Cytokine Growth Factor Rev. 15:197–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Betsholtz C, Johnsson A, Heldin CH,
Westermark B, Lind P, Urdea MS, Eddy R, Shows TB, Philpott K and
Mellor AL: cDNA sequence and chromosomal localization of human
platelet-derived growth factor A-chain and its expression in tumour
cell lines. Nature. 320:695–699. 1986. View
Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Heldin CH and Westermark B: Mechanism of
action and in vivo role of platelet-derived growth factor. Physiol
Rev. 79:1283–1316. 1999.PubMed/NCBI
|
|
5.
|
Li X, Pontén A, Aase K, Karlsson L,
Abramsson A, Uutela M, Bäckström G, Hellström M, Boström H, Li H,
Soriano P, Betsholtz C, Heldin CH, Alitalo K, Ostman A and Eriksson
U: PDGF-C is a new protease-activated ligand for the PDGF
alpha-receptor. Nat Cell Biol. 2:302–309. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
LaRochelle WJ, Jeffers M, McDonald WF,
Chillakuru RA, Giese NA, Lokker NA, Sullivan C, Boldog FL, Yang M,
Vernet C, Burgess CE, Fernandes E, Deegler LL, Rittman B, Shimkets
J, Shimkets RA, Rothberg JM and Lichenstein HS: PDGF-D, a new
protease-activated growth factor. Nat Cell Biol. 3:517–521. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Reigstad LJ, Varhaug JE and Lillehaug JR:
Structural and functional specificities of PDGF-C and PDGF-D, the
novel members of the platelet-derived growth factors family. FEBS
J. 272:5723–5741. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Betsholtz C: Biology of platelet-derived
growth factors in development. Birth Defects Res C Embryo Today.
69:272–285. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Andrae J, Gallini R and Betsholtz C: Role
of platelet-derived growth factors in physiology and medicine.
Genes Dev. 22:1276–1312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Betsholtz C, Lindblom P, Bjarnegard M,
Enge M, Gerhardt H and Lindahl P: Role of platelet-derived growth
factor in mesangium development and vasculopathies: lessons from
platelet-derived growth factor and platelet-derived growth factor
receptor mutations in mice. Curr Opin Nephrol Hypertens. 13:45–52.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Floege J, Eitner F and Alpers CE: A new
look at platelet-derived growth factor in renal disease. J Am Soc
Nephrol. 19:12–23. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Bonner JC: Regulation of PDGF and its
receptors in fibrotic diseases. Cytokine Growth Factor Rev.
15:255–273. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Raines EW: PDGF and cardiovascular
disease. Cytokine Growth Factor Rev. 15:237–254. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Floege J, Eitner F, Van Roeyen C and
Ostendorf T: PDGF-D and renal disease: yet another one of those
growth factors? J Am Soc Nephrol. 14:2690–2691. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Ostendorf T, van Roeyen CR, Peterson JD,
Kunter U, Eitner F, Hamad AJ, Chan G, Jia XC, Macaluso J,
Gazit-Bornstein G, Keyt BA, Lichenstein HS, LaRochelle WJ and
Floege J: A fully human monoclonal antibody (CR002) identifies
PDGF-D as a novel mediator of mesangioproliferative
glomerulonephritis. J Am Soc Nephrol. 14:2237–2247. 2003.
View Article : Google Scholar
|
|
16.
|
van Roeyen CR, Ostendorf T, Denecke B,
Bokemeyer D, Behrmann I, Strutz F, Lichenstein HS, LaRochelle WJ,
Pena CE, Chaudhuri A and Floege J: Biological responses to PDGF-BB
versus PDGF-DD in human mesangial cells. Kidney Int. 69:1393–1402.
2006.PubMed/NCBI
|
|
17.
|
Rios A, Vargas-Robles H, Gámez-Méndez AM
and Escalante B: Cyclooxygenase-2 and kidney failure.
Prostaglandins Other Lipid Mediat. 98:86–90. 2012. View Article : Google Scholar
|
|
18.
|
Schneider A and Stahl RA: Cyclooxygenase-2
(COX-2) and the kidney: current status and potential perspectives.
Nephrol Dial Transplant. 13:10–12. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Krämer BK, Kammerl MC and Kömhoff M: Renal
cyclooxygenase-2 (COX-2). Physiological, pathophysiological, and
clinical implications. Kidney Blood Press Res. 27:43–62.
2004.PubMed/NCBI
|
|
20.
|
Giovanni G and Giovanni P: Do
non-steroidal anti-inflammatory drugs and COX-2 selective
inhibitors have different renal effects? J Nephrol. 15:480–488.
2002.PubMed/NCBI
|
|
21.
|
Smith WL, DeWitt DL and Garavito RM:
Cyclooxygenases: structural, cellular, and molecular biology. Annu
Rev Biochem. 69:145–182. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Smith WL and DeWitt DL: Prostaglandin
endoperoxide H synthases-1 and -2. Adv Immunol. 62:167–215. 1996.
View Article : Google Scholar
|
|
23.
|
Hla T and Neilson K: Human
cyclooxygenase-2 cDNA. Proc Natl Acad Sci USA. 89:7384–7388. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Mitchell JA, Belvisi MG, Akarasereenont P,
Robbins RA, Kwon OJ, Croxtall J, Barnes PJ and Vane JR: Induction
of cyclooxygenase-2 by cytokines in human pulmonary epithelial
cells: regulation by dexamethasone. Br J Pharmacol. 113:1008–1014.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Wadleigh DJ, Reddy ST, Kopp E, Ghosh S and
Herschman HR: Transcriptional activation of the cyclooxygenase-2
gene in endotoxin-treated RAW 264.7 macrophages. J Biol Chem.
275:6259–6266. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Jang BC: Induction of COX-2 in human
airway cells by manganese: role of PI3K/PKB, p38 MAPK, PKCs, Src,
and glutathione depletion. Toxicol In Vitro. 23:120–126. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Xu K, Chang CM, Gao H and Shu HK:
Epidermal growth factor-dependent cyclooxygenase-2 induction in
gliomas requires protein kinase C-delta. Oncogene. 28:1410–1420.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Wang D and Dubois RN: The role of COX-2 in
intestinal inflammation and colorectal cancer. Oncogene.
29:781–788. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Hirose S, Yamamoto T, Feng L, Yaoita E,
Kawasaki K, Goto S, Fujinaka H, Wilson CB, Arakawa M and Kihara I:
Expression and localization of cyclooxygenase isoforms and
cytosolic phospholipase A2 in anti-Thy-1 glomerulonephritis. J Am
Soc Nephrol. 9:408–416. 1998.PubMed/NCBI
|
|
30.
|
Khan KN, Stanfield KM, Harris RK and Baron
DA: Expression of cyclooxygenase-2 in the macula densa of human
kidney in hypertension, congestive heart failure, and diabetic
nephropathy. Ren Fail. 23:321–330. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Wang JL, Cheng HF, Shappell S and Harris
RC: A selective cyclooxygenase-2 inhibitor decreases proteinuria
and retards progressive renal injury in rats. Kidney Int.
57:2334–2342. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Cheng HF, Wang CJ, Moeckel GW, Zhang MZ,
McKanna JA and Harris RC: Cyclooxygenase-2 inhibitor blocks
expression of mediators of renal injury in a model of diabetes and
hypertension. Kidney Int. 62:929–939. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Goppelt-Struebe M, Rehm M and Schaefers
HJ: Induction of cyclooxygenase-2 by platelet-derived growth factor
(PDGF) and its inhibition by dexamethasone are independent of
NF-kappaB/IkappaB transcription factors. Naunyn Schmiedebergs Arch
Pharmacol. 361:636–345. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Xu K, Kitchen CM, Shu HK and Murphy TJ:
Platelet-derived growth factor-induced stabilization of
cyclooxygenase 2 mRNA in rat smooth muscle cells requires the c-Src
family of protein-tyrosine kinases. J Biol Chem. 282:32699–32709.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Nakao S, Ogata Y, Yamamoto Y, Furuyama S
and Sugiya H: Platelet-derived growth factor-induced arachidonic
acid release for enhancement of prostaglandin E(2) synthesis in
human gingival fibroblasts pretreated with interleukin-1beta. J
Cell Biochem. 92:579–590. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Dean JL, Brook M, Clark AR and Saklatvala
J: p38 mitogen-activated protein kinase regulates cyclooxygenase-2
mRNA stability and transcription in lipopolysaccharide-treated
human monocytes. J Biol Chem. 274:264–269. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Guan Z, Buckman SY, Miller BW, Springer LD
and Morrison AR: Interleukin-1beta-induced cyclooxygenase-2
expression requires activation of both c-Jun NH2-terminal kinase
and p38 MAPK signal pathways in rat renal mesangial cells. J Biol
Chem. 273:28670–28676. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Ammoun S, Flaiz C, Ristic N, Schuldt J and
Hanemann CO: Dissecting and targeting the growth factor-dependent
and growth factor-independent extracellular signal-regulated kinase
pathway in human schwannoma. Cancer Res. 68:5236–5245. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Okada A, Yaguchi T, Kanno T, Gotoh A,
Nakano T and Nishizaki T: PDGF-D/PDGF-ββ receptor-regulated
chemotaxis of malignant mesothelioma cells. Cell Physiol Biochem.
29:241–250. 2012.
|
|
40.
|
Borkham-Kamphorst E, van Roeyen CR,
Ostendorf T, Floege J, Gressner AM and Weiskirchen R:
Pro-fibrogenic potential of PDGF-D in liver fibrosis. J Hepatol.
46:1064–1074. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Menè P, Abboud HE and Dunn MJ: Regulation
of human mesangial cell growth in culture by thromboxane A2 and
prostacyclin. Kidney Int. 38:232–239. 1990.PubMed/NCBI
|
|
42.
|
Stahl RA, Thaiss F, Haberstroh U, Kahf S,
Shaw A and Schoeppe W: Cyclooxygenase inhibition enhances rat
interleukin 1 beta-induced growth of rat mesangial cells in
culture. Am J Physiol. 259:419–424. 1990.PubMed/NCBI
|
|
43.
|
Zahner G, Wolf G, Ayoub M, Reinking R,
Panzer U, Shankland SJ and Stahl RA: Cyclooxygenase-2
overexpression inhibits platelet-derived growth factor-induced
mesangial cell proliferation through induction of the tumor
suppressor gene p53 and the cyclin-dependent kinase inhibitors
p21waf-1/cip-1 and p27kip-1. J Biol Chem. 277:9763–9771. 2002.
View Article : Google Scholar
|
|
44.
|
Trifan OC, Smith RM, Thompson BD and Hla
T: Overexpression of cyclooxygenase-2 induces cell cycle arrest.
Evidence for a prostaglandin-independent mechanism. J Biol Chem.
274:34141–34147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Shibata T, Tamura M, Kabashima N, Serino
R, Tokunaga M, Matsumoto M, Miyamoto T, Miyazaki M, Furuno Y,
Takeuchi M, Abe H, Okazaki M and Otsuji Y: Fluvastatin attenuates
IGF-1-induced ERK1/2 activation and cell proliferation by mevalonic
acid depletion in human mesangial cells. Life Sci. 84:725–731.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Mitchell D, Rodgers K, Hanly J, McMahon B,
Brady HR, Martin F and Godson C: Lipoxins inhibit Akt/PKB
activation and cell cycle progression in human mesangial cells. Am
J Pathol. 164:937–946. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Bassa BV, Noh JW, Ganji SH, Shin MK, Roh
DD and Kamanna VS: Lysophosphatidylcholine stimulates EGF receptor
activation and mesangial cell proliferation: regulatory role of Src
and PKC. Biochim Biophys Acta. 1771:1364–1371. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Tokuyama H, Kim S, Zhang Y, Langham RG,
Cox AJ, Gow RM, Kelly DJ and Gilbert RE: Protein kinase C β
inhibition ameliorates experimental mesangial proliferative
glomerulonephritis. Nephrology (Carlton). 16:649–655. 2011.
|