Introduction
Bisphosphonates (BPs) remain the most widely used
and effective antiresorptive agents for the treatment of diseases
in which there is an increase in the number or activity of
osteoclasts, including tumor-associated osteolysis and
hypercalcemia (1). BPs are a
class of pharmacological compounds that are used in the treatment
of postmenopausal osteoporosis; however, they are associated with a
number of side-effects such as osteonecrosis of the jaw. Following
BP administration, there was an increase in circulating endothelial
cell apoptosis in multiple myeloma patients and osteonecrosis
subjects (2).
Nitrogen-containing bisphosphonate (N-BP) has been
reported to induce antiproliferative and apoptotic effects in human
pancreatic cancer cells in vitro (3) and to exert antiangiogenic effects
in vivo and in vitro (4). Alendronate (Aln) acts on bone marrow
stromal cells to stimulate osteogenic differentiation and to
inhibit adipogenic differentiation in a dose-dependent manner;
these effects are mediated by activating extracellular
signal-regulated kinase (ERK) and c-Jun-N-terminal kinase (JNK)
(5). In addition, Aln inhibits
the cell survival pathway stimulated by the phosphoinositide
3-kinase (PI3K)/Akt/nuclear factor-κB (NF-κB) pathway, thus causing
apoptosis of osteosarcoma cells (6). Zoledronate (Zol) inhibits human
umbilical vein endothelial cell (HUVEC) adhesion, survival,
migration and actin stress fiber formation by interfering with
protein prenylation and it affects ERK1/2, JNK, Rock, FAK and PKB
as kinases in a prenylation-dependent manner (7); it also downregulates vascular
endothelial growth factor, endothelial nitric oxide synthase, Akt,
and matrix metalloproteinase (MMP)-9 activities in ischemic tissues
(8).
Sphingolipid metabolites play key roles in the
regulation of numerous cellular processes important for health and
disease. One of the most significant of these metabolites is
sphingosine-1-phosphate (S1P). This signaling molecule regulates
cell growth and suppresses apoptosis (9), suggesting that it may play a role in
cancer. The biological effects of S1P are mediated by activation of
G protein-coupled S1P receptors, leading to the activation of
downstream effectors such as p44/42, MAPK and Akt (10). S1P has been shown to regulate the
migratory behavior of osteoclast precursors in bone tissues
(11), and it regulates a wide
range of cellular activities involved in angiogenesis, wound
healing, apoptosis, and atherosclerosis in vascular endothelial
cells (12). Thus, S1P induces
cell migration, expression of several cell adhesion molecules, DNA
synthesis and cell survival.
We previously showed that BP was able to induce cell
death in normal endothelial cells and activated protein C (APC) was
able to inhibit BP-mediated endothelial cell damage via inhibition
of NF-κB and enhancement of MMP-2 activity (13). In the present study, we
investigated the protective effect of S1P and the possible
mechanism against BP-induced endothelial cell damage in HUVECs.
Materials and methods
Cell culture
The HUVECs were purchased from Lonza (Walkersville,
MD, USA). Cells were grown in Biorich containing 20% fetal bovine
serum (FBS) with 50 μg/ml endothelial cell growth supplement (BD
Biosciences) and 50 μg/ml heparin (Sigma). Cells were cultured at
37°C in a humidified atmosphere of 5% CO2 and 95% air.
HUVECs were used at passages 3–8.
Lactate dehydrogenase (LDH) assay
Cytotoxicity was assessed by the LDH assay in the
supernatant medium using an LDH Cytotoxicity Detection kit (Takara
Bio, Inc., Tokyo, Japan) according to the manufacturer’s protocol.
The LDH activity was determined by measuring the absorbance at 490
nm using a microplate reader (Spectra Max M2; Molecular Devices,
USA).
Cell viability assay
Cell survival was counted by Guava easyCyte HT
(Millipore). Cells were stained by Annexin V in the detached cells
using an Annexin V assay kit (Santa Cruz Biotechnology, Inc.,)
according to the manufacturer’s protocol. Annexin V measurement was
determined by measuring the fluorescence at excitation 488 nm and
emission 525/30 using a Guava easyCyte HT (Millipore).
Protein extraction and western blot
analyses
HUVECs were washed two times with PBS and lysed in a
cell lysis buffer (25 mM HEPES; pH 7.4, 100 mM NaCl, 1 mM EDTA, 5
mM MgCl2, 0.1 mM DTT and protease inhibitor mixture).
Proteins were electrophoretically resolved on a 10–15% sodium
dodecyl sulfate (SDS) gel, and immunoblotting was performed in a
routine manner. Equal amounts of lysate protein were resolved on a
10–15% SDS-polyacrylamide gel and electrophoretically transferred
to a nitrocellulose membrane. Immunoreactivity was detected through
sequential incubation with horseradish peroxidase-conjugated
secondary antibodies and ECL reagents. The antibodies used for
immunoblotting were caspase-3, phospho-JNK (Cell Signaling
Technology), Rap-1A (Santa Cruz Biotechnology, Inc.,) and β-actin
(Sigma). Images were examined using an Fusion-FX7 imaging system
(Vilber Lourmat).
Immunofluorescent staining
Cultured HUVECs on 8-chamber culture slides were
fixed with cold acetone and blocked by 5% FBS in TBST and incubated
with mouse p-JNK antibody (Cell Signaling Technology) and rabbit
active caspase-3 antibody (R&D Systems) overnight at 4°C. After
washing with TBST, cells were incubated with goat anti-mouse IgG
conjugated with Alexa Fluor® 488 (green) and goat
anti-rabbit IgG conjugated with Alexa Fluor® 546 (red).
Cells were washed with TBST, mounted with fluorescence mounting
medium (Dako) and observed under a fluorescence microscope (Nikon
Eclipse 80i; Nikon Corporation). Images were acquired and processed
using a Nikon digital camera and software (Diagnostic Instruments,
Australia) and ImageJ.
Statistical analyses
Data are expressed as the means ± standard deviation
(SD), and were compared using the Student’s t-test and the ANOVA
Duncan test with the SAS statistical package (SAS Institute, Cary,
NC, USA). The results were considered to indicate statistically
significant differences at P<0.05 or P<0.01, and P<0.01 or
P<0.005.
Results
S1P protects BP-induced endothelial cell
death
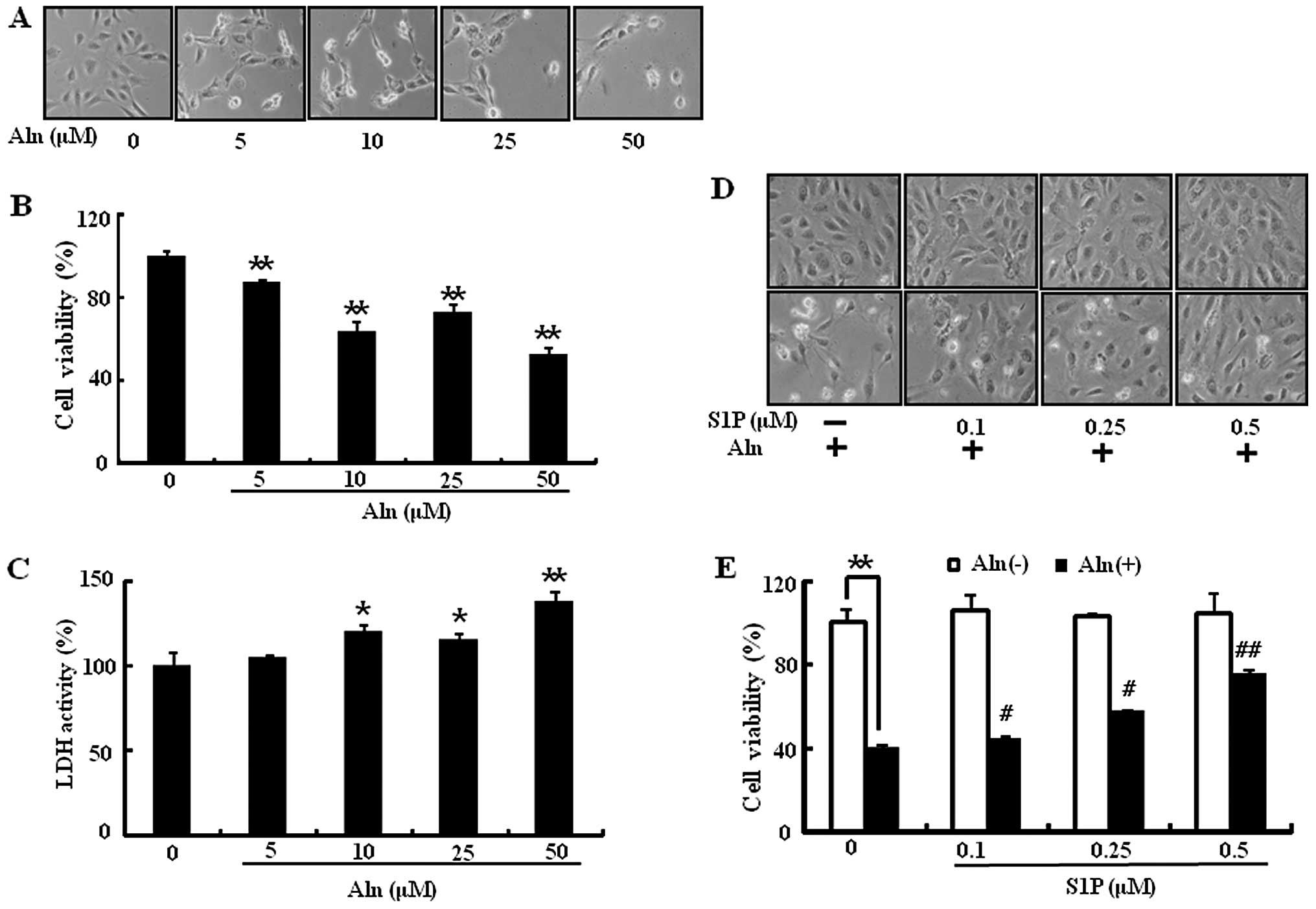
It is well known that BP induces endothelial cell
death (13). Since S1P has been
described to induce cell survival, in the present study we examined
whether S1P could protect BP-induced endothelial cell death. HUVECs
were exposed to Aln dose-dependently for 24 h prior to assessment
of cell viability. Aln induces a decrease in cell viability up to
50% (Fig. 1A and B) and an
increase in cytotoxicity (Fig.
1C). When HUVECs were pretreated with S1P for 1 h and then
exposed to Aln (50 μM), S1P significantly prevented Aln-induced
endothelial cell death in a dose-dependent manner (Fig. 1D and E). Examination of cell
morphology also supported the protective effect of the S1P and Aln
cotreatment in HUVECs (Fig. 1D).
HUVECs were pretreated with or without 0.5 μM S1P and then exposed
to Aln (50 μM), Zol (100 μM) or risedronate (Ris) (100 μM) for 24
h. Zol and Ris treatments induced cell death as well as Aln and
were significantly reversed by treatment with S1P (Fig. 2A and B). To determine the ability
of S1P to inhibit BP-induced apoptosis, TUNEL assays were performed
following exposure of BP in the presence or absence of S1P
(Fig. 2C). A high amount of
apoptotic cells was observed in BP-treated cells but S1P
pretreatment inhibited cell damage by reducing the amount of
apoptotic cells. Overall, we found that the order of potency of
N-BPs for inducing apoptosis of HUVECs and S1P could inhibit
BP-mediated endothelial cell damage.
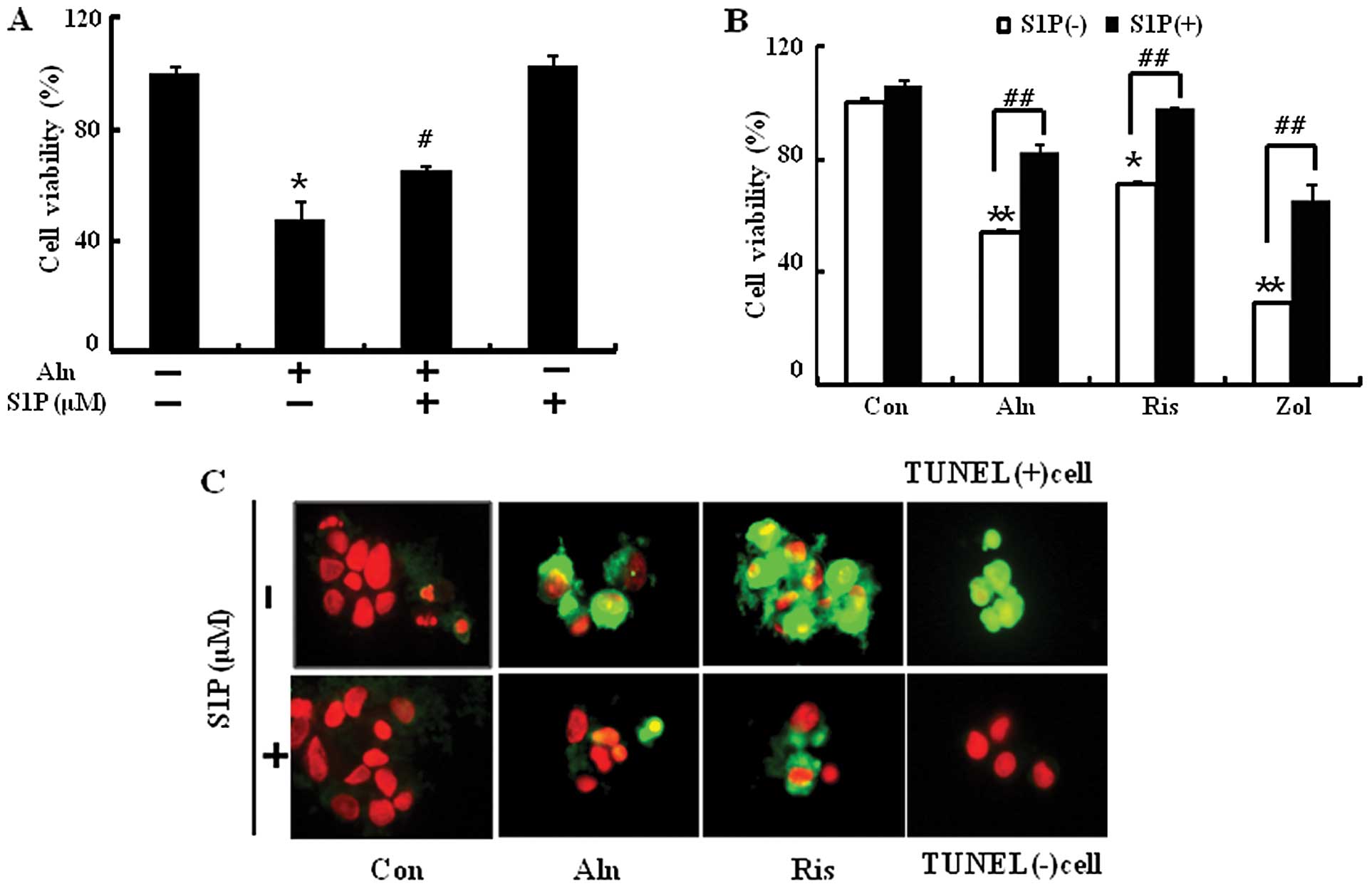 | Figure 2S1P protects against BP-induced
endothelial cell death. HUVECs were pretreated with 0.5 μM of S1P
(1 h) and then exposed to 500 μM Aln for 24 h. (A) Cell viability
was measured after Annexin V assay by flow cytometry. (B) Cells
were pretreated with 0.5 μM of S1P (1 h) and then exposed to Aln
(500 μM), Zol (100 μM) or Ris (100 μM) and cell viability was
measured. (C) Representative immunofluorescence images of
TUNEL-positive (green) HUVECs at 12 h after exposure to 500 μM of
Aln in the absence and presence of S1P. The cells were
counterstained with propidium iodide (red) to show all cell nuclei.
Magnification, ×400. Bar graph indicates the mean ± SEM (n=3).
*P<0.05, **P<0.01, significant
differences between control and each treatment group.
#P<0.01, ##P<0.005, significant
differences between 500 μM Aln and cotreatment with S1P group. BP,
bisphosphonate; S1P, sphingosine-1-phosphate; Aln, alendronate;
Ris, risedronate; Zol, zoledronate. |
Inhibitory effect of S1P on BP-induced
cleaved caspase-3 and NF-κB activation in endothelial cells
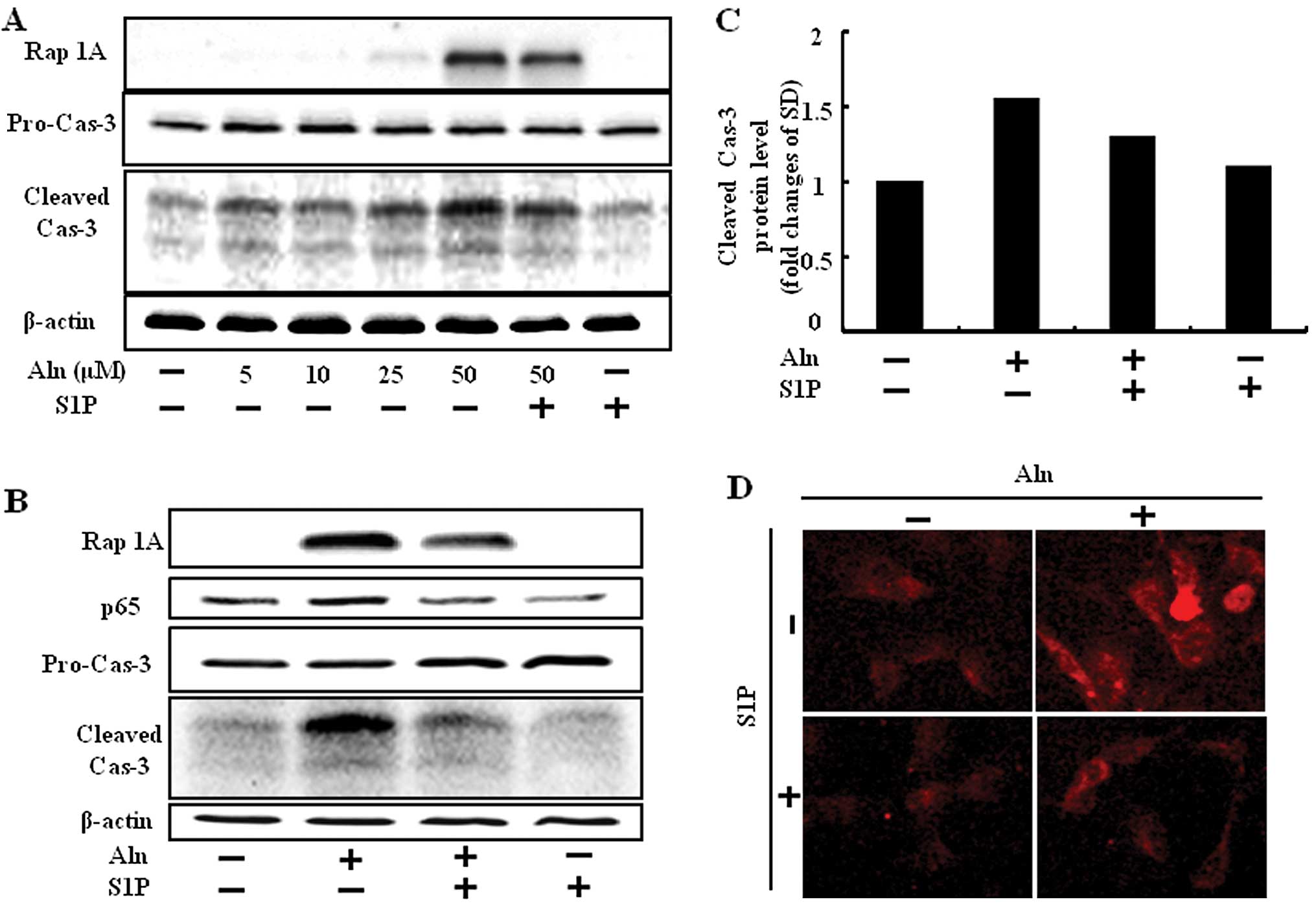
Since N-BPs induced accumulation of a toxic by
preventing protein prenylation, unprenylated protein increase for
cell has been confirmed using Rap-1A, a geranylgeranylation marker
that binds to the non-geranylgeranylated form. To evaluate whether
BP accumulates cytotoxicity by protein unprenylation, HUVECs were
treated with Aln in a dose-dependent manner for 24 h following
treatment with or without 0.5 μM S1P and then an experiment to
measure Rap-1A was performed. We found that 25 μM of Aln inhibited
geranylgeranylation slightly and 50 μM of Aln did so strongly,
whereas cotreatment with S1P and Aln reduced the unprenylated form
of Rap-1A protein compared to Aln treatment alone (Fig. 3A and B).
Caspases play a key role in the execution phase of
apoptosis. Caspase-3 subsequently cleaved cellular substrates,
thereby precipitating the marked morphological changes of
apoptosis. Herein, we assessed whether S1P has inhibitory effects
on Aln-induced caspase-3 activity with western blotting and
immunofluorescence analysis. HUVECs activated caspase-3 cleavage in
the treatment of Aln. By contrast, cleaved caspase-3 decreased
Aln-induced protein level in the presence of S1P (Fig. 3).
Aln inhibits the cell survival pathway stimulated by
the PI3K/Akt/NF-κB pathway, thus causing apoptosis of osteosarcoma
cells (6), and zoledronic acid
induces the activation of NF-κB in dendritic cells (14). We previously demonstrated that BPs
induce cell death in normal endothelial cells and that APC could
inhibit BP-mediated endothelial cell damage via inhibition of NF-κB
(13). We studied the expression
of NF-κB in response to S1P, and found that NF-κB was markedly
increased by Aln in HUVECs and they were blocked in response to S1P
treatment (Fig. 3B).
These results confirmed S1P has inhibitory effects
on BP-induced caspase-3 and NF-κB activation.
S1P protects BP-induced endothelial cell
death via inhibition of JNK
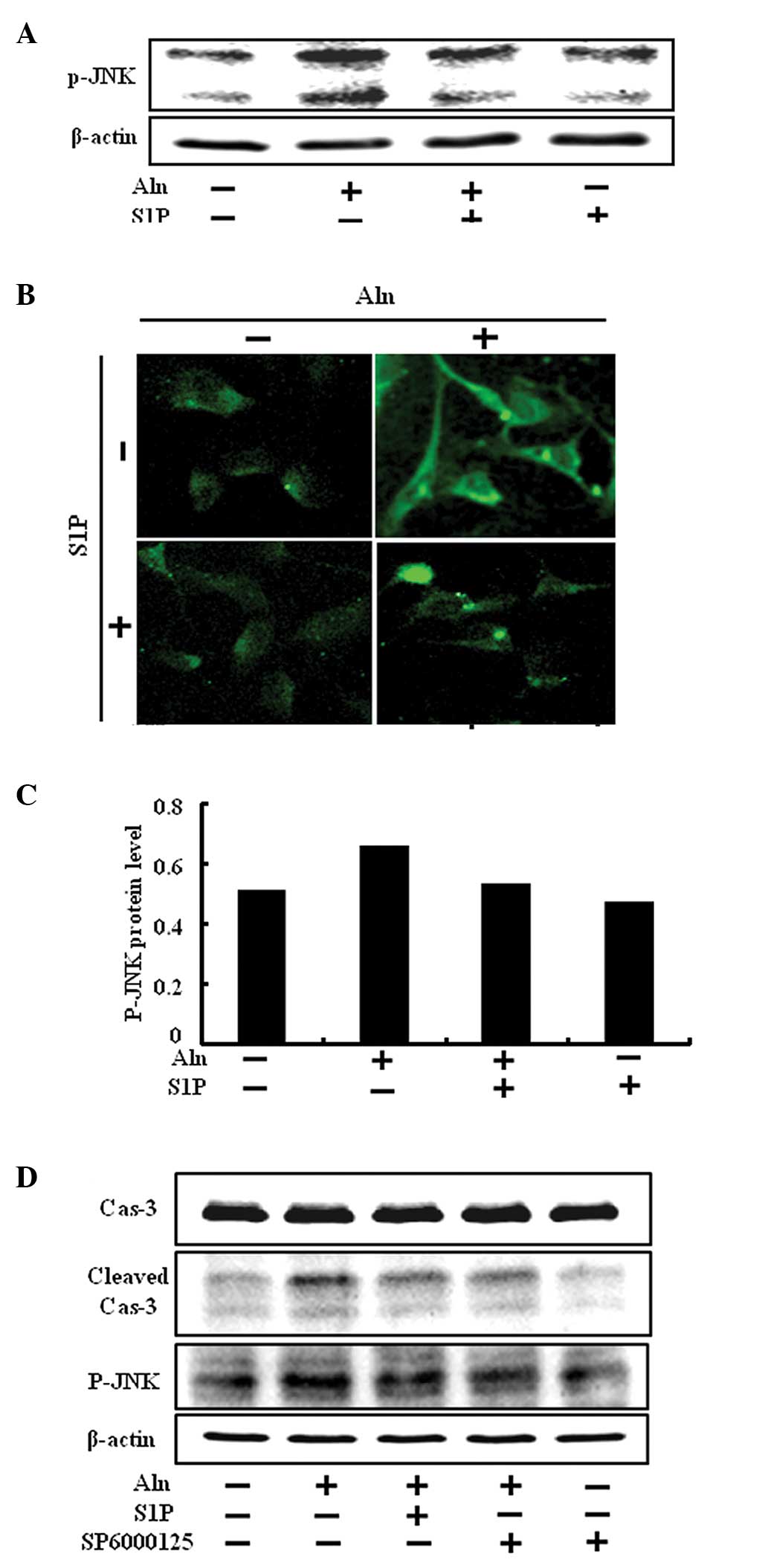
JNK belongs to the mitogen-activated protein kinase
family and also plays a role in the cellular apoptosis pathway.
HUVECs were treated with Aln and the role of p-JNK activity on
BP-induced endothelial cell death in the presence or absence of S1P
was evaluated by western blotting and immunofluorescence analysis.
A significant increase in p-JNK levels was observed in HUVECs
treated with Aln. On the contrary, the presence of S1P
significantly reduced the level of p-JNK (Fig. 4A–C). The result showed that the
level of p-JNK was upregulated by Aln, and S1P inhibited
Aln-induced phosphorylation of JNK.
In order to confirm this result, we tested whether
SP600125, a JNK inhibitor, was able to inhibit BP-induced
endothelial cell death. The inhibitor was added to HUVECs in the
presence of 0.5 μM S1P for 12 h at concentrations of 1 μM. SP600125
attenuated reduced p-JNK activation that was induced by Aln
(Fig. 4D and E). Furthermore,
SP600125 decreased cleaved caspase-3 (Fig. 4D) and increased the cell viability
compared with Aln treatment only (Fig. 4F and G). Therefore, we suggest
that S1P protects against BP-induced endothelial cell death via
inhibition of JNK.
Discussion
Bisphosphonates (BPs), stable analogs of
pyrophosphate, strongly inhibit bone resorption and have been used
to treat various diseases caused by increased bone resorption, such
as postmenopausal osteoporosis, and tumor bone metastases (15). They are divided into two groups,
according to the structure of the side chains; a
nitrogen-containing type and a non-nitrogen-containing type.
Non-nitrogen-containing BPs are reported to act through the
intracellular accumulation of nonhydrolyzable ATP analogs that
exert cytotoxic effects on osteoclasts (16), while nitrogen-containing BPs are
known to inhibit the mevalonate pathway and reduce the prenylation
of small GTP-binding proteins, such as Ras, Rho, Rac and Cdc42
(17).
N-BPs may cause apoptosis by inhibiting the
mevalonate pathway and, hence, by preventing protein prenylation
(17,18); it is possible that the delay
before the appearance of apoptotic cells is dependent on the rate
of loss of prenylated proteins that may promote cell survival or
maintain normal cell function, such as Ras or nuclear lamins
(19). BP has been reported to
exert antiangiogenic effects in vivo and in vitro
(4). Zoledronate inhibits HUVEC
adhesion, survival, migration and it affects ERK1/2, JNK, Rock, FAK
and PKB as kinases in a prenylation-dependent manner (7). In this study, we confirmed that
alendronate induced endothelial cell death through activation of
JNK as well as on the caspase-3 and NF-κB signaling pathway known
to regulate apoptotic cell death in a prenylation-dependent manner
(Figs. 3A and B and 4A). However, this mechanism does not
fully determine the effects of unprenylated proteins on cell death,
and it remains elusive in nitrogen-containing BP-induced
endothelial cell apoptosis in connection with unprenylation of
protein.
Sphingolipids are ubiquitous components of cell
membranes and their metabolites ceramide (Cer), sphingosine (Sph),
and Sphingosine-1-phosphate (S1P). S1P is the terminal product of
sphingolipid metabolism, a process that occurs in all mammalian
cells. It is formed by the phosphorylation of sphingosine, by two
kinases, sphingosine kinase 1 and 2 (SphK1 and SphK2) and its
levels are tightly controlled both by the SphKs and the enzymes
which include S1P lyase (SPL) (9). S1P in serum is mainly released from
stores in activated platelets (20), and, when released, it can interact
with endothelial cells thus playing a role in endothelial cell
migration, proliferation and angiogenesis. Serum starvation-induced
HUVEC cell death is enhanced by inhibiting the intracellular
pathway through overexpression of SphK1 (21).
JNK can activate apoptotic signaling either through
nuclear or mitochondrial signaling pathways. Activated JNK
translocates to the nucleus where it induces transcription of
pro-apoptotic genes such as Bax and Bak, and decreases the
expression of pro-survival genes including c-Jun (22). The sphingolipid degradation
product trans-2-hexadecenal induces cytoskeletal
reorganization and apoptosis in a JNK-dependent manner (22). S1P is known for relevant bioactive
lipid in functions, such as endothelial cell proliferation,
adhesion and inhibition of permeability.
In this study, we found that S1P induces a decrease
in the expression of cleaved caspase-3 and NF-κB in HUVECs.
Caspase-3 cleavage and NF-κB are members of the activator of
apoptosis protein family that is expressed in BP-induced
endothelial cell death (13). BPs
also cause apoptosis and activate JNK signaling pathways in
osteoclasts and J774 macrophages in vitro (23). Collectively, we suggested that
BP-induced apoptosis is accompanied by activation of JNK in
endothelial cells and S1P, the terminal product of sphingolipid
metabolism, prevents JNK activation. Furthermore, S1P attenuated
the BP-induced apoptosis of HUVECs by inhibition of JNK (Fig. 4) and we suggest that S1P has the
potential to be a therapeutic drug in various vascular diseases
induced by endothelial cell damage. Further studies are required to
improve the cotreatment with S1P and BP conferring endothelial
protection with few side-effects in various therapies.
Acknowledgements
This study was supported by the National Research
Foundation of the Korea Grant funded by the Korean Government
(2010-E00019, 2010-21492).
References
|
1
|
Roelofs AJ, Thompson K, Gordon S and
Rogers MJ: Molecular mechanisms of action of bisphosphonates:
current status. Clin Cancer Res. 12:6222s–6230s. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Allegra A, Alonci A, Penna G, et al:
Bisphosphonates induce apoptosis of circulating endothelial cells
in multiple myeloma patients and in subjects with
bisphosphonate-induced osteonecrosis of the jaws. Acta Haematol.
124:79–85. 2010. View Article : Google Scholar
|
|
3
|
Tassone P, Tagliaferri P, Viscomi C, et
al: Zoledronic acid induces antiproliferative and apoptotic effects
in human pancreatic cancer cells in vitro. Br J Cancer.
88:1971–1978. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hashimoto K, Morishige K, Sawada K, et al:
Alendronate suppresses tumor angiogenesis by inhibiting Rho
activation of endothelial cells. Biochem Biophys Res Commun.
354:478–484. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fu L, Tang T, Miao Y, Zhang S, Qu Z and
Dai K: Stimulation of osteogenic differentiation and inhibition of
adipogenic differentiation in bone marrow stromal cells by
alendronate via ERK and JNK activation. Bone. 43:40–47. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inoue R, Matsuki NA, Jing G, Kanematsu T,
Abe K and Hirata M: The inhibitory effect of alendronate, a
nitrogen-containing bisphosphonate on the PI3K-Akt-NFkappaB pathway
in osteosarcoma cells. Br J Pharmacol. 146:633–641. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hasmim M, Bieler G and Ruegg C:
Zoledronate inhibits endothelial cell adhesion, migration and
survival through the suppression of multiple, prenylation-dependent
signaling pathways. J Thromb Haemost. 5:166–173. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsai SH, Huang PH, Chang WC, et al:
Zoledronate inhibits ischemia-induced neovascularization by
impairing the mobilization and function of endothelial progenitor
cells. PLoS One. 7:e410652012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maceyka M, Harikumar KB, Milstien S and
Spiegel S: Sphingosine-1-phosphate signaling and its role in
disease. Trends Cell Biol. 22:50–60. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morales-Ruiz M, Lee MJ, Zollner S, et al:
Sphingosine 1-phosphate activates Akt, nitric oxide production, and
chemotaxis through a Gi protein/phosphoinositide 3-kinase pathway
in endothelial cells. J Biol Chem. 276:19672–19677. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ishii M and Kikuta J:
Sphingosine-1-phosphate signaling controlling osteoclasts and bone
homeostasis. Biochim Biophys Acta. 1831:223–227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kimura T, Sato K, Kuwabara A, et al:
Sphingosine 1-phosphate may be a major component of plasma
lipoproteins responsible for the cytoprotective actions in human
umbilical vein endothelial cells. J Biol Chem. 276:31780–31785.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Seol JW, Lee YJ, Jackson CJ, Sambrook PN
and Park SY: Activated protein C inhibits bisphosphonate-induced
endothelial cell death via the endothelial protein C receptor and
nuclear factor-κB pathways. Int J Mol Med. 27:835–840.
2011.PubMed/NCBI
|
|
14
|
Wolf AM, Rumpold H, Tilg H, Gastl G,
Gunsilius E and Wolf D: The effect of zoledronic acid on the
function and differentiation of myeloid cells. Haematologica.
91:1165–1171. 2006.PubMed/NCBI
|
|
15
|
Russell RG, Watts NB, Ebetino FH and
Rogers MJ: Mechanisms of action of bisphosphonates: similarities
and differences and their potential influence on clinical efficacy.
Osteoporos Int. 19:733–759. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rogers MJ, Russell RG, Blackburn GM,
Williamson MP and Watts DJ: Metabolism of halogenated
bisphosphonates by the cellular slime mould Dictyostelium
discoideum. Biochem Biophys Res Commun. 189:414–423. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luckman SP, Hughes DE, Coxon FP, Graham R,
Russell G and Rogers MJ: Nitrogen-containing bisphosphonates
inhibit the mevalonate pathway and prevent post-translational
prenylation of GTP-binding proteins, including Ras. J Bone Miner
Res. 13:581–589. 1998. View Article : Google Scholar
|
|
18
|
Luckman SP, Coxon FP, Ebetino FH, Russell
RG and Rogers MJ: Heterocycle-containing bisphosphonates cause
apoptosis and inhibit bone resorption by preventing protein
prenylation: evidence from structure-activity relationships in J774
macrophages. J Bone Miner Res. 13:1668–1678. 1998. View Article : Google Scholar
|
|
19
|
Perez-Sala D and Mollinedo F: Inhibition
of isoprenoid biosynthesis induces apoptosis in human promyelocytic
HL-60 cells. Biochem Biophys Res Commun. 199:1209–1215. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yatomi Y, Igarashi Y, Yang L, et al:
Sphingosine 1-phosphate, a bioactive sphingolipid abundantly stored
in platelets, is a normal constituent of human plasma and serum. J
Biochem. 121:969–973. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Limaye V, Li X, Hahn C, et al: Sphingosine
kinase-1 enhances endothelial cell survival through a
PECAM-1-dependent activation of PI-3K/Akt and regulation of Bcl-2
family members. Blood. 105:3169–3177. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kumar A, Byun HS, Bittman R and Saba JD:
The sphingolipid degradation product trans-2-hexadecenal
induces cytoskeletal reorganization and apoptosis in a
JNK-dependent manner. Cell Signal. 23:1144–1152. 2011.
|
|
23
|
Dunford JE, Rogers MJ, Ebetino FH, Phipps
RJ and Coxon FP: Inhibition of protein prenylation by
bisphosphonates causes sustained activation of Rac, Cdc42, and Rho
GTPases. J Bone Miner Res. 21:684–694. 2006. View Article : Google Scholar : PubMed/NCBI
|