Introduction
Cigarettes are possibly the single most significant
source of toxic chemical exposure and chemically-mediated illness
in humans. The World Health Organization forecasts that cigarettes
will kill approximately 10 million individuals per year globally by
the year 2020 (1). Indeed,
tobacco-assoicated cancers account for a considerable proportion of
cancer-related deaths. In addition, cigarette smoking is a powerful
risk factor for atherosclerotic cardiovascular disease and is
associated with the increased incidence of stroke and coronary
artery disease (2–4). Among the toxic compounds contained
in tobacco smoke are polycyclic aromatic hydrocarbons (PAHs), such
as benzo[a]pyrene (BaP) (5). BaP
is readily absorbed following inhalation and is rapidly distributed
to several tissues, including the kidneys, small intestine,
trachea, stomach, testes, liver and oesophagus. BaP is metabolised
by cytochrome P450 enzymes, resulting in the formation of a number
of metabolites, including the reactive epoxide metabolite, BaP
7,8-diol-9,10-epoxide, which may bind to DNA and is believed to be
responsible for its carcinogenicity (6). In addition, BaP may impair lysosomes
and break down cellular membranes (7).
There is strong evidence to suggest oxidative stress
is one of the most potent inductors of vascular inflammation in
atherogenesis (8,9). Reactive oxygen species (ROS) are
known to change the oxidation-reduction (redox) state of exposed
cells, and it is known that several inflammatory genes and the
related transcription factors are regulated through redox-sensitive
mechanisms (10). Nuclear
factor-κB (NF-κB) may respond directly to oxidative stress and the
activation of NF-κB is a key redox-sensitive event associated with
vascular dysfunction. Endothelial dysfunction is an early hallmark
of atherosclerosis. Endothelial progenitor cells (EPCs) play an
important role in restoring vascular tone in response to vascular
injury. Mature endothelial cells have a very low regenerative
capacity, compared with circulating EPCs, which can proliferate,
migrate and differentiate into mature endothelial cells (11–13). EPCs have been shown to enhance the
formation of new endothelium in animal models, in which vessel
injury occurs after balloon injury, myocardial infarction, or heart
transplantation (14). Thus, the
presence of healthy EPCs is crucial to post-event vascular
reconstruction (15).
In animal models, BaP alone has been shown to induce
atherosclerotic lesions (16,17), and repeated cycles of vascular
injury by BaP increase the onset and progression of atherosclerotic
lesions in animals. This atherogenic response is partly mediated by
the activation of cis-acting antioxidant/electrophile response
elements that enhance c-Ha-ras transcription in vascular smooth
muscle cells. The activation of antioxidant/electrophile responsive
cis-acting elements may depend on the metabolism of BaP by
cytochrome P450 enzymes to intermediates that induce oxidative
stress and modulate gene expression. It remains unclear whether BaP
affects the function of EPCs. Since BaP has been implicated in the
initiation and progression of atherosclerotic vascular lesions and
EPCs have been implicated in the repair of vascular lesions, in
this study, we investigated the effects of BaP on the function of
EPCs. We also evaluated the role of redox mechanisms and NF-κB
modulation during this process.
Materials and methods
Reagents
BaP was purchased from Sigma (St. Louis, MO, USA).
For the fluorescence-activated cell sorter (FACS) analyses, the
mouse IgG antibodies (FITC, perCP, and PE) and anti-human CD34-PE
antibody were from Caltag Laboratories. The anti-human
VE-Cadherin-FITC antibody was from Bender MedSystem™ Austria. The
anti-human VEGFR-2-PE and AC133-FITC antibodies were from R&D
System. The Cell Counting Kit-8 (CCK-8) was obtained from Dojindo
Laboratories (Kumamoto, Japan).
Cells and culture conditions
Informed consent was obtained from all participating
mothers. The study was approved by the Institutional Review Board
of the Second Affiliated Hospital, Wenzhou Medical College,
Wenzhou, China. Umbilical cord blood (~50 ml) from a normal
delivery was collected by needle/syringe from the placental side of
the umbilical vein after the newborn was delivered but prior to
placental delivery. Samples were processed within 2 h, and cord
blood mononuclear cells were isolated from umbilical cord blood by
Ficoll gradient centrifugation. Cells were seeded at
5×106/cm2 into 6-well culture dishes which
were pre-coated with fibronectin (Roche Applied Science,
Indianapolis, IN, USA). The culture medium was endothelial growth
medium-2 (EGM-2; Lonza, Basel, Switzerland), which contains fetal
calf serum (FCS, 10%, w/v), and antibiotics. Cells were cultured in
humidified incubators with 5% CO2 and initially allowed
to adhere for 24 h, followed by medium change every 3 days. When
cultures reached over 90% confluence, adherent cells were detached
with 0.05% trypsin-EDTA (Gibco, Carlsbad, CA, USA) and
replated.
Flow cytometry
Second passage cells were collected, washed twice in
ice-cold FACS buffer containing phosphate buffer (pH 7.2) with 5 mm
EDTA and 5% fetal bovine serum (FBS), and adjusted to
1×106 cells/ml. At least 50,000 cells were incubated
with fluorescence-labeled monoclonal antibodies or the respective
isotype control (1/20 diluted, 4°C, 30 min). After the washing
steps, the labeled cells were analyzed by flow cytometry using a
FACSCalibur flow cytometer and CellQuest Pro software (BD
Biosciences, Bedford, MA, USA). In addition, GSC7901 (a gastric
cancer cell line) was used as the control.
Immunocytochemistry
After culture for 6 days, adherent cells were
incubated with the fluorescent probe
1,1′-dioctadecyl-1-3,3,3′,3′-tetramethyl-indo-carbocyanine
perchlorate-acetylated-LDL (DiI-Ac-LDL; 2.4 μg/ml; Molecular
Probes) or rhodamine conjugated lectin (Lectin Kit; Sigma) at 37°C
for 4 h. After staining, samples were viewed under an inverted
fluorescent microscope (Leica, Wetzlar, Germany). Surface binding
of rhodamine conjugated lectins was green and ac-LDL in cytoplasm
was red.
Factor VIII-related antigen
immunohistochemistry
After the cultured cells were fixed with 95% (v/v)
alcohol for 30 min, 0.5% (v/v) H2O2 methanol
was added to inactivate endogenous peroxidase. The cells were then
incubated with diluted primary antibody (1:1,000) overnight at 4°C,
followed by incubation with the secondary antibody and strept
actividin-biotin complex (SABC) for 20 min in the oven at 37°C
according to standard protocols. Positive cells were stained brown.
Gastric cancer cells (GSC7901) acted as the negative control.
Cell proliferation assay
Cell proliferation was quantified using CCK-8.
Briefly, cells were plated in flat-bottomed 96-well microplates at
1×104 cells/well and incubated in EGM-2 medium
containing 2% carboxyfluorescein (CFS) for 24 h (3 wells/group).
The culture medium was then changed to EGM-2 medium containing 10%
CFS and cultured for an additional 2 h. Cells were then exposed to
BaP at concentrations of 10, 20 and 50 μmol/l for 24 h to examine
the effects of BaP. BaP was dissolved in DMSO and diluted to the
desired final concentrations in culture medium. The control cells
were left untreated and the solvent control cells were incubated
with DMSO alone (1 ml/l). CCK-8 (10 μl/well) was added to the wells
at the end of the experiment. After incubation at 37°C for 4 h, the
absorbance of each well was determined using a microplate reader at
450 nm. The degree of cell proliferation was determined as the
percentage of absorbance of the treated cells to that of the
control cells.
Migration assay
After the cells were cultured in the absence or
presence of BaP (10, 20 and 50 μmol/l) for 24 h as described above,
adhesive cells were digested, collected and resuspended in EBM-2
medium containing 2% FBS and 0.1 bovine serum albumin (BSA). Cells
were adjusted to 5×105 cells/ml. Cell migration was
quantified by a Transwell chemotaxis assay using a Boyden chamber
(18). Briefly, 5×104
cells (100 μl) were plated in the upper of 2 chambers divided by a
membrane with 8 μm pores (Transwell; Corning). EBM-2 medium
containing 10% FBS (600 μl) was added to the lower chamber. After
24 h, the membranes were washed twice in D-Hank’s solution (Gibco)
and fixed in 4% formaldehyde. After wiping the cells off the upper
side of the membrane with a cotton swab (Q-tip), the membranes were
detached and mounted on glass slides with 0.25% crystal violet.
Migrated cells were counted under a microscope. Each experimental
condition was performed in triplicate, and the number of migrated
cells was determined from 10 random ×400 high-power
fields/membrane.
Cell adhesion assay
After the cells were cultured in the absence or
presence of BaP (10, 20 and 50 μmol/l) for 24 h as described above,
adhesive cells were digested, collected and resuspended in EGM-2
medium containing 2% FBS. Cell-matrix adhesion was investigated in
96-well plates coated overnight (4°C) with fibronectin (1 μg/ml).
Cells were seeded at 5×103 cells/well in 100 μl.
Adhesion was carried out for 30 min at 37°C, 5% CO2.
After the removal of non-adherent cells by 2 washing steps with
EGM-2 medium containing 2% FBS at room temperature, adhesion was
quantified by counting the adherent cells from 10 random ×200
high-power fields under an inverted microscope.
Tube formation assay
Matrigel (BD Biosciences) was dissolved at 4°C
overnight, and 96-well plates were prepared with 50 μ Matrigel in
each well after coating and incubating at 37°C for 60 min as
previously described (19). After
the cells were cultured in the absence or presence of BaP (10, 20
and 50 μmol/l) for 24 h as described above, adhesive cells were
digested, collected and resuspended in EGM-2 medium containing 2%
FBS and 0.1 BSA. Cells were adjusted to 5×105 cells/ml.
EPCs of 1×105 were added to the gel. After 24 h of
incubation at 37°C, the number of tubes formed was determined from
5 random ×400 high-power fields under a light microscope.
Measurement of intracellular ROS
The oxidation of 2′,7′-dichlorofluorescein diacetate
(DCFH-DA; Sigma-Aldrich) to 2′,7′-dichlorofluorescein (DCF) was
used to estimate ROS levels as previously described (20). Cells at passage 3 were digested,
collected and plated in a 6-well plate with EGM-2 medium containing
2% FBS for 24 h. There was a slide pre-coated with fibronectin in
each well. The cells (5×106/ml) were then cultured in
the absence or presence of BaP (10, 20 and 50 μmol/l) for an
additional 24 h as described above. Thereafter, the cells were
incubated with 10 μmol/l chloromethyl-dihydrodichlorofluorescein
diacetate (CM-H2DCF-DA) for 3 h at 37°C. After removal of the
medium and washing of the cells twice, the slide was removed and
the fluorescence intensity (relative fluorescence units) was
measured at an excitation and emission wavelength of 488 and 522
nm, respectively, using a spectrofluorometer (Hitachi). The
fluorescence intensity was measured by image analysis Image-Pro
Plus 6.0 software.
Measurement of malondialdehyde (MDA) and
superoxide dismutase (SOD) in supernatants
MDA in the supernatants was measured using a
commercial kit (Jiancheng Bioengineering Co., Ltd., Nanjing, China)
according to the manufacturer’s instructions that utilizes the
measurement of thiobarbituric acid-reactive substances (TBARS).
SOD activity was measured using a superoxide
dismutase assay kit (Jiancheng Bioengineering Co., Ltd., Nanjing,
China) according to the manufacturer’s instructions.
Real-time RT-PCR analysis
Total RNA was isolated and transcribed to cDNA using
the RT-PCR kit (MBI Fermentas, Burlington, ON, Canada). RNA (1 μg)
was reverse transcribed in a total volume of 20 μl. An aliquot of 1
μl of the reverse transcription reaction was used in the real-time
PCR reactions (20 μl final volume) and performed in a LightCycle
480 Thermocycler (Roche). Fold inductions were calculated using the
cycle threshold ΔΔCt method as previously described (21,22). Briefly, PCR was performed at 95°C
(30 sec) followed by 40 cycles at 95°C (5 sec)/60°C (20 sec).
SYBR-Green intercalating dye was used for signal detection. For
each sample, the number of cycles required to generate a given
threshold signal (Ct) was recorded. With a standard curve generated
from serial dilutions of sample cDNA, the ratio of NF-κB p65 or p50
expression relative to GAPDH expression was calculated for each
experimental group and normalized relative to an average of ratios
from the control group. The sequences of the primers used in this
study were as follows: p65 forward, 5′-CACCGGATTGAGGAGAAACGT-3′ and
reverse primer, 5′-ATCTGCCCAGAAGGAAACACC-3′; p50 forward,
5′-GGATTTCGTTTCCGTTATGTATGT-3′ and reverse primer,
5′-TGTCCTTGGGTCCAGCAGTT-3′; and ACTB forward,
5′-CGTGGACATCCGCAAAGAC-3′ and reverse primer,
5′-AAGAAAGGGTGTAACGCAACTAAG-3′.
Enzyme-linked immunosorbent assay
(ELISA)
Cells at passage 3 were digested, collected and
plated in EBM-2 medium containing 2% FBS for 24 h. Cells were then
cultured in the absence or presence of BaP (10, 20 and 50 μmol/l)
for an additional 24 h as described above. Using supernatant
samples, ELISA was performed with ELISA kits (Westang
Biotechnological Co., Ltd., Shanghai, China) for interleukin
(IL)-1β and tumor necrosis factor (TNF)-α, as indicated in the
manufacturer’s instructions.
NF-κB translocation assay
NF-κB translocation in the cells was examined using
a NF-κB activation kit (Beyotime Institute of Biotechnology,
Shanghai, China) which contains DAPI, anti-NF-κB p65 monoclonal
antibody (mAb) and secondary Cy3-conjugated mAB dyes. Cells at
passage 3 were digested, collected and plated in EBM-2 medium
containing 2% FBS for 24 h. The cells were then cultured in the
absence or presence of BaP (10, 20 and 50 μmol/l) for an additional
24 h as described above. Fixation, permeabilization, and
immunofluorescence staining of the cells were performed according
to the manufacturer’s instructions. The difference between the
intensity of nuclear and cytoplasmic NF-κB-associated fluorescence
was reported as translocation parameter.
Effect of NF-κB inhibition on BaP-induced
EPC dysfunction
To evaluate the role of NF-κB in BaP-induced
dysfunction, pyrrolidine dithiocarbamate (PDTC, 25, 50 and 75
μmol/l), an antioxidant that has been shown to selectively inhibit
NF-κB activation, was added to the culture 2 h before BaP was added
in the above experiments (23).
Statistical analysis
Data are expressed as the means ± standard deviation
(SD). Differences between data sets were assessed by one-way ANOVA.
A P-value <0.05 was considered to indicate a statistically
significant difference.
Results
Characterization of EPCs from human
umbilical cord blood
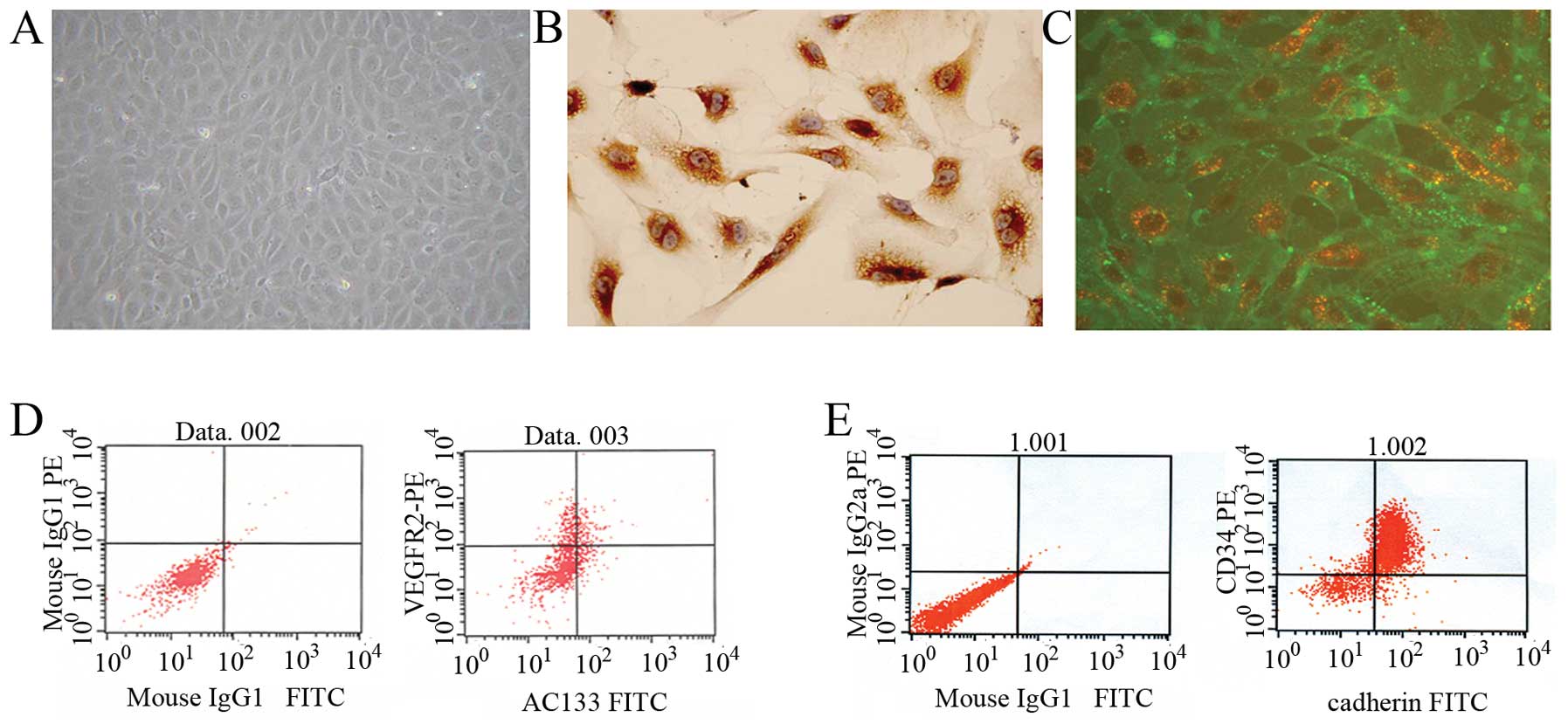
EPCs were isolated from human umbilical cord blood
mononuclear cells. At day 4, cell colonies appeared and the
colonies had a typical cobblestone shape under a phase-contrast
inverted microscope (Fig. 1A).
Immunochemistry showed that the cells were positive for factor VIII
(Fig. 1B). To confirm that the
cells acquired endothelial phenotypes after becoming adherent,
fluorescent staining was used to detect double-positive cell
binding of FITC-labeled Ulex europaeus agglutinin-1 (UEA-1) lectin
and DiI-labeled acetylated low-density lipoprotein (Fig. 1C). To further characterize the
EPCs, FACS analysis was performed. Immunophenotyping revealed that
ex vivo expanded EPCs expressed VEGFR-2 (60.95±0.88%), AC133
(16.08±0.44%), CD34 (77.19±3.32%) and VE-cadherin (25.54±0.73%)
(Fig. 1D and E). This cell
population is consistent with the early pro-angiogenic EPC type
described by other authors (24,25).
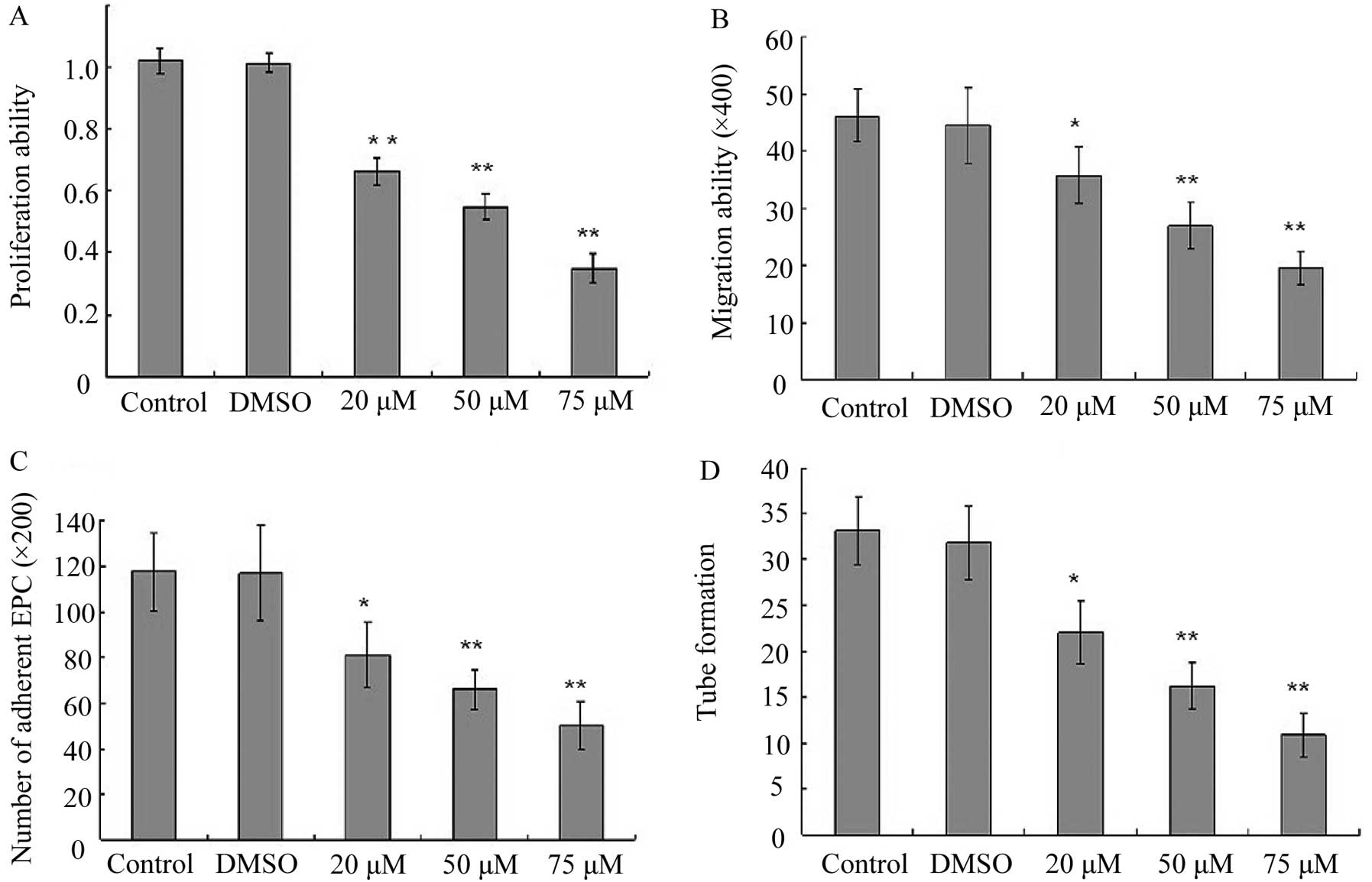
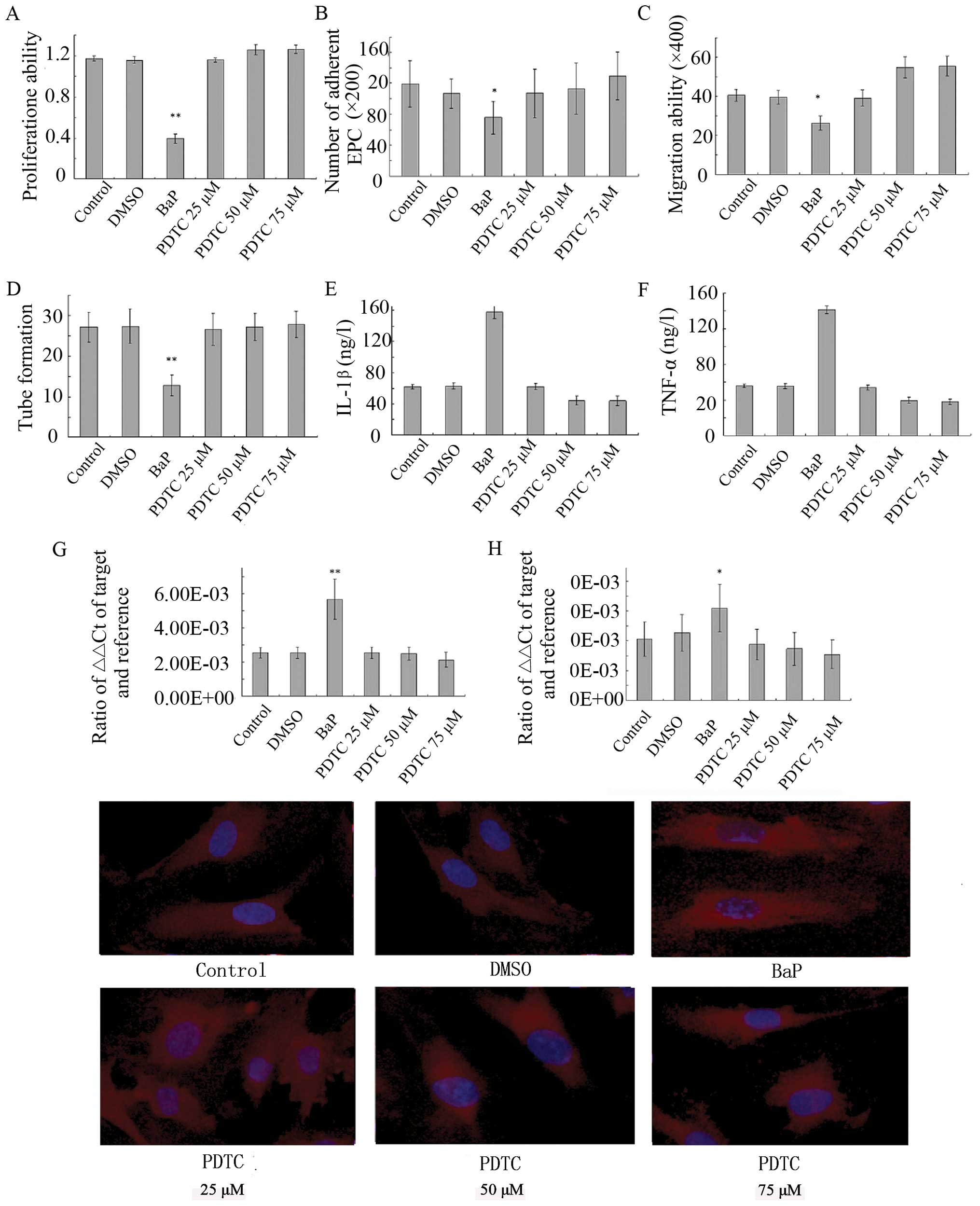
Effect of BaP on EPC proliferation
In order to investigate the cytotoxic effects of BaP
on EPCs, cell proliferation assay was performed. BaP inhibited the
cell proliferation in a dose-dependent manner for 24 h (Fig. 2A). Therefore, the concentration of
BaP of 20 μM was selected for the experiments.
BaP inhibits migration of EPCs
Considering that the migratory capability of EPCs is
crucial for the mobilization of EPCs from the bone marrow into
peripheral blood, we then examined the effect of BaP on the
migratory capability of EPCs using Transwell assay. BaP
significantly reduced the migratory capability of EPCs in a
dose-dependent manner (Fig.
2B).
BaP inhibits adhesion of EPCs
Adhesion assays were performed to examine the
adhesion of EPCs to fibronectin. The cell-matrix adhesion of EPCs
to immobilized fibronection was substantially downregulated by BaP
in a dose-dependent manner (Fig.
2C).
BaP inhibits in vitro capillary-like
structure formation of EPCs
To examine the effects of BaP on the capacity of
EPCs to form capillary-like structures, EPCs were seeded on
Matrigel® for 24 h after they were treated with BaP. BaP
induced significant decreases in the number of capillary-like
structures in a dose-dependent manner (Fig. 2D).
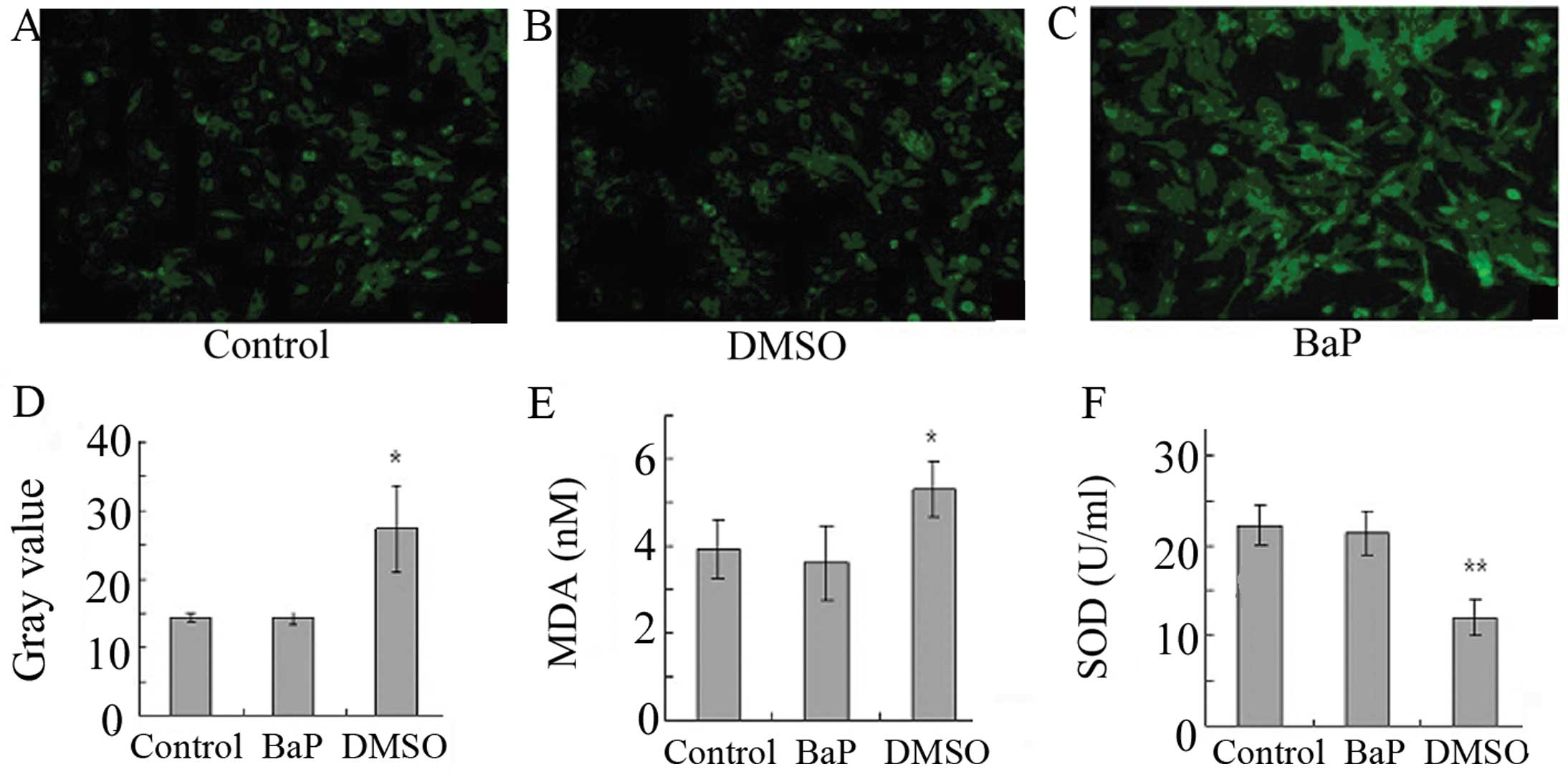
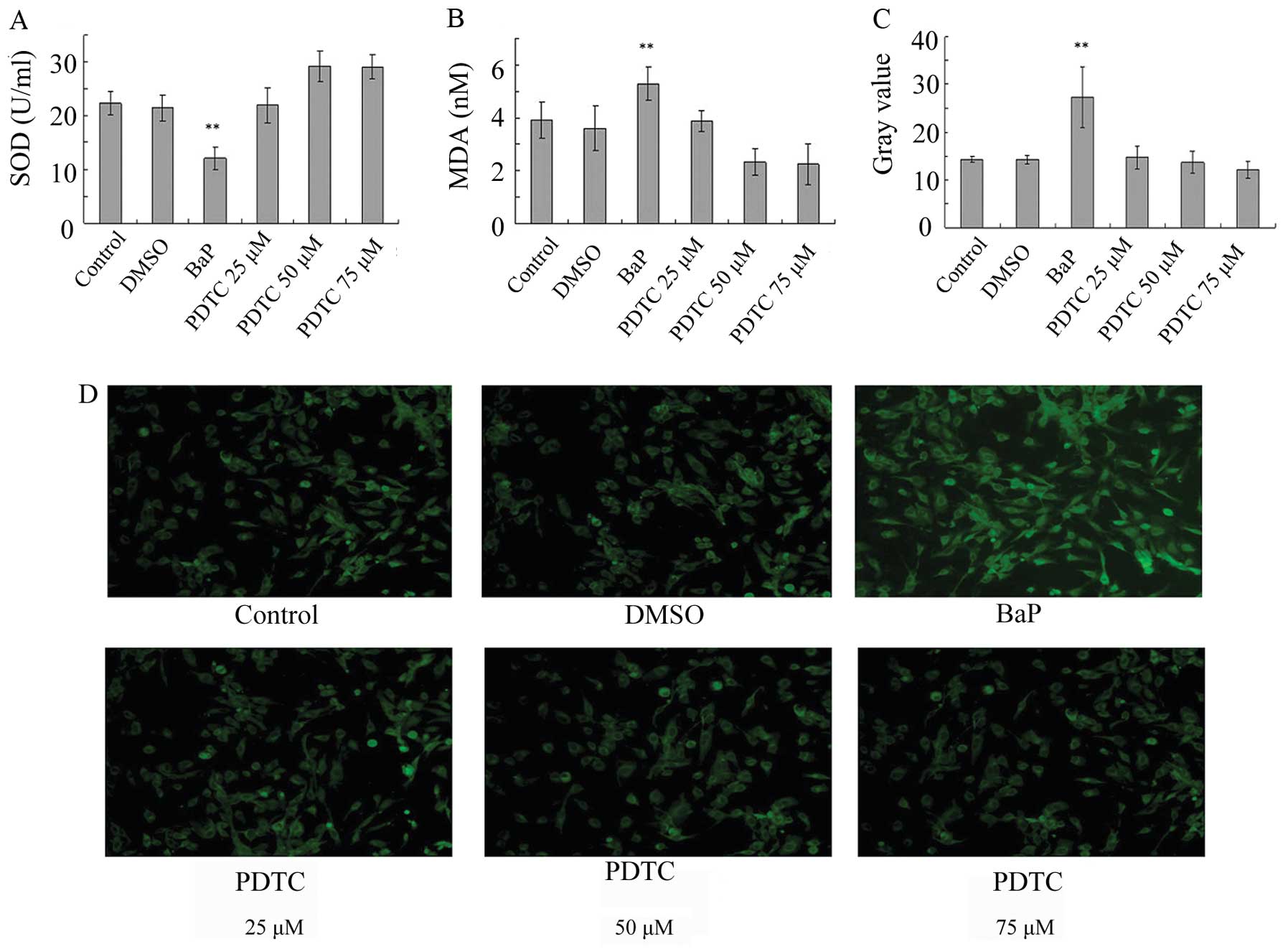
BaP increases oxidative stress in
EPCs
To determine whether oxidative stress is involved in
the action of BaP, ROS levels were analyzed. The treatment of EPCs
with BaP (20 μM) for 45 min significantly increased cellular ROS
levels, as indicated by the gray value of green fluorescence
(Fig. 3A-D). Furthermore, BaP
increased the production of MDA and suppressed the production of
SOD (Fig. 3E and F). These data
demonstrate that BaP increases oxidative stress in EPCs.
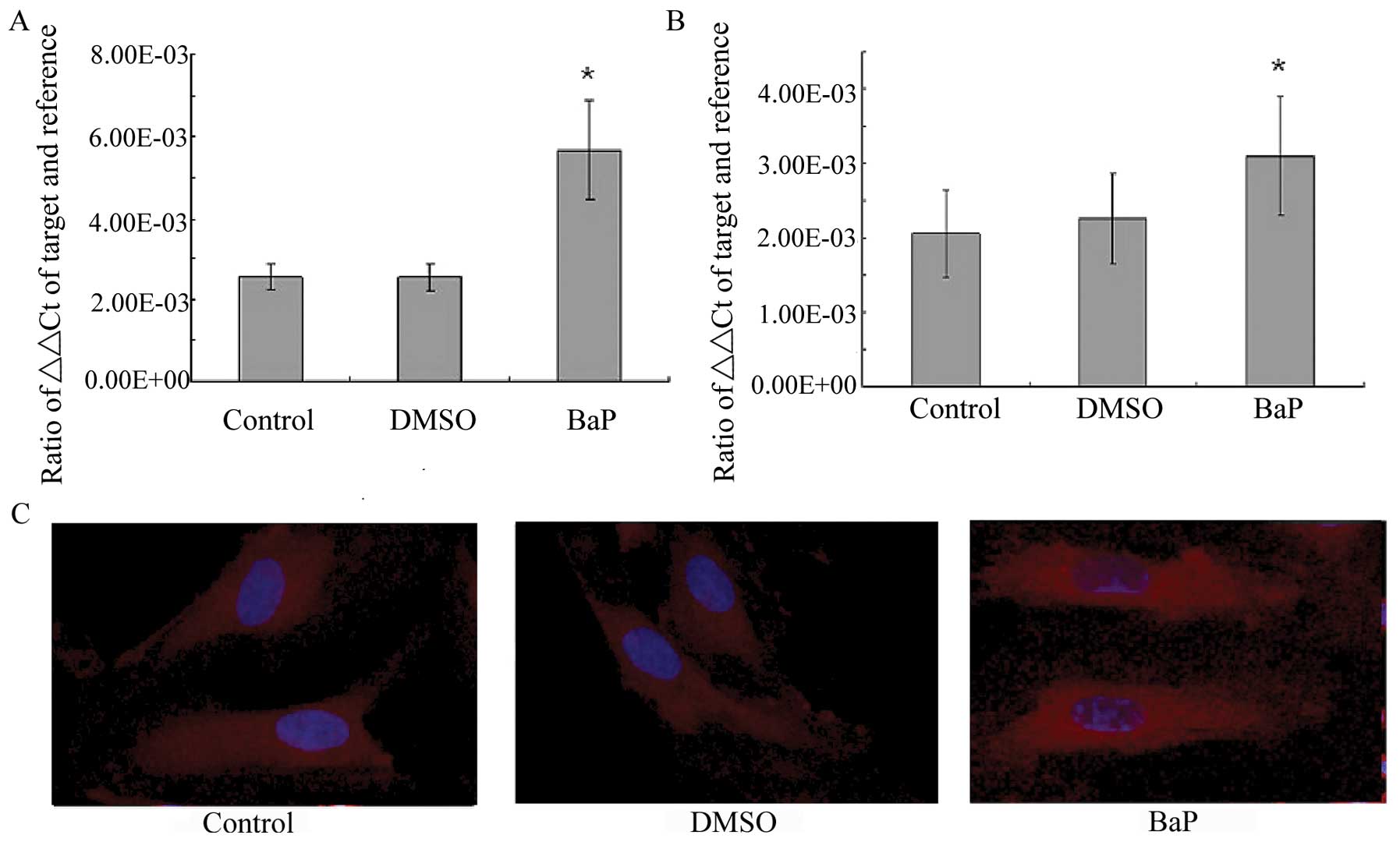
BaP increases NF-κB activity
BaP was then examined for its effect on NF-κB
transcription and translocation. RT-PCR demonstrated that the
treatment of EPCs with BaP (20 μM) significantly increased the mRNA
levels of the p65 and p50 subunits of NF-κB (Fig. 4A and B). The morphological changes
of NF-κB translocation indicated by immunofluorescence staining
(Fig. 4C) showed that BaP induced
NF-κB translocation. When the cells were left untreated, most of
the fluorescence staining for NF-κB was in the cytoplasm and rare
NF-κB staining was observed in the nucleus. When the cells were
stimulated with BaP, NF-κB staining significantly increased in the
nucleus, suggesting that NF-κB translocated from the cytoplasm into
the nucleus.
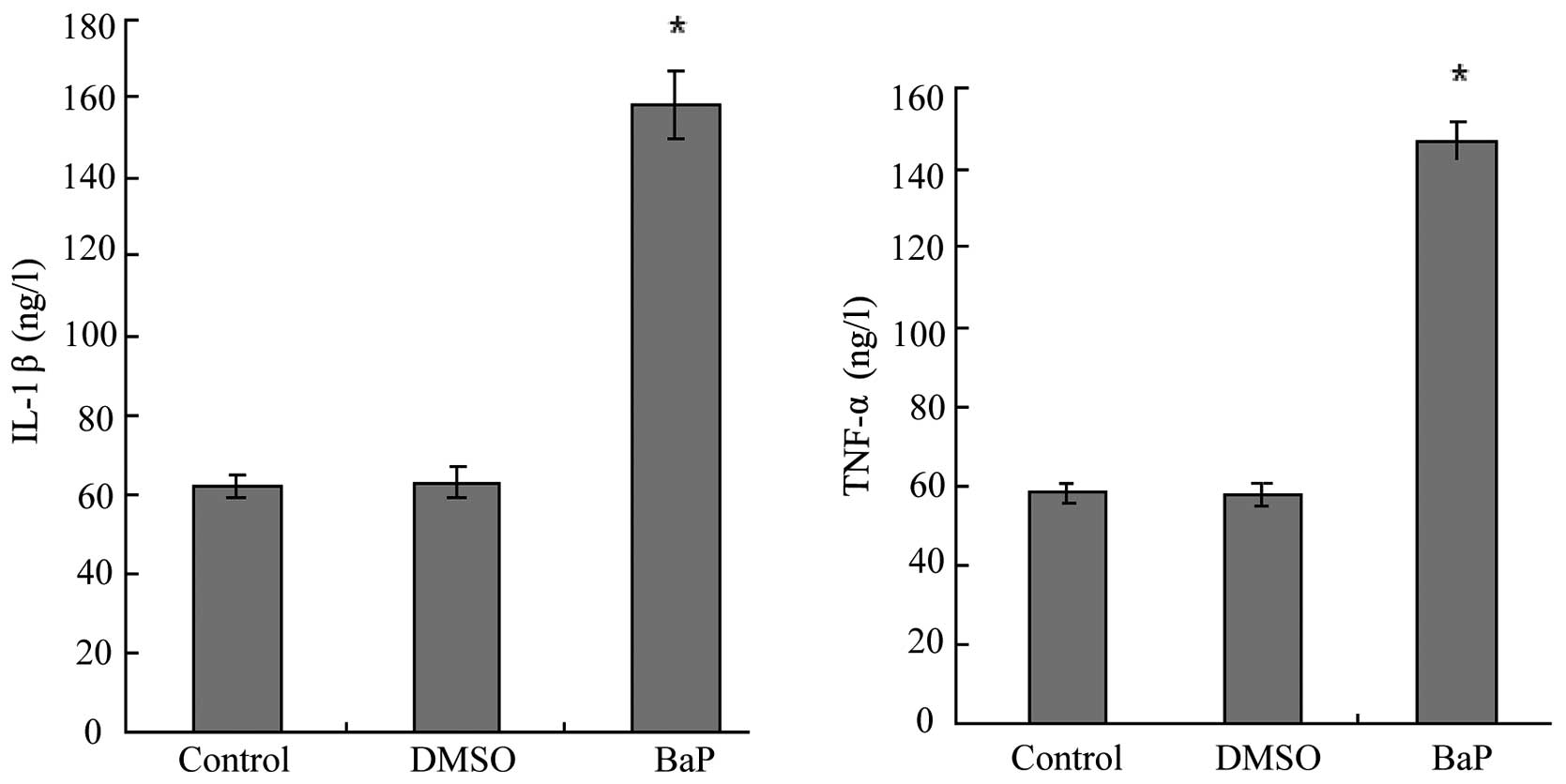
BaP increases the production of IL-1β and
TNF-α
IL-1β and TNF-α are downstream targets of NF-κB. We
then examined the effects of BaP on the production of IL-1β and
TNF-α by EPCs. The results of ELISA for EPC culture supernatants
showed that BaP (20 μM) increased the secretion of IL-1β and TNF-α
by EPCs (Fig. 5).
Effects of BaP on EPCs are abolished by
PDTC
As shown above, BaP significantly promoted NF-κB
activation, increased intracellular ROS production and inhibited
the proliferation, migration, adhesion and angiogenesis of EPCs
in vitro in a dose-dependent manner. To evaluate the role of
NF-κB, the effects of PDTC, a NF-κB inhibitor, on BaP-induced EPC
dysfunction were examined. The pre-treatment of EPCs with PDTC
prior to BaP treatment significantly inhibited the stimulatory
effect of BaP on oxidative stress (Fig. 6) and reversed BaP-induced EPC
dysfunction in a dose-dependent manner (Fig. 7). These results suggest that BaP
stimulates oxidative stress, at least in part, through the
activation of NF-κB.
Discussion
To our knowledge, our study demonstrates for the
first time that BaP hampers the function of EPCs. The major
findings of our study are that BaP: i) inhibits EPC proliferation;
ii) BaP modulates angiogenic-related properties of EPCs in
vitro; iii) BaP increases oxidative stress in EPCs and iv)
inhibits EPC proliferation at least in part, through the NF-κB
pathway. Taken together, these findings provide a novel mechanism
underlying the detrimental effects of BaP on vascular function, as
EPCs play a critical role in angiogenesis and potentially aid in
the repair of the injured endothelium (24–28).
A number of studies have shown that EPCs form a
heterogenous population of cells. The most widely accepted
phenotype for the identification of EPCs by FACS comprises
CD34+/kinase insert domain containing receptor
(KDR)+ and CD133+/KDR+ cells
(29–31). Bone marrow-derived EPCs possess
the capacity to proliferate, migrate and differentiate into
endothelial lineage cells (32).
EPCs are mobilized from bone marrow into the circulation during
vascular injury, and then home to the site of neovascularization
and thereby contribute to postnatal neovascularization (33,34). Moreover, the findings that
decreased levels of EPCs correlate with subsequent increases in
cardiovascular events suggest that EPCs are important predictors of
cardiovascular mortality and morbidity (15,35). Indeed, the number of circulating
EPCs and their function have been reported to be reduced in
patients with coronary artery disease (36), diabetes (37), metabolic syndrome (38) and severe heart failure (39).
Excessive generation of ROS and reactive nitrogen
species may contribute to endothelial dysfunction and plays a
critical role in the progressive deterioration of vessel structure
and function (40). While low
levels of ROS are essential and participate in important
intracellular signaling pathways (41), excessive generation of ROS may
result in cytotoxic oxidative stress. Cigarette smoke contains high
concentrations of ROS, nitric oxide, peroxynitrite and free
radicals of organic compounds (42,43). In addition to these short-lived,
highly reactive substances, previous studies have shown that
aqueous cigarette tar extracts also contain pro-oxidant substances
that have the potential to increase the cellular production of ROS
(44). It has thus been
hypothesized that water-soluble components of cigarette smoke that
are likely to reach the systemic circulation can directly promote
oxidative stress in the vasculature and blood cells (42).
NF-κB is a ubiquitous nuclear transcription factor
that plays a major regulatory role in inflammation. It resides in
the inactive state in the cytoplasm as a heterotrimer consisting of
p50, p65 and IκBα subunits. This transcription factor is a dimeric
complex composed of different members of the Rel/NF-κB family of
polypeptides. The p50-p65 heterodimer is retained in the cytoplasm
by the inhibitory subunit, IκBα. Upon activation of the complex,
IκBα sequentially undergoes phosphorylation, ubiquitination and
degradation, thus releasing the p50-p65 heterodimer for
translocation to the nucleus. An IκBα kinase, IKK, has been
identified that phosphorylates serine residues in IκBα at position
32 and 36. The treatment of cells with various inflammatory and
oxidative stress stimuli activates IKK, thus leading to the
degradation of IκBα and activation of the transcription factor.
Experimental data support the activation of the transcription
factor NF-κB as a key redox-sensitive event associated with
vascular dysfunction (45). The
results of this study also demonstrate that the activation of NF-κB
in EPCs is associated with a functional consequence, i.e., the
decrease in the proliferation, migration and adhesion of EPCs. To
further investigate the pro-inflammatory effect of BaP on EPCs, we
examined the expression of NF-κB target genes (IL-1β and TNF-α).
IL-1β and TNF-α were upregulated by BaP, and pre-treatment with
NF-κB inhibitors diminished this overexpression, suggesting an
NF-κB-mediated transcriptional mechanism.
In conclusion, the results of this study show that
in cultured EPCs, BaP activates the NF-κB pathway, upregulating the
expression of pro-inflammatory cytokines involved in
atherosclerosis. This means that water-soluble components of
cigarette smoke can promote oxidative stress not only of
endothelial cells but possibly also of EPCs. This study also
revealed that NF-κB p65 participates in the negative regulation of
the oxidative stress of EPCs, and may provide a new insight into
the possible role of NF-κB in suppressing EPC function. This
pathway may be a novel therapeutic target in atherosclerosis.
Further studies are required to confirm whether its inhibition is
useful in the treatment of this disorder.
Acknowledgements
This study was supported by grants from the
Zhenjiang Province Natural Science Foundation (no. Y2110550), the
Research Project of Wenzhou Science and the Technology Bureau (nos.
H20100056 and H20080027).
References
|
1
|
World Health Organisation Tobacco Free
Initiative. Oslo, Norway. WHO Workshop on Advancing Knowledge on
Regulating Tobacco Products; February 2000;
|
|
2
|
Rogot E and Murray JL: Smoking and causes
of death among U.S. veterans: 16 years of observation. Public
Health Rep. 95:213–222. 1980.PubMed/NCBI
|
|
3
|
Howard G, Wagenknecht LE, Burke GL, et al:
Cigarette smoking and progression of atherosclerosis: The
Atherosclerosis Risk in Communities (ARIC) Study. JAMA.
279:119–124. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang CQ, Xu L, Lam TH, Lin JM, Cheng KK
and Thomas GN: Smoking cessation and carotid atherosclerosis: the
Guangzhou Biobank Cohort Study-CVD. J Epidemiol Community Health.
64:1004–1009. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rodgman A, Smith CJ and Perfetti TA: The
composition of cigarette smoke: a retrospective, with emphasis on
polycyclic components. Hum Exp Toxicol. 19:573–595. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baird WM, Hooven LA and Mahadevan B:
Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and
mechanism of action. Environ Mol Mutagen. 45:106–114. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi Z, Dragin N, Miller ML, et al: Oral
benzo[a]pyrene-induced cancer: two distinct types in different
target organs depend on the mouse Cyp1 genotype. Int J Cancer.
127:2334–2350. 2010.
|
|
8
|
Griendling KK, Sorescu D and Ushio-Fukai
M: NAD(P)H oxidase: role in cardiovascular biology and disease.
Circ Res. 86:494–501. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ross R: Atherosclerosis is an inflammatory
disease. Am Heart J. 138:S419–S420. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Libby P: Inflammation in atherosclerosis.
Nature. 420:868–874. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li DW, Liu ZQ, Wei J, Liu Y and Hu LS:
Contribution of endothelial progenitor cells to neovascularization
(Review). Int J Mol Med. 30:1000–1006. 2012.PubMed/NCBI
|
|
12
|
Rabelink TJ, de Boer HC, de Koning EJ and
van Zonneveld AJ: Endothelial progenitor cells: more than an
inflammatory response? Arterioscler Thromb Vasc Biol. 24:834–838.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hristov M, Zernecke A, Bidzhekov K, et al:
Importance of CXC chemokine receptor 2 in the homing of human
peripheral blood endothelial progenitor cells to sites of arterial
injury. Circ Res. 100:590–597. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hutter R, Carrick FE, Valdiviezo C, et al:
Vascular endothelial growth factor regulates reendothelialization
and neointima formation in a mouse model of arterial injury.
Circulation. 110:2430–2435. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hill JM, Zalos G, Halcox JP, et al:
Circulating endothelial progenitor cells, vascular function, and
cardiovascular risk. N Engl J Med. 348:593–600. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ramos KS, Zhang Y, Sadhu DN and Chapkin
RS: The induction of proliferative smooth muscle cell phenotypes by
benzo[a]pyrene is characterized by up-regulation of inositol
phospholipid metabolism and c-Ha-ras gene expression. Arch Biochem
Biophys. 332:213–222. 1996.
|
|
17
|
Curfs DM, Lutgens E, Gijbels MJ, Kockx MM,
Daemen M and van Schooten FJ: Chronic exposure to the carcinogenic
compound benzo[a]pyrene induces larger and phenotypically different
atherosclerotic plaques in ApoE-knockout mice. Am J Pathol.
164:101–108. 2004.PubMed/NCBI
|
|
18
|
Smaniotto S, Martins-Neto AA, Dardenne M
and Savino W: Growth hormone is a modulator of lymphocyte
migration. Neuroimmunomodulation. 18:309–313. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kleinman HK and Martin GR: Matrigel:
basement membrane matrix with biological activity. Semin Cancer
Biol. 15:378–386. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cominacini L, Garbin U, Pasini AF, et al:
Oxidized low-density lipoprotein increases the production of
intracellular reactive oxygen species in endothelial cells:
inhibitory effect of lacidipine. J Hypertens. 16:1913–1919. 1998.
View Article : Google Scholar
|
|
21
|
Heid CA, Stevens J, Livak KJ and Williams
PM: Real time quantitative PCR. Genome Res. 6:986–994. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gibson UE, Heid CA and Williams PM: A
novel method for real time quantitative RT-PCR. Genome Res.
6:995–1001. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ziegler-Heitbrock HW, Sternsdorf T, Liese
J, et al: Pyrrolidine dithiocarbamate inhibits NF-kappa B
mobilization and TNF production in human monocytes. J Immunol.
151:6986–6993. 1993.PubMed/NCBI
|
|
24
|
Kalka C, Masuda H, Takahashi T, et al:
Transplantation of ex vivo expanded endothelial progenitor cells
for therapeutic neovascularization. Proc Natl Acad Sci USA.
97:3422–3427. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hur J, Yoon CH, Kim HS, et al:
Characterization of two types of endothelial progenitor cells and
their different contributions to neovasculogenesis. Arterioscler
Thromb Vasc Biol. 24:288–293. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grunewald M, Avraham I, Dor Y, et al:
VEGF-induced adult neovascularization: recruitment, retention, and
role of accessory cells. Cell. 124:175–189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamahara K and Itoh H: Potential use of
endothelial progenitor cells for regeneration of the vasculature.
Ther Adv Cardiovasc Dis. 3:17–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sirker AA, Astroulakis ZM and Hill JM:
Vascular progenitor cells and translational research: the role of
endothelial and smooth muscle progenitor cells in endogenous
arterial remodelling in the adult. Clin Sci. 116:283–299. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Peichev M, Naiyer AJ, Pereira D, et al:
Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells
identifies a population of functional endothelial precursors.
Blood. 95:952–958. 2000.
|
|
30
|
Urbich C and Dimmeler S: Endothelial
progenitor cells: characterization and role in vascular biology.
Circ Res. 95:343–353. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Salvatore P, Casamassimi A, Sommese L, et
al: Detrimental effects of Bartonella henselae are
counteracted by L-arginine and nitric oxide in human endothelial
progenitor cells. Proc Natl Acad Sci USA. 105:9427–9432. 2008.
|
|
32
|
Asahara T and Kawamoto A: Endothelial
progenitor cells for postnatal vasculogenesis. Am J Physiol Cell
Physiol. 287:C572–C579. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kamihata H, Matsubara H, Nishiue T, et al:
Implantation of bone marrow mononuclear cells into ischemic
myocardium enhances collateral perfusion and regional function via
side supply of angioblasts, angiogenic ligands, and cytokines.
Circulation. 104:1046–1052. 2001. View Article : Google Scholar
|
|
34
|
Szmitko PE, Fedak PW, Weisel RD, Stewart
DJ, Kutryk MJ and Verma S: Endothelial progenitor cells: new hope
for a broken heart. Circulation. 107:3093–3100. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Werner N, Kosiol S, Schiegl T, et al:
Circulating endothelial progenitor cells and cardiovascular
outcomes. N Engl J Med. 353:999–1007. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vasa M, Fichtlscherer S, Aicher A, et al:
Number and migratory activity of circulating endothelial progenitor
cells inversely correlate with risk factors for coronary artery
disease. Circ Res. 89:E1–E7. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tepper OM, Galiano RD, Capla JM, et al:
Human endothelial progenitor cells from type II diabetics exhibit
impaired proliferation, adhesion, and incorporation into vascular
structures. Circulation. 106:2781–2786. 2002. View Article : Google Scholar
|
|
38
|
Fadini GP, De Kreutzenberg SV, Coracina A,
et al: Circulating CD34+ cells, metabolic syndrome, and
cardiovascular risk. Eur Heart J. 27:2247–2255. 2006.PubMed/NCBI
|
|
39
|
Heeschen C, Lehmann R, Honold J, et al:
Profoundly reduced neovascularization capacity of bone marrow
mononuclear cells derived from patients with chronic ischemic heart
disease. Circulation. 109:1615–1622. 2004. View Article : Google Scholar
|
|
40
|
Cai H and Harrison DG: Endothelial
dysfunction in cardiovascular diseases: the role of oxidant stress.
Circ Res. 87:840–844. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sundaresan M, Yu ZX, Ferrans VJ, Irani K
and Finkel T: Requirement for generation of
H2O2 for platelet-derived growth factor
signal transduction. Science. 270:296–299. 1995.
|
|
42
|
Pryor WA, Prier DG and Church DF:
Electron-spin resonance study of mainstream and sidestream
cigarette smoke: nature of the free radicals in gas-phase smoke and
in cigarette tar. Environ Health Perspect. 47:345–355. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pryor WA, Stone K, Zang LY and Bermudez E:
Fractionation of aqueous cigarette tar extracts: fractions that
contain the tar radical cause DNA damage. Chem Res Toxicol.
11:441–448. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zang LY, Stone K and Pryor WA: Detection
of free radicals in aqueous extracts of cigarette tar by electron
spin resonance. Free Radic Biol Med. 19:161–167. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Haddad JJ: Oxygen sensing and
oxidant/redox-related pathways. Biochem Biophys Res Commun.
316:969–977. 2004. View Article : Google Scholar : PubMed/NCBI
|