Introduction
Alzheimer’s disease (AD) is characterized by
cerebrovascular damage and neuronal dysfunction leading to
progressive cognitive decline. The hallmark pathologies of AD
include amyloid β-peptide (Aβ) deposition, neurofibrillary tangles
containing hyperphosphorylated tau and increased microglial
reactivity as well as inflammatory processes. Hypercholesterolemia,
a well-established vascular risk factor, is associated with the
pathophysiology of AD (1,2) and it has also been shown to increase
the production of Aβ in animal and cellular models (3–7).
However, the mechanism by which hypercholesterolemia causes Aβ
accumulation in the brain and contributes to the pathogenesis of AD
remains unclear.
Hypercholesterolemia and AD share inflammation as a
pathophysiological trait. It was considered that high levels of
serum cholesterol would induce vascular changes similar to early
inflammatory lesions of atherosclerosis and blood brain barrier
permeability as well as localized neuroinflammatory changes
(8). Furthermore, chronic
inflammatory responses including microglial activation have been
detected in AD brains (9,10). It is likely that
hypercholesterolemia resulting from either dietary practice or
genetic disposition facilitates an exacerbated neuroinflammatory
response in association with increased Aβ generation, leading to
AD-associated degeneration. To test this theory, we administered a
high-fat diet to a hypercholesterolemic strain (Apo E KO mice) for
4 weeks prior to Aβ25–35 injection and for 4 weeks
following Aβ25–35 injection, for a total of 8 weeks of
treatment. This was followed by assessment of cognitive function
with respect to neuroinflammatory and AD-related biochemical
changes.
We hypothesized that hypercholesterolemia
accelerates amyloid pathology, tau pathology and neuroinflammation,
subsequently exacerbating the cognitive decline in AD. We
investigated the effects of hypercholesterolemia on AD-related
neuropathological markers including Aβ, phosphorylatedtau (p-tau)
and microglial activation in the brains of normal and
hypercholesterolemic mice injected with Aβ25–35 (a toxic
fragment of full-length Aβ). Spatial learning and memory in
Aβ25–35-injected mice was assessed by the Morris water
maze test. Exploring the interplay between hypercholesterolemia and
AD may enable development of a rational basis for the prevention
and treatment of AD underlying clinically detected dementia.
Materials and methods
Aβ25–35 injection model
Wild-type male C57BL/6J and apolipoprotein E knock
out (Apo E KO) mice (Japan SLC, Shizuoka, Japan) were housed under
diurnal lighting conditions and allowed food and tap water ad
libitum. All animal procedures were conducted in accordance
with the institutional guidelines for animal research, and approved
by the University Animal Care and Use Committee (PNU-2011-000419).
An Aβ-induced cognitive impairment mouse model was produced based
on modification of previously reported methods (11). Anesthesia was achieved by
isoflurane (2% induction and 1.5% maintenance, in 80%
N2O and 20% O2) administered via a face mask.
The depth of anesthesia was confirmed by the absence of
cardiovascular changes in response to tail pinch. Rectal
temperature was kept at 36.5–37.5°C using a Panlab™
thermostatically controlled heating mat (Harvard Apparatus,
Holliston, MA, USA). Aβ25–35 (10 nmol in 5 μl of
saline; Sigma-Aldrich, St. Louis, MO, USA) was injected
intracerebroventricularly (icv) into the mice, aimed at 1 mm
lateral to the midline, 0.5 mm posterior to the bregma and 3 mm
deep using a 25 μl Hamilton syringe with a 26-gauge needle
(Hamilton, Reno, NV, USA) at a rate of 0.5 μl/min using a
stereotaxic injector (KD Scientific, Holliston, MA, USA). The
vehicle group of mice received icv injections of an equal volume of
saline. Hypercholesterolemia was induced by providing a
Western-type high-fat diet (42% of total calories from fat; 0.15%
cholesterol; Research Diets, New Brunswick, NJ, USA) for 4 weeks in
Apo E KO mice prior to Aβ25–35 injection and for 4 weeks
following Aβ25–35 injection, for a total of 8 weeks.
Measurement of plasma cholesterol
Blood was collected from the left ventricle under
light anesthesia and stored on ice for 30 min prior to
centrifugation at 13,000 rpm and 4°C for 10 min, after which the
plasma was separated and kept at −80°C until assayed. The
lipoprotein cholesterol distribution of plasma samples was
determined enzymatically with reagents from Kyowa Medex (Tokyo,
Japan) using a TBA-200FR NEO apparatus (Toshiba Medical Systems,
Tokyo, Japan).
Immunohistochemistry
Four weeks after Aβ25–35 injection, mice
were deeply anesthetized with thiopental sodium and subsequently
perfused transcardially with cold PBS followed by 4%
paraformaldehyde for fixation. The brain of each mouse was then
removed and further fixed for 24 h in 4% paraformaldehyde at 4°C
followed by cryoprotection in 20% sucrose for 72 h at 4°C. Next,
the isolated brains were frozen in an optical cutting temperature
medium for frozen tissue specimens (Sakura Finetek, Torrance, CA,
USA) and then stored in the freezer at −80°C until examined. The
frozen brains were subsequently cut at a thickness of 10 μm
using a Leica CM 3050 Cryostat (Leica Microsystems, Wetzlar,
Germany), after which the sections were immunostained with
antibodies against Aβ (Covance, Emeryville, CA, USA), p-tau
(Sigma-Aldrich) and CD11b (Serotec, Oxford, UK). Following
additional incubation with biotinylated secondary antibody, samples
were incubated in ABC reagent (Vector Laboratories, Burlingame, CA,
USA). All reactions were visualized by development in 3,3′
diaminobenzidine substrates (Vector Laboratories), and all samples
were visualized using a light microscope (Carl Zeiss, Jena,
Germany). The immunoreaction products were quantified using the
iSolution full image analysis software (Image & Microscope
Technology, Vancouver, BC, Canada).
Morris water maze task
Spatial learning and memory deficits were assessed
using the Morris water maze task as previously described (11) with minor modification. The
experiment was performed on mice after a 1- or 4-week
Aβ25–35 injection period. The maze consisted of a
1.15-m-diameter pool that was painted flat white and had a
10-cm-diameter platform halfway between the center of the pool and
the edge, 1 cm below the surface of the water. The water in the
pool was made opaque by the addition of powdered milk. The water
temperature was 19–21°C. The water tank was located in a test room
that contained several cues external to the maze. The position of
the cues remained unchanged throughout the water-maze task. Each
mouse was subjected to a series of 5 trials/day. For each trial,
mice were randomized to 1 of 4 directional starting locations
(north, south, east and west) and placed in the pool facing the
wall. During the 4 subsequent training days, the mice were given 5
trial sessions/day with the platform in place before an
Aβ25–35 injection. When a mouse located the platform, it
was permitted to remain on it for 10 sec. If the mouse did not
locate the platform within 180 sec, it was placed on the platform
for 10 sec. Mice were given a maximum of 180 sec to find the
submerged platform. Swimming was videotaped, and the escape latency
time to reach the platform was analyzed using the Smart software
(Panlab, Barcelona, Spain).
Data analysis
All data are expressed as the means ± SEM.
Statistical comparisons were performed using paired or unpaired
Student’s t-tests, and one-way analysis of variance (ANOVA) or
two-way ANOVA for repeated measures followed by Fisher’s protected
least significant difference test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Hypercholesterolemia increases Aβ, p-tau
and CD11b expression in Aβ-injected mice
Plasma cholesterol levels were significantly higher
in Apo E KO mice that were fed a high-fat diet than in control mice
fed normal chow (737.56±27.58 vs. 99.4±7.58 mg/dl, respectively,
P<0.01), indicating that administration of the high-fat diet for
8 weeks induced hypercholesterolemia. To validate the function of
hypercholesterolemia on AD in vivo, Aβ25–35 was
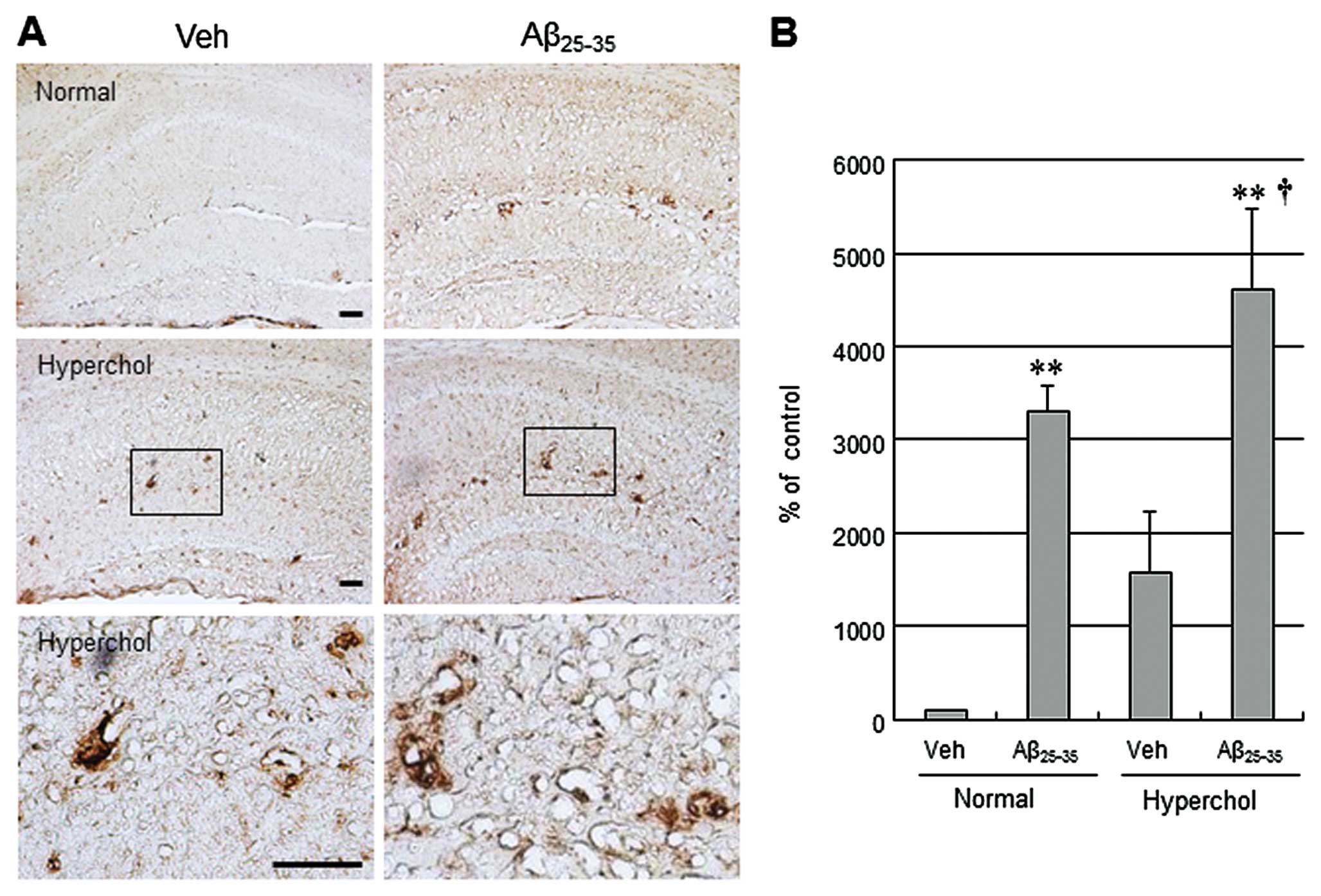
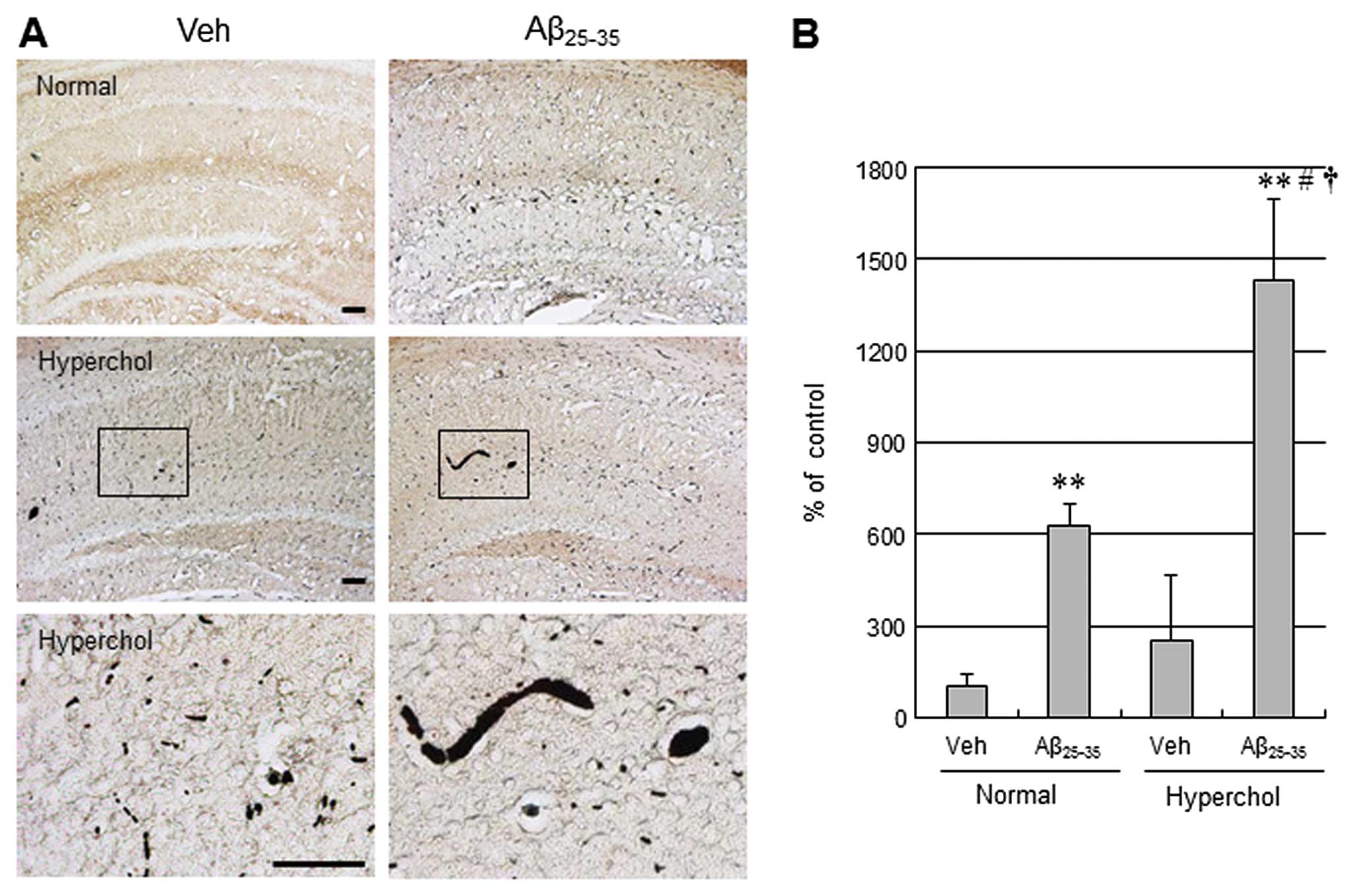
used for the AD-like pathological model in this study. Unilateral
icv injection of Aβ25–35 led to increased
immunoreactivity for the AD-related neuropathological markers Aβ
and p-tau in the hippocampus (Figs.
1 and 2). Moreover,
hypercholesterolemia tended to increase Aβ and p-tau levels in the
vehicle group and further increased Aβ and p-tau levels in
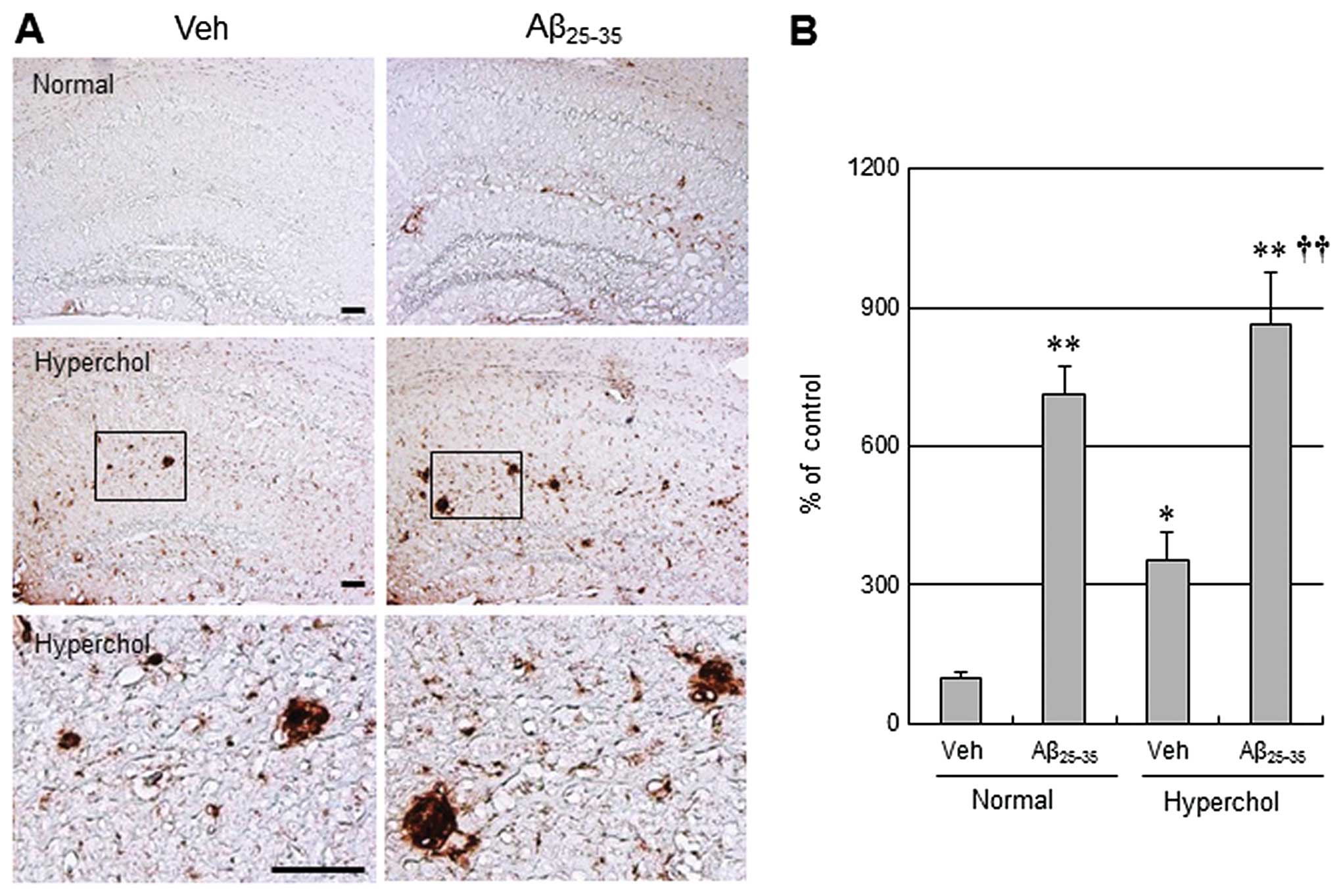
Aβ25–35-injected mice. CD11b (also known as
αmβ2 integrin and Mac1) is a sensitive marker
of microglial activation. Immunostaining of CD11b was evident in
the hippocampus of normal mice receiving icv injections of
Aβ25–35 (Fig. 3).
Consistent with Aβ and p-tau, hypercholesterolemia led to a more
significant increase in CD11b immunostaining in both the vehicle
group and the Aβ25–35-injected group of
hypercholesterolemic mice. However, there were no significant
changes in GFAP immunoreactivity in normal and hypercholesterolemic
mice treated following Aβ25–35 icv injections (data not
shown).
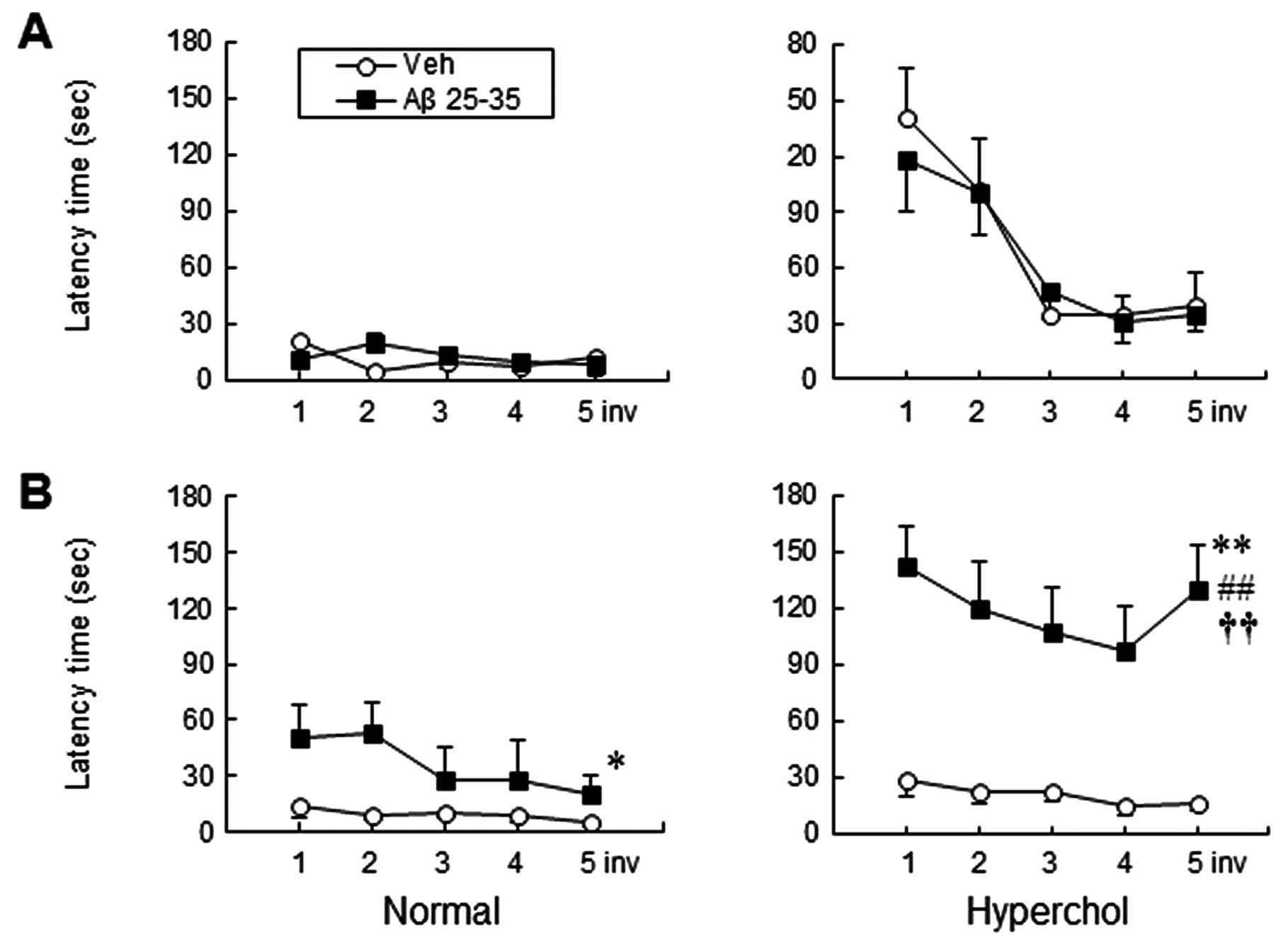
Hypercholesterolemia aggravates cognitive
impairment in Aβ-injected mice
To verify whether hypercholesterolemia has any
effect on cognitive deficits, we subjected the mice to a water
maze. As shown in Fig. 4, icv
injection of Aβ25–35 resulted in a significantly
increased escape latency time in the target quadrant when compared
to the vehicle group at 4 weeks after Aβ injection (P<0.05 vs.
vehicle) (Fig. 4B). Conversely,
no significant difference was observed among groups at 1 week after
Aβ injection (Fig. 4A). Moreover,
hypercholesterolemia significantly exacerbated the cognitive
decline when compared to the Aβ25–35-injected group of
normal mice at 4 weeks after Aβ injection (P<0.01, two-way ANOVA
for repeated measures). Thus, decreased spatial learning in
response to hypercholesterolemia was coincident with accelerated
expression of Aβ and p-tau and activation of microglia in the
Aβ25–35-injected group of hypercholesterolemic mice.
Discussion
In the present study, we examined the effects of
hypercholesterolemia on the cognitive deficits in
Aβ25–35-injected mice with respect to neuroinflammatory
and AD-related biochemical changes. Our results demonstrated that
hyper-cholesterolemia strongly accelerated Aβ accumulation, p-tau
expression and microglial activation in the brains of
Aβ25–35-injected mice. In addition, hypercholesterolemia
significantly aggravated spatial learning and memory in
Aβ25–35-injected mice as assessed by the Morris water
maze test. These data suggest that hypercholesterolemia accelerates
the AD-related pathology, which was accompanied by
neuroinflammation and subsequent memory impairment in this mouse
model. Thus, we propose that diet can be used to modulate the risk
of developing AD.
Clinical studies indicate that patients with
elevated cholesterol have increased susceptibility to AD (12–14) and that lowering plasma cholesterol
with statin therapy during middle age is likely to benefit
cognitive function in later life (15,16). Experimental studies using rabbits
(3) and a transgenic mouse model
of AD (4–6) have demonstrated that diet-induced
hypercholesterolemia increased Aβ levels in the brain and thus
accelerated extracellular Aβ deposition. Aβ depositions and
neurofibrillary tangles are distinctive hallmarks of AD. A
disturbed cholesterol homeostasis within lipid rafts might
influence amyloid precursor protein (APP) processing, which results
in increased Aβ production and deposition (17,18). Conversely, little is known about
the function of cholesterol in the formation of neurofibrillary
tangles. Our data revealed that hypercholesterolemia enhanced Aβ as
well as p-tau levels in the brain, which was confirmed by
descriptive immunohistochemical analysis. Our findings are in
accordance with previous studies showing elevated concentrations of
Aβ (4,19,20) and an enhanced level of
hyperphosphorylated tau (20)
after consumption of a cholesterol-enhanced diet.
AD is characterized by cerebrovascular damage and
neuronal dysfunction, which leads to progressive cognitive decline.
Despite clear evidence of hypercholesterolemia-induced amyloid
pathology, the relationship between hypercholesterolemia and memory
impairment in animals is somewhat controversial (21–23). In this study, hypercholesterolemia
led to significant exacerbation of cognitive decline when compared
to the Aβ25–35-injected group of normal mice at 4 weeks
after Aβ injection. Similar to our findings, aggravation of
learning deficits in association with Aβ deposition in APP
transgenic mice with low density lipoprotein receptor
(LDLR)-deficiency has been reported (24).
The exact mechanisms responsible for the cognitive
deficit induced by hypercholesterolemia in mice remain unknown. One
possible explanation is that a oxidative-inflammatory cycle in the
vasculature could have deleterious consequences for brain function
and cognition (25,26). It is well known that inflammation
occurs in AD brains and hypercholesterolemic animal models
(8,26). Our data demonstrate that
hypercholesterolemia induced elevated immunohistochemical-positive
staining for the microglial marker CD11b. There is now convincing
evidence that inflammatory cascades primarily mediated by activated
microglia play a significant role in AD-like changes (9,27).
The mechanism of microglial activation and the role of
hypercholesterolemia in the process of neuroinflammation are not
fully known. It is possible that the systemic inflammation under
hypercholesterolemic conditions, which causes dysfunctional
cerebrovasculature, may trigger activation of the perivascular
microglia (28) and recruitment
of microglia to the inflamed brain arterioles could then initiate a
cascade of events leading to AD-like pathology and cognitive
dysfunction.
The effects of hypercholesterolemia in vivo
have been studied in different animal models, and experimental
studies have shown that hypercholesterolemia increases brain Aβ
deposition and neuroinflammation in rabbits (3), soluble APP transgenic mice (4) and LDLR deficit mice (26). In the present study, we used Apo E
KO mice on a high-fat diet to induce hypercholesterolemia and
Aβ25–35, a toxic core fragment of full-length
Aβ1–40 (29), to model
AD-like pathology. Although it remains unclear whether the
Aβ25–35 fragment occurs in AD brains, a number of
studies have demonstrated that the acute injection of this peptide
into the cerebral ventricle results in some pathological events
observed in AD, such as neurotoxic effects similar to those
produced by Aβ1–40 (30) or impairment of memory in mice
(31). To the best of our
knowledge, the present study is the first to examine the
hypercholesterolemic effects in an Aβ25–35-injected
AD-like pathological mouse model.
In conclusion, the present study supports the
findings of clinical studies suggesting that a high-fat diet
increases the risk of developing AD and therefore suggests that
diet could be used to reduce the risk of developing the disease.
These findings suggest that dietary methods might offer a path to
the prevention of AD without resorting to drugs that are expensive
and may have harmful side-effects.
Acknowledgements
This work was supported by a 2-Year
Research Grant of Pusan National University.
References
|
1
|
Solomon A and Kivipelto M:
Cholesterol-modifying strategies for Alzheimer’s disease. Expert
Rev Neurother. 9:695–709. 2009.PubMed/NCBI
|
|
2
|
Stefani M and Liguri G: Cholesterol in
Alzheimer’s disease: unresolved questions. Curr Alzheimer Res.
6:15–29. 2009.
|
|
3
|
Sparks DL, Scheff SW, Hunsaker JC III, Liu
H, Landers T and Gross DR: Induction of Alzheimer-like beta-amyloid
immuno-reactivity in the brains of rabbits with dietary
cholesterol. Exp Neurol. 126:88–94. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Refolo LM, Malester B, LaFrancois J, et
al: Hypercholesterolemia accelerates the Alzheimer’s amyloid
pathology in a transgenic mouse model. Neurobiol Dis. 7:321–331.
2000.
|
|
5
|
Levin-Allerhand JA, Lominska CE and Smith
JD: Increased amyloid- levels in APPSWE transgenic mice treated
chronically with a physiological high-fat high-cholesterol diet. J
Nutr Health Aging. 6:315–319. 2002.PubMed/NCBI
|
|
6
|
Shie FS, Jin LW, Cook DG, Leverenz JB and
LeBoeuf RC: Diet-induced hypercholesterolemia enhances brain A beta
accumulation in transgenic mice. Neuroreport. 13:455–459. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Simons M, Keller P, De Strooper B,
Beyreuther K, Dotti CG and Simons K: Cholesterol depletion inhibits
the generation of beta-amyloid in hippocampal neurons. Proc Natl
Acad Sci USA. 95:6460–6464. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xue QS, Sparks DL and Streit WJ:
Microglial activation in the hippocampus of hypercholesterolemic
rabbits occurs independent of increased amyloid production. J
Neuroinflammation. 4:202007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Akiyama H, Barger S, Barnum S, et al:
Inflammation and Alzheimer’s disease. Neurobiol Aging. 21:383–421.
2000.
|
|
10
|
Rogers J: The inflammatory response in
Alzheimer’s disease. J Periodontol. 79(Suppl 8): S1535–S1543.
2008.
|
|
11
|
Takeda S, Sato N, Niisato K, et al:
Validation of Abeta1-40 administration into mouse cerebroventricles
as an animal model for Alzheimer disease. Brain Res. 1280:137–147.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Casserly I and Topol E: Convergence of
atherosclerosis and Alzheimer’s disease: inflammation, cholesterol,
and misfolded proteins. Lancet. 363:1139–1146. 2004.
|
|
13
|
Sjogren M, Mielke M, Gustafson D, Zandi P
and Skoog I: Cholesterol and Alzheimer’s disease - is there a
relation? Mech Ageing Dev. 127:138–147. 2006.
|
|
14
|
Canevari L and Clark JB: Alzheimer’s
disease and cholesterol: the fat connection. Neurochem Res.
32:739–750. 2007.
|
|
15
|
van Vliet P, van de Water W, de Craen AJ
and Westendorp RG: The influence of age on the association between
cholesterol and cognitive function. Exp Gerontol. 44:112–122.
2009.
|
|
16
|
Cedazo-Minguez A, Ismail MA and Mateos L:
Plasma cholesterol and risk for late-onset Alzheimer’s disease.
Expert Rev Neurother. 11:495–498. 2011.
|
|
17
|
Ghribi O: Potential mechanisms linking
cholesterol to Alzheimer’s disease-like pathology in rabbit brain,
hippocampal organotypic slices, and skeletal muscle. J Alzheimers
Dis. 15:673–684. 2008.
|
|
18
|
Simons M, Keller P, Dichgans J and Schulz
JB: Cholesterol and Alzheimer’s disease: is there a link?
Neurology. 57:1089–1093. 2001.
|
|
19
|
Sharma S, Prasanthi RPJ, Schommer E, Feist
G and Ghribi O: Hypercholesterolemia-induced Abeta accumulation in
rabbit brain is associated with alteration in IGF-1 signaling.
Neurobiol Dis. 32:426–432. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ghribi O, Larsen B, Schrag M and Herman
MM: High cholesterol content in neurons increases BACE,
beta-amyloid, and phosphorylated tau levels in rabbit hippocampus.
Exp Neurol. 200:460–467. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fitz NF, Cronican A, Pham T, et al: Liver
X receptor agonist treatment ameliorates amyloid pathology and
memory deficits caused by high-fat diet in APP23 mice. J Neurosci.
30:6862–6872. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ullrich C, Pirchl M and Humpel C:
Hypercholesterolemia in rats impairs the cholinergic system and
leads to memory deficits. Mol Cell Neurosci. 45:408–417. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schreurs BG: The effects of cholesterol on
learning and memory. Neurosci Biobehav Rev. 34:1366–1379. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cao D, Fukuchi K, Wan H, Kim H and Li L:
Lack of LDL receptor aggravates learning deficits and amyloid
deposits in Alzheimer transgenic mice. Neurobiol Aging.
27:1632–1643. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Evola M, Hall A, Wall T, Young A and
Grammas P: Oxidative stress impairs learning and memory in apoE
knockout mice. Pharmacol Biochem Behav. 96:181–186. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thirumangalakudi L, Prakasam A, Zhang R,
et al: High cholesterol-induced neuroinflammation and amyloid
precursor protein processing correlate with loss of working memory
in mice. J Neurochem. 106:475–485. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Heneka MT and O’Banion MK: Inflammatory
processes in Alzheimer’s disease. J Neuroimmunol. 184:69–91.
2007.
|
|
28
|
Perry VH, Newman TA and Cunningham C: The
impact of systemic infection on the progression of
neurodegenerative disease. Nat Rev Neurosci. 4:103–112. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yankner BA, Duffy LK and Kirschner DA:
Neurotrophic and neurotoxic effects of amyloid beta protein:
reversal by tachykinin neuropeptides. Science. 250:279–282. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamada K and Nabeshima T: Animal models of
Alzheimer’s disease and evaluation of anti-dementia drugs.
Pharmacol Ther. 88:93–113. 2000.
|
|
31
|
Kim DH, Kim S, Jeon SJ, et al: The effects
of acute and repeated oroxylin A treatments on Abeta(25–35)-induced
memory impairment in mice. Neuropharmacology. 55:639–647.
2008.PubMed/NCBI
|