Introduction
Microglial cells, which are regarded as the most
important immune cells in the central nervous system (CNS), are
activated by brain injuries. Following activation by bacterial
toxins, microglial cells secrete a wide range of inflammatory
mediators, such as tumor necrosis factor-α (TNF-α), interleukin
(IL)-1β, nitric oxide (NO) and prostanoids (1,2).
Lipopolysaccharide (LPS) activates microglial cells
and plays vital roles in the pathogenesis of inflammatory responses
by inducing the production of inflammatory mediators (3,4).
TNF-α and IL-1β are the most important mediators, and they are
known to be secreted during the early phase of inflammatory disease
(5).
Prostaglandin E2 (PGE2), which
is one of the most prominent prostanoids produced by astrocytes and
microglia that have been exposed to noxious stimuli (6,7),
is produced by the conversion of arachidonic acid by cyclooxygenase
(COX). There are 2 isoforms of COX, COX-1 and COX-2. COX-2 is
expressed by pro-inflammatory mediators and mitogenic stimuli
(8).
NO is an important physiological messenger and
effector molecule in a number of biological systems, including
immunological, neuronal and cardiovascular tissues (9). NO is endogenously generated from
L-arginine by NO synthase (NOS). There are 3 types of NOS,
endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible NOS
(iNOS). Of these, iNOS plays an important role in inflammation and
host-defense responses (10).
Nuclear factor-κB (NF-κB) regulates the expression
of genes involved in cellular proliferation, inflammatory responses
and cell adhesion (8,11). In unstimulated cells, NF-κB is
held in the cytoplasm by inhibitory IκB protein (IκB). The
activation of NF-κB is then induced by the phosphorylation,
ubiquitination and proteasome-mediated degradation of the IκB
protein. Following activation, NF-κB undergoes nuclear traslocation
and DNA binding (12).
Urinary trypsin inhibitors are widely used for the
treatment of patients with acute inflammatory disorders, such as
pancreatitis (13), septic shock
(14), hemorrhagic shock
(15) and ischemia-reperfusion
injury (16,17). Additionally, urinary trypsin
inhibitors are known to ameliorate the enhanced production of
pro-inflammatory molecules (18).
Ulinastatin is one of the urinary trypsin inhibitors and is an
intrinsic serine-protease inhibitor that can be extracted and
purified from human urine (19).
Ulinastatin has been shown to decrease the occurrence of coronary
artery lesions in patients with acute Kawasaki disease by the
suppression of neutrophils (20)
and to reduce LPS-induced pulmonary injury (21). Diverse effects of ulinastatin have
been documented; however, the mechanisms underlying the
anti-inflammatory activity of ulinastatin are not yet fully
understood.
In the present study, we investigated the effect of
ulinastatin on LPS-induced inflammation in relation with NF-κB
activation. To accomplish this, we used a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay, reverse transcription-polymerase chain reaction (RT-PCR),
western blot analysis, electrophoretic mobility gel shift assay
(EMSA), PGE2 immunoassay, and NO detection in BV2 mouse
microglial cells.
Materials and methods
Cell culture
BV2 mouse microglial cells were cultured in
Dulbecco’s modified Eagle’s Medium (DMEM; Gibco BRL, Grand Island,
NY, USA) supplemented with 10% heat-inactivated fetal bovine serum
(FBS; Gibco BRL) at 37°C under 5% CO2-95% O2
in a humidified cell incubator. The cells were then plated onto
culture dishes at a density of 2×104
cells/cm2 24 h prior to drug treatments.
MTT cytotoxicity assay
BV2 mouse microglial cells were grown in 100 μl of
culture medium per well in 96-well plates. Ulinastatin was
purchased from Wakamoto Pharmaceutical Co., Ltd. (Tokyo, Japan). To
determine the cytotoxicity of ulinastatin, cells were treated with
ulinastatin at concentrations of 1, 10, 100, 1,000 and 10,000 U/ml
for 24 h. The cells in the control group were left untreated. After
incubation, 10 μl of MTT labeling reagent containing 5 mg/ml MTT in
phosphate-buffered saline were added to each well, and the plates
were then incubated for an additional 2 h. Subsequently, 100 μl of
solubilization solution containing 10% sodium dodecyl sulfate (SDS)
in 0.01 M hydrochloric acid were added to each well, after which
the cells were incubated for a further 12 h. The absorbance was
then measured using a microtiter plate reader (Bio-Tek, Winooski,
VT, USA) at a test wavelength of 595 nm and a reference wavelength
of 690 nm. The optical density (OD) was then calculated as the
difference between the absorbance at the reference wavelength and
that of the test wavelength. The percentage viability was then
calculated as follows: (OD of drug-treated sample/control OD)
×100.
RNA isolation and RT-PCR
RT-PCR was conducted to determine the mRNA
expression of COX-2 and iNOS. Briefly, total RNA was isolated from
the BV2 mouse microglial cells using RNAzol™B (Tel-Test Inc.,
Friendswood, TX, USA). Subsequently, 2 μg of RNA and 2 μl of random
hexamers (Promega, Madison, WI, USA) were combined, after which the
mixture was heated at 65°C for 15 min. A total of 1 μl of AMV
reverse transcriptase (Promega), 5 μl of 2.5 mM dNTP (Promega), 0.5
μl of RNasin (Promega) and 8 μl of 5× AMV RT buffer (Promega) were
then added to the mixture, which was then brought up to a final
volume of 40 μl using diethylpyrocarbonate (DEPC)-treated water.
The reaction mixture was then incubated at 42°C for 2 h.
PCR amplification was performed in a reaction
mixture with a final volume of 40 μl that contained 1 μl of the
appropriate cDNA, 0.5 μl of each set of primers at a concentration
of 10 pM, 4 μl of 10× RT buffer, 1 μl of 2.5 mM dNTP and 0.2 U of
Taq DNA polymerase (Takara, Shiga, Japan). The primers used to
amplify COX-2 were 5′-CCAGATGCTATCTTTGG GGAGAC-3′ (a 23-mer
sense oligonucleotide) and 5′-CTTGCA TTGATGGTGGCTG-3′ (a 19-mer
antisense oligonucleotide). The primers used to amplify iNOS
were 5′-CAAGAGTTTGA CCAGAGGACC-3′ (a 21-mer sense oligonucleotide)
and 5′-TGGAACCACTCGTACTTGGGA-3′ (a 21-mer antisense
oligonucleotide). Finally, to amplify cyclophilin as the
internal control, the primer sequences were 5′-ACCCCACCGTGTTC
TTCGAC-3′ (a 20-mer sense oligonucleotide) and 5′-CATTTG
CCATGGACAAGATG-3′ (a 20-mer antisense oligonucleotide).
To amplify the COX-2 and iNOS genes,
PCR was conducted using a PTC-0150 MiniCycler (Bio-Rad, Hercules,
CA, USA) subjecting the reaction mixture to the following
conditions: initial denaturation at 94°C for 5 min, followed by 32
cycles of denaturation at 94°C for 30 sec, annealing at 60°C for 30
sec and extension at 72°C for 45 sec, with a final extension at
72°C for 10 min. For cyclophilin, PCR was conducted under
same conditions, except only 25 amplification cycles were
conducted.
Preparation of whole cell extract
To prepare the whole cell extract, cells were
incubated in ice-cold whole cell lysate buffer that contained 50 mM
HEPES (pH 7.5), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM
magnesium chloride hexahydrate, 1 mM
ethyleneglycol-bis-(β-aminoethyl ether)-N,N′-tetraacetic acid
(EGTA), 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 μg/ml
leupeptin, 1 μg/ml pepstatin, 1 mM sodium orthovanadate and 100 mM
sodium fluoride (NaF) for 15 min. The cells were then centrifuged
at 14,000 rpm for 15 min at 4°C, and the supernatant was
stored.
Preparation of nuclear and cytosolic
extracts
The cells were collected and suspended in hypotonic
buffer [10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM
potassium chloride (KCl), 0.2 mM PMSF, 0.5 mM dithiothreitol (DTT)
and 10 μg/ml aprotinin], after which they were incubated on ice for
10 min. The cells were then lysed by the addition of 0.1% Nonidet
P-40 and vigorous vortexing for 10 sec. The cells were then
centrifuged at 4,000 rpm for 5 min at 4°C, after which the
supernatants containing protein were collected. The pellets
acquired from the cytosolic protein extraction were then
resuspended in high salt buffer (20 mM HEPES, pH 7.9, 25% glycerol,
400 mM KCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 1 mM
NaF and 1 mM sodium orthovanadate). Finally, the cells were
centrifuged at 14,000 rpm for 5 min at 4°C, and the supernatants
were stored.
Western blot analysis
Whole protein extract was used to evaluate the
protein expression of COX-2 and iNOS. In addition, cytosolic
extract was used for the detection of IκB-α, while nuclear extract
was used for the detection of NF-κB (p65) protein expression. Prior
to analysis, the protein concentrations were measured using a
Bio-Rad colorimetric protein assay kit (Bio-Rad). Subsequently, 40
μg of protein were separated on SDS-polyacrylamide gels and then
transferred onto a nitrocellulose membrane (Schleicher &
Schuell GmbH, Dassel, Germany). Goat COX-2 antibody (1:1,000; Santa
Cruz Biotechnology, Santa Cruz, CA, USA), rabbit iNOS antibody
(1:500; Santa Cruz Biotechnology), rabbit NF-κB (p65) antibody
(1:500; Santa Cruz Biotechnology) and rabbit IκB-α antibody (1:500;
Santa Cruz Biotechnology) were used as the primary antibodies.
Horseradish peroxidase-conjugated anti-goat antibody (1:4,000;
Santa Cruz Biotechnology) was used to probe for COX-2. Anti-rabbit
antibody (1:2,000; Vector Laboratories, Burlingame, CA, USA) was
used as the secondary antibody for iNOS, NF-κB (p65) and IκB-α.
Bands were detected using the enhanced chemiluminescence (ECL)
detection system (Santa Cruz Biotechnology).
EMSA
EMSA was performed using a commercially available
gel shift kit (Panomics, Inc., Redwood City, CA, USA) according to
the manufacturer’s instructions. Briefly, 10 μg of nuclear extract
were incubated with biotin end-labeled 22-mer double-stranded NF-κB
oligonucleotide 5′-AGTTGAGG GGACTTTCCCAGGC-3′ (underlined
letters indicate NF-κB-binding site) for 30 min at 15°C. The
specificity of NF-κB DNA binding was then determined by evaluating
the effects of competition with a 33-fold unlabeled
oligonucleotide. The DNA-protein complexes were then analyzed by
electrophoresis on a 6% non-denaturing polyacrylamide gel and
subsequently transferred onto a neutrally charged nylon membrane.
The oligonuleotide on the membrane was then fixed using a UV
crosslinker for 3 min, after which band detection was performed
using the detection system provided with the kit.
Measurement of PGE2
synthesis
PGE2 synthesis was assessed using a
commercially available PGE2 competitive enzyme
immunoassay kit (Amersham Biosciences Corp., Piscataway, NJ, USA).
Briefly, 100 μl of supernatant from the culture medium and the
standards were added to different wells on the goat anti-mouse
IgG-coated microtiter plate provided with the kit. Mouse
anti-PGE2 antibody and peroxidase-conjugated
PGE2 were then added to each well, after which the plate
was incubated at room temperature with shaking for 2 h. The wells
were then drained and washed, after which
3,3′,5,5′-tetramethylbenzidine/hydrogen peroxide solution was
added. The plate was then incubated at room temperature with
shaking for 30 min, after which the reaction was stopped by the
addition of H2SO4. The absorbance of the
content of each well was then measured at a wavelength of 450
nm.
Determination of NO production
To determine the effect of ulinastatin on NO
production, the amount of nitrite in the supernatant was measured
using a commercially available NO detection kit (Intron, Inc.,
Seoul, Korea). After collection of 100 μl of supernatant, 50 μl of
N1 buffer were added to each well, and the plate was then incubated
at room temperature for 10 min. N2 buffer was then added, after
which the plate was incubated at room temperature for 10 min. The
absorbance of the content of each well was then measured at a
wavelength of 540 nm and the nitrite concentration was calculated
from a nitrite standard curve.
Statistical analysis
The results are presented as the means ± standard
error of the mean (SEM). The data were analyzed by one-way ANOVA
followed by Duncan’s post hoc test using SPSS software. P<0.05
was considered to indicate a statistically significant
difference.
Results
Effect of ulinastatin on the viability of
BV2 mouse microglial cells
To assess the cytotoxic effect of ulinastatin on BV2
mouse microglial cells, the cells were cultured with ulinastatin at
final a concentration of 1, 10, 100, 1,000 and 10,000 U/ml for 24
h, after which MTT assay was conducted. Cells cultured in
ulinastatin-free medium were used as the control. The viability of
cells incubated with ulinastatin at concentrations of 1, 10, 100,
1,000 and 10,000 U/ml for 24 h was 96.50±0.94%, 97.22±1.03%,
95.00±1.41%, 92.45±1.38% and 56.96±1.13% of the control value,
respectively. These results demonstrate that ulinastatin exerted no
cytotoxic effects at concentrations <1,000 U/ml and that 10,000
U/ml of ulinastatin significantly reduced cell viability.
Therefore, we used 10, 100 and 1,000 U/ml of ulinastatin for all
subsequent experiments.
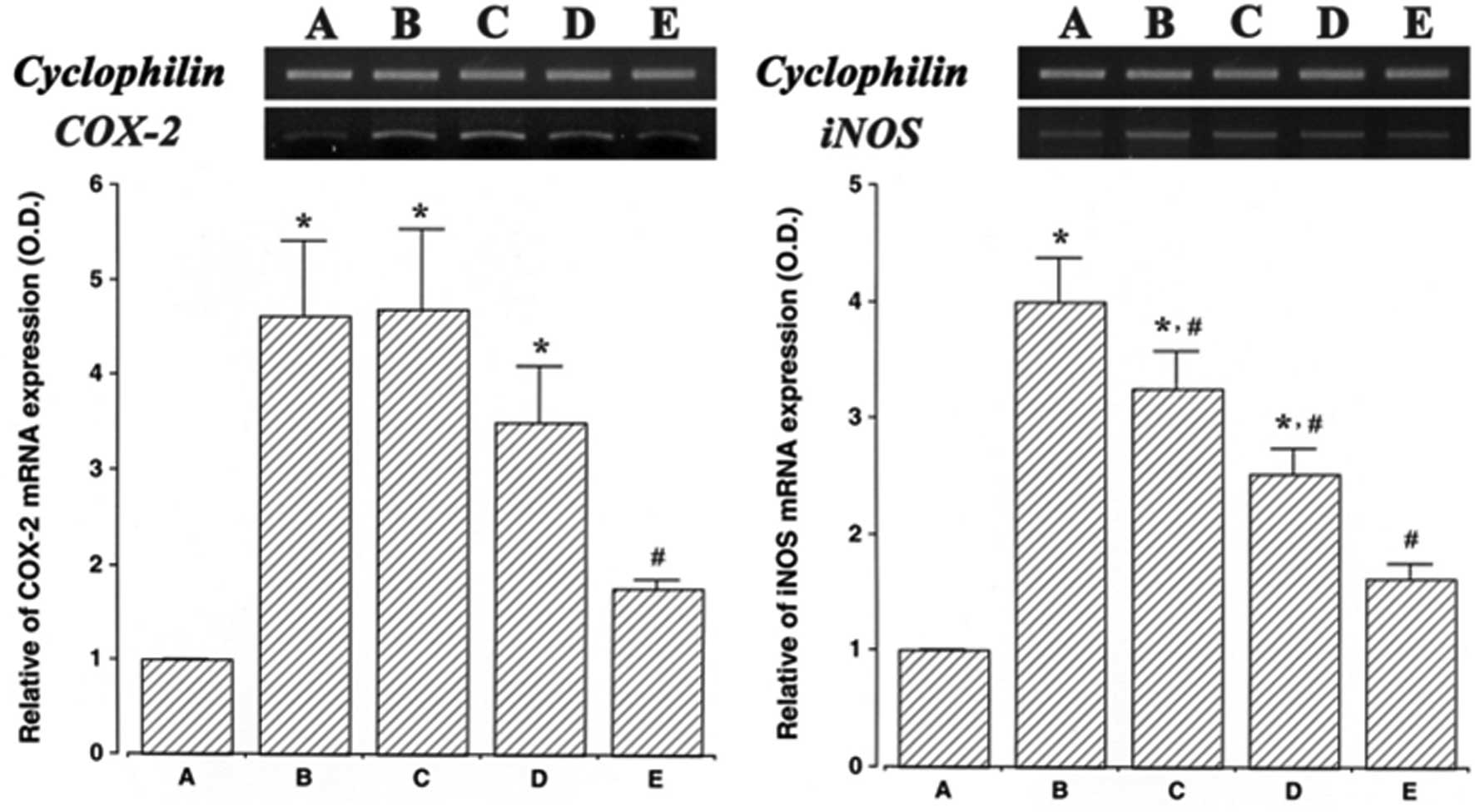
Effect of ulinastatin on the mRNA
expression of COX-2 and iNOS
The levels of COX-2 and iNOS mRNA in ulinastatin
treated cells were evaluated and then compared to the levels in the
LPS-treated cells to determine the effects of ulinstatin on the
expression of these genes. In the present study, the levels of
COX-2 and iNOS mRNA in the control cells were set at 1.00.
The level of COX-2 mRNA was markedly increased to
4.61±0.78 following treatment with 2 μg/ml LPS for 24 h. However,
these increased COX-2 mRNA levels decreased to 4.68±0.85, 3.50±0.58
and 1.76±0.97 in the cells that were pre-treated for 1 h with 10,
100 and 1,000 U/ml ulinastatin, respectively, and then treated with
2 μg/ml LPS for 24 h.
The level of iNOS mRNA following treatment with 2
μg/ml LPS for 24 h was markedly increased to 3.99±0.37. However,
the levels of iNOS mRNA were only 3.24±0.31, 2.52±0.21 and
1.62±0.11 when the cells were pre-treated for 1 h with 10, 100 and
1,000 U/ml ulinastatin, respectively, and then treated with 2 μg/ml
LPS for 24 h. These findings demonstrate that treatment with LPS
enhanced COX-2 and iNOS mRNA expression in BV2 mouse microglial
cells, and that pre-treatment with ulinastatin suppressed the
LPS-induced COX-2 and iNOS mRNA expression (Fig. 1).
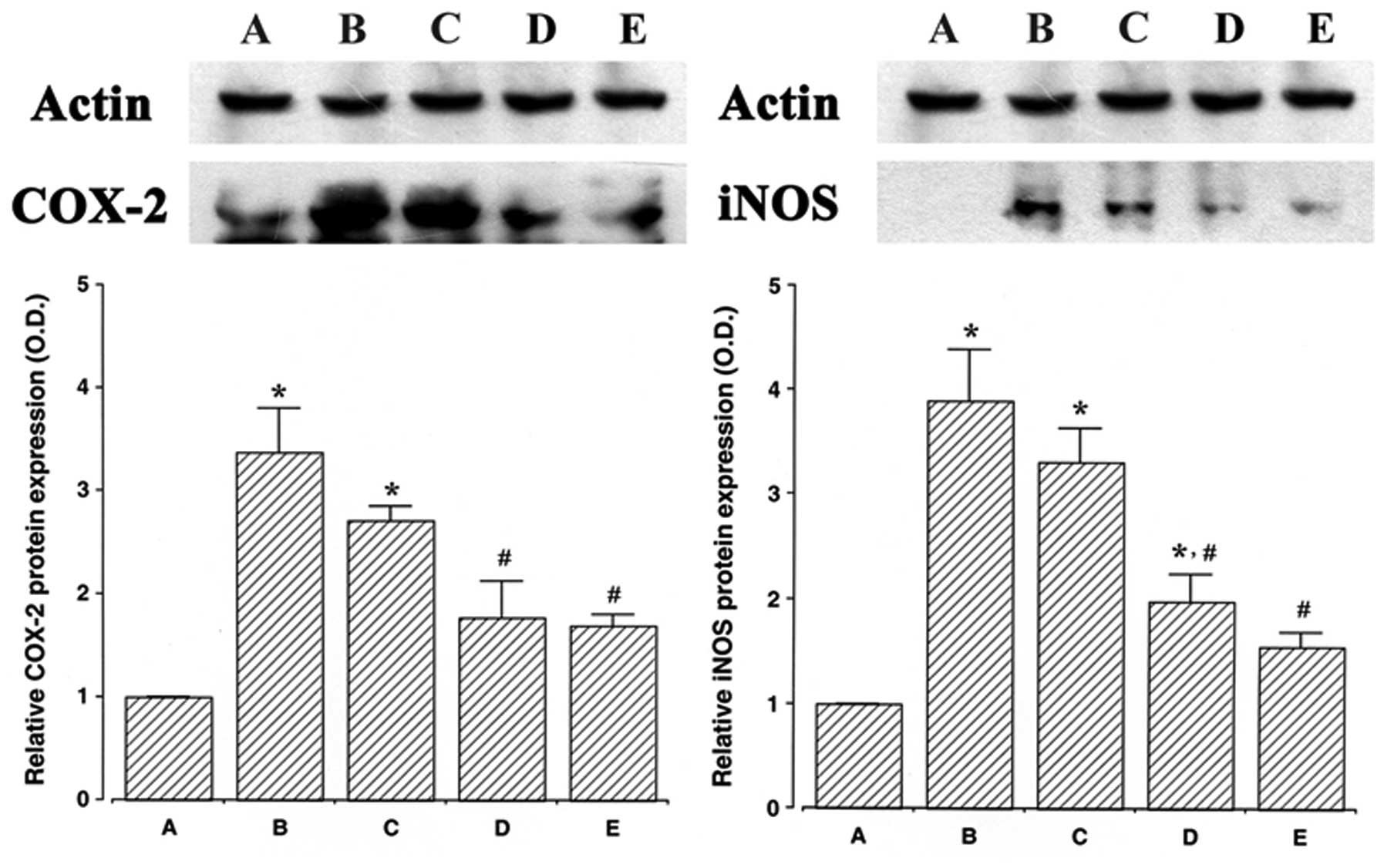
Effect of ulinastatin on the protein
expression of COX-2 and iNOS
The protein levels of COX-2 and iNOS in the cells
that were treated with ulinastatin were evaluated and compared to
the levels in the control cells to determine the effects of
ulinastatin on the expression of these proteins. In the present
study, the protein levels of COX-2 and iNOS in the control cells
were set at 1.00.
The protein level of COX-2 markedly increased to
3.37±0.41 following treatment with 2 μg/ml LPS for 24 h. However,
the protein levels of COX-2 decreased to 2.70±0.14, 1.77±0.35 and
1.69±0.11 in the cells that were pre-treated for 1 h with 10, 100
and 1,000 U/ml of ulinastatin, respectively, and then treated with
2 μg/ml LPS for 24 h.
Following treatment with 2 μg/ml LPS for 24 h, the
protein level of iNOS markedly increased to 3.89±0.48. However, the
protein levels of iNOS decreased to 3.31±0.32, 1.98±0.26 and
1.54±0.14 in the cells that were pre-treated with 10, 100 and 1,000
U/ml ulinastatin, respectively, and then treated with 2 μg/ml LPS
for 24 h. These results demonstrate that treatment with LPS
enhanced COX-2 and iNOS protein expression in BV2 mouse microglial
cells, and that pre-treatment with ulinastatin suppressed the
LPS-induced protein expression of COX-2 and iNOS (Fig. 2).
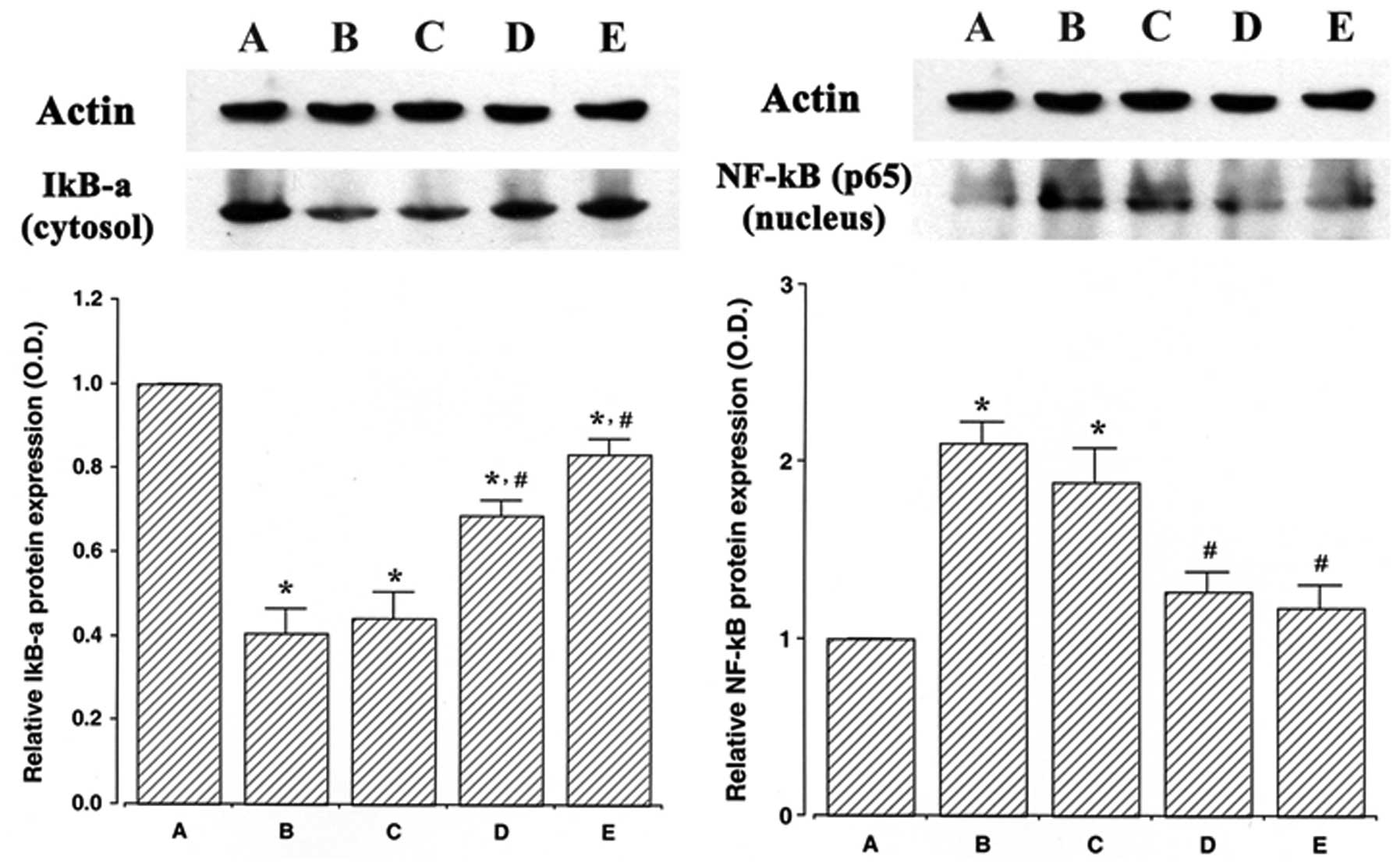
Effect of ulinastatin on NF-κB protein in
the nuclear fraction and IκB-α protein in the cytosolic
fraction
The protein levels of NF-κB and IκB-α were evaluated
to determine the effects of ulinastatin on the expression of these
proteins. In the present study, the protein levels of NF-κB and
IκB-α in the control cells were set at 1.00.
The protein level of NF-κB in the nuclear fraction
markedly increased to 2.10±0.12 following treatment with 2 μg/ml
LPS for 30 min. However, the protein levels of NF-κB in the nuclear
fraction decreased to 1.88±0.19, 1.26±0.10 and 1.17±0.12 in the
cells that were pre-treated for 1 h with 10, 100 and 1,000 U/ml
ulinastatin, respectively, and then treated with 2 μg/ml LPS.
The protein level of IκB-α in the cytosolic fraction
markedly decreased to 0.40±0.05 following treatment with 2 μg/ml
LPS for 30 min. However, the protein levels of IκB-α in the
cytosolic fraction increased to 0.44±0.06, 0.68±0.03 and 0.83±0.03
in the cells that were pre-treated for 1 h with 10, 100 and 1,000
U/ml of ulinastatin, respectively, and then treated with 2 μg/ml
LPS for 30 min. The results of this study demonstrate that
treatment with LPS increased NF-κB protein expression in the
nucleus and decreased IκB-α protein expression in the cytosol of
BV2 mouse microglial cells, whereas pre-treatment with ulinastatin
suppressed NF-κB protein expression and increased IκB-α protein
expression (Fig. 3).
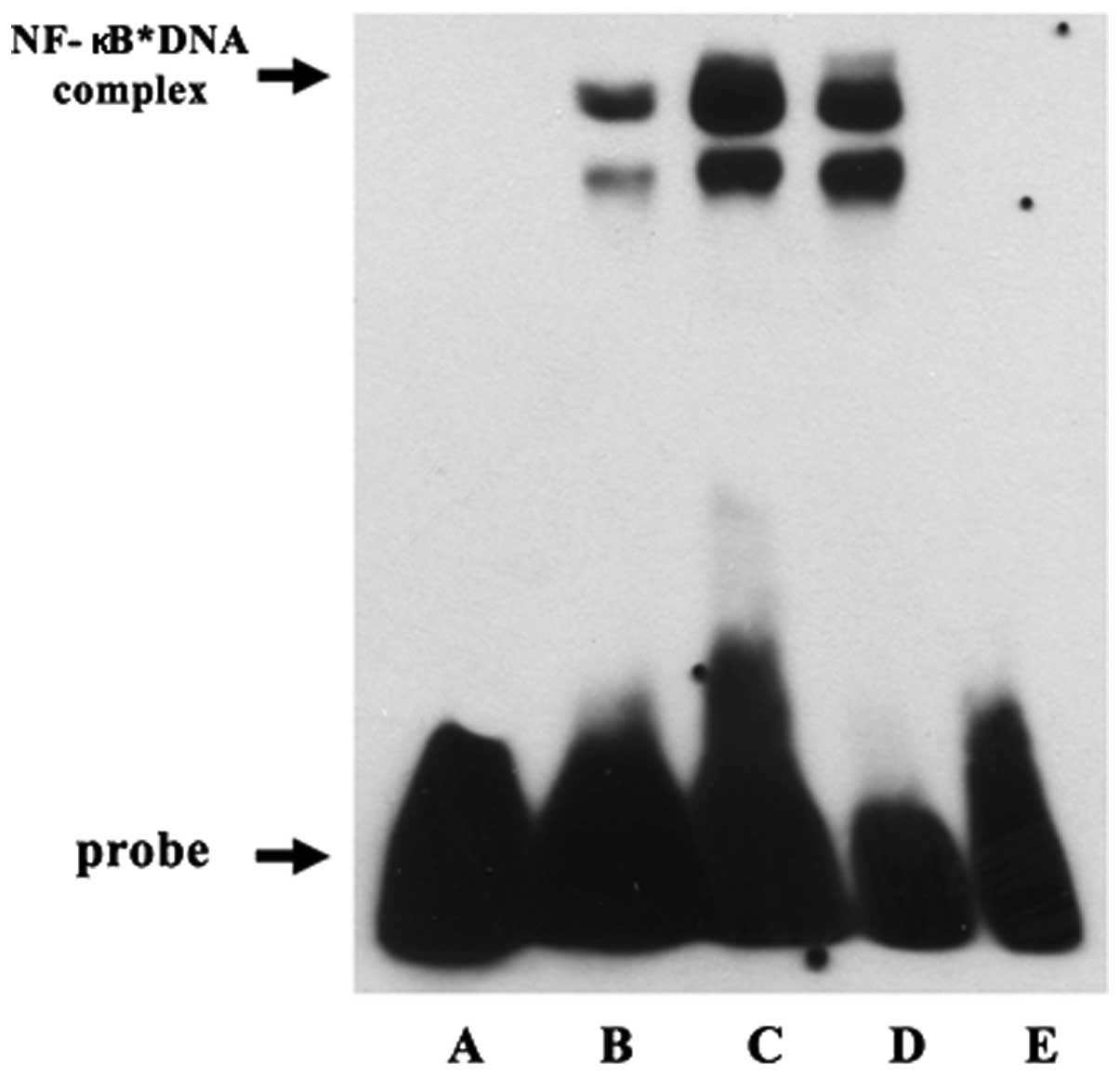
Effect of ulinastatin on DNA binding
activity of NF-κB
To confirm the effect of ulinastatin on NF-κB
activation, we evaluated the DNA binding activity of NF-κB using
EMSA. Treatment with 2 μg/ml LPS for 30 min increased the DNA
binding activity of NF-κB. However, the LPS-induced DNA binding
activity of NF-κB decreased in response to pre-treatment with 1,000
U/ml ulinastatin. These findings demonstrate that treatment with
LPS enhanced the DNA binding activity of NF-κB in BV2 mouse
microglial cells and that pre-treatment with ulinastatin suppressed
the LPS-induced increase in the DNA binding activity of NF-κB
(Fig. 4).
Effect of ulinastatin on PGE2
synthesis and NO production
The results of a PGE2 immunoassay
revealed that the amount of PGE2 present in the culture
medium increased from 26.58±0.82 pg/ml to 94.34±7.04 pg/ml
following 24 h of exposure to LPS. However, the levels of
PGE2 synthesis decreased to 90.90±7.61, 69.27±4.15,
62.51±4.92 and 38.91±1.49 pg/ml in the cells that were pre-treated
for 1 h with 10, 100, 1,000 U/ml ulinastatin and 500 μM
acetylsalicylic acid (ASA), respectively, prior to treatment with 2
μg/ml LPS for 24 h.
The results of the NO detection assay revealed that
the concentration of nitrite increased from 3.00±0.38 μM to
19.97±0.93 μM following 24 h of exposure to LPS. However, the
levels of NO production decreased to 17.14±0.78, 16.15±0.81,
11.91±0.44 and 13.05±0.79 μM in the cells that were pre-treated for
1 h with 10, 100, 1,000 U/ml ulinastatin and 500 μM ASA,
respectively, prior to treatment with 2 μg/ml LPS for 24 h.
These results demonstrate that LPS enhanced
PGE2 synthesis and NO production in BV2 mouse microglial
cells and that pre-treatment with ulinastatin suppressed the
LPS-induced PGE2 synthesis and NO production (Fig. 5).
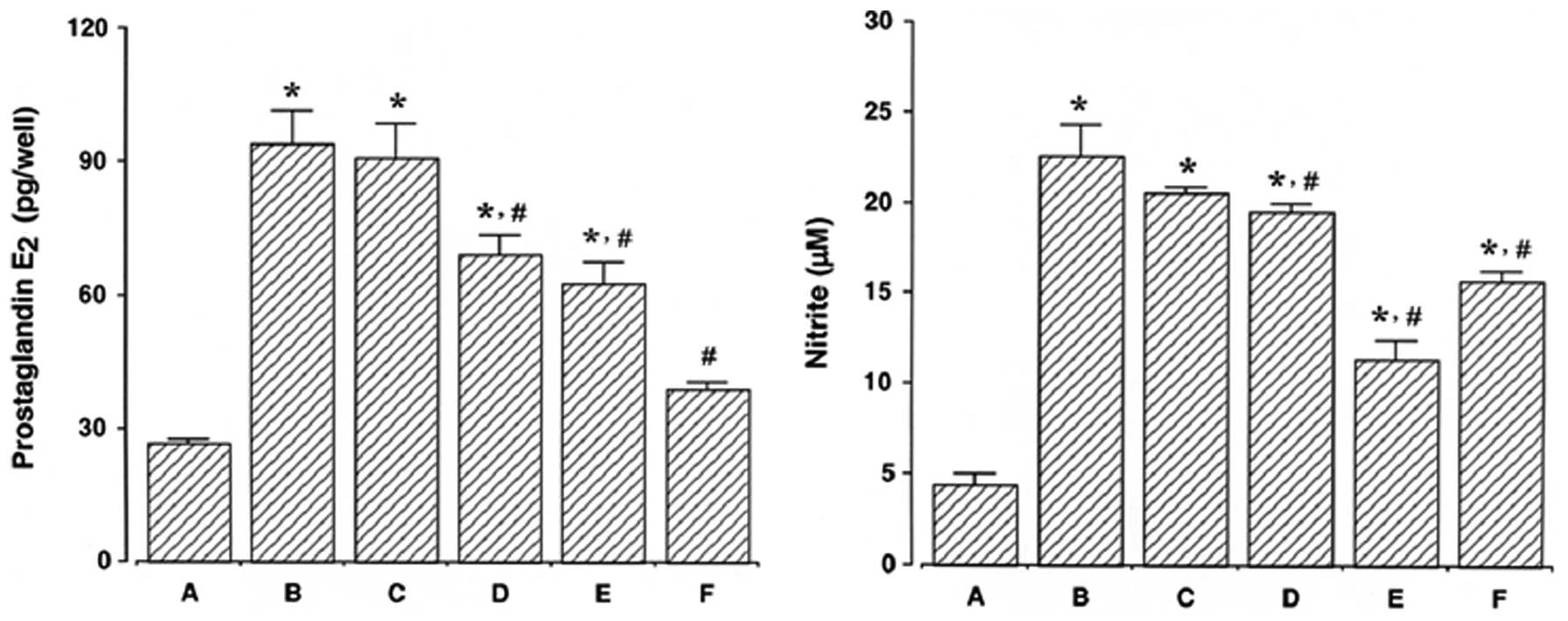 | Figure 5Measurement of prostaglandin
E2 (PGE2) synthesis and nitric oxide (NO)
production in BV2 mouse microglial cells. BV2 mouse microglial
cells were pre-treated with ulinastatin for 1 h at a concentration
of 10, 100, and 1,000 U/ml, and then treated with 2 μg/ml
lipopolysaccharide (LPS) for 24 h. Left panel, PGE2
synthesis; right panel, NO production. (A) Control group, (B)
LPS-treated group, (C) LPS- and 10 U/ml ulinastatin pre-treated
group, (D) LPS- and 100 U/ml ulinastatin pre-treated group, (E)
LPS- and 1,000 U/ml ulinastatin pre-treated group, (F) LPS- and 500
μM acetylsalicylic acid pre-treated group. The results are
presented as the means ± standard error of the mean (SEM).
*P<0.05 compared to the control group.
#P<0.05 compared to the LPS-treated group. |
Discussion
Organ protection is a routine therapy in patients
with severe trauma, infection, and even multiple organ dysfunction
syndrome. Urinary trypsin inhibitors have been widely used for the
treatment of acute inflammatory disorders (22). A number of studies have reported
that urinary trypsin inhibitors suppress the enhanced production of
pro-inflammatory molecules such as prostaglandin H2
synthase-2 (23), IL-8 (24) and TNF-α (25). Recently, Qiu et al
(26) demonstrated that
ulinastsatin exerted protective effect against smoke
inhalation-induced acute lung injury and the subsequent development
of pulmonary fibrosis in rats. Pre-treatment with ulinastatin has
also been shown to ameliorate oxidative injury in rats (27). Inflammation, which is a complex
process that commences with a primary reaction in tissues, is
involved in multiple pathologies, including arthritis, asthma,
multiple sclerosis, colitis, inflammatory bowel disease and
atherosclerosis. COX-2 and iNOS are 2 primary inflammatory markers
produced by microglia in the CNS.
In the present study, pre-treatment with ulinastatin
significantly suppressed LPS-induced COX-2 expression and
PGE2 synthesis in BV2 mouse microglial cells. A high
level of COX-2 activity is closely associated with the occurrence
of arthritis. In addition, it is well known that specific COX-2
inhibitors attenuate the symptoms of inflammation (28). PGE2 is a major
metabolite of the COX-2 pathway that has emerged as an important
lipid mediator of inflammatory and immune regulatory processes
(29).
As shown in the present study, pre-treatment with
ulinastatin significantly inhibited LPS-induced iNOS expression as
well as NO production in BV2 mouse microglial cells. During
macrophage activation, iNOS has been shown to mediate NO-induced
cell injury through the generation of reactive radicals, such as
peroxynitrite (4). The excessive
production of NO by iNOS has been implicated in a variety of
pathological processes, including septic shock, rheumatoid
arthritis and carcinogenesis (30,31). In addition, NO is known to
modulate the activity of COX-2 in a cyclic guanosine monophosphate
(cGMP)-independent manner, as well as to play a critical role in
the release of PGE2 by the direct activation of COX-2
(32). This synergistic induction
of NO and PGE2 is also related to the overproduction of
the earliest expressed pro-inflammatory cytokines (33,34).
The ubiquitous NF-κB signaling pathway plays an
important role in the regulation of inflammation through the
transcription of the COX-2, iNOS and cytokine genes (8). In this study, we investigated the
DNA binding activity of NF-κB to determine whether the inhibition
of COX-2 and iNOS is mediated by the NF-κB signaling pathway.
Pre-treatment with ulinastatin significantly suppressed the
LPS-induced nuclear translocation of NF-κB, and this inhibition
corresponded to the inhibition of COX-2 and iNOS expression in BV2
mouse microglial cells. These findings are supported by previous
studies, indicating that the blockage of NF-κB transcriptional
activity suppresses COX-2 and iNOS expression and pro-inflammatory
cytokine production (2,35,36). NF-κB is located in the cytoplasm
as an inactive complex bound to IκB-α, and then it is degraded upon
phosphorylation, after which it dissociates to produce activated
NF-κB (37). The results from the
present study indicate that ulinastatin blocks the LPS-induced
translocation of NF-κB via the inhibition of the degradation of
IκB-α. The blockage of NF-κB activation has potential as a
therapeutic modality for the treatment of inflammatory bowel
disease and arthritis (38,39). The inhibitory effect of
ulinastatin on NF-κB signal transduction has been shown to suppress
the proliferation and induce the apoptosis of human breast cancer
cells (40).
In this study, we demonstrated that ulinastatin
exerts analgesic and anti-inflammatory effects by suppressing COX-2
and iNOS expression, which results in the inhibition of
PGE2 synthesis and NO production. The present study also
reveals that the analgesic and anti-inflammatory effects of
ulinastatin involve the blockage of NF-κB activation in
LPS-stimulated BV2 mouse microglial cells. These results suggest
that the analgesic and anti-inflammatory effects of ulinastatin
possibly occur via the suppression of COX-2 and iNOS expression
through the downregulation of NF-κB activity.
Acknowledgements
This study was supported by the Program of Kyung Hee
University for the Young Researcher of Medical Science in 2007
(KHU-20071478).
References
|
1
|
Liu B and Hong JS: Role of microglia in
inflammation-mediated neurodegenerative diseases: mechanisms and
strategies for therapeutic intervention. J Pharmacol Exp Ther.
304:1–7. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moon DO, Choi YH, Kim ND, Park YM and Kim
GY: Anti-inflammatory effects of β-lapachone in
lipopolysaccharide-stimulated BV2 microglia. Int Immunopharmacol.
7:506–514. 2007.
|
|
3
|
Kubes P and McCafferty DM: Nitric oxide
and intestinal inflammation. Am J Med. 109:150–158. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee AK, Sung SH, Kim YC and Kim SG:
Inhibition of lipopolysaccharide-inducible nitric oxide synthase,
TNF-α and COX-2 expression by sauchinone effects on I-κBα
phosphorylation, C/EBP and AP-1 activation. Br J Pharmacol.
139:11–20. 2003.
|
|
5
|
Palladino MA, Bahjat FR, Theodorakis EA
and Moldawer LL: Anti-TNF-α therapies: the next generation. Nat Rev
Drug Discov. 2:736–746. 2003.
|
|
6
|
Kalmar B, Kittel A, Lemmens R, Kornyei Z
and Madarasz E: Cultured astrocytes react to LPS with increased
cyclooxygenase activity and phagocytosis. Neurochem Int.
38:453–461. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park HJ, Kim IT, Won JH, Jeong SH, Park
EY, Nam JH, Choi J and Lee KT: Anti-inflammatory activities of
ent-16αH,17-hydroxy-kauran-19-oic acid isolated from
the roots of Siegesbeckia pubescens are due to the
inhibition of iNOS and COX-2 expression in RAW 264.7 macrophages
via NF-κB inactivation. Eur J Pharmacol. 558:185–193.
2007.PubMed/NCBI
|
|
8
|
Surh YJ, Chun KS, Cha HH, Han SS, Keum YS,
Park KK and Lee SS: Molecular mechanisms underlying chemopreventive
activities of anti-inflammatory phytochemicals: down-regulation of
COX-2 and iNOS through suppression of NF-κB activation. Mutat Res.
480–481:243–268. 2001.PubMed/NCBI
|
|
9
|
Bredt DS and Snyder SH: Nitric Oxide: a
physiologic messenger molecule. Annu Rev Biochem. 63:175–195. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xie Q and Nathan C: The high-output nitric
oxide pathway: role and regulation. J Leukoc Biol. 56:576–582.
1994.PubMed/NCBI
|
|
11
|
Lappas M, Permezel M, Georgiou HM and Rice
GE: Nuclear factor kappa B regulation of proinflammatory cytokines
in human gestational tissues in vitro. Biol Reprod. 67:668–673.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Karin M: The beginning of the end: IκB
kinase (IKK) and NF-κB activation. J Biol Chem. 274:27339–27342.
1999.
|
|
13
|
Maciejewski R, Burdan F, Burski K, Madej
B, Ziemiakowicz R, Dabrowski A and Wallner G: Selected biochemical
parameters and ultrastructural picture of pancreas due to
Ulinastatin treatment of experimental acute pancreatitis. Exp
Toxicol Pathol. 56:305–311. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tani T, Aoki H, Yoshioka T, Lin KJ and
Kodama M: Treatment of septic shock with a protease inhibitor in a
canine model: a prospective, randomized, controlled trial. Crit
Care Med. 21:925–930. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Masuda T, Sato K, Noda C, Ikeda KM,
Matsunaga A, Ogura MN, Shimizu K, Nagasawa H, Matsuyama N and Izumi
T: Protective effect of urinary trypsin inhibitor on myocardial
mitochondria during hemorrhagic shock and reperfusion. Crit Care
Med. 31:1987–1992. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yano T, Anraku S, Nakayama R and Ushijima
K: Neuroprotective effect of urinary trypsin inhibitor against
focal cerebral ischemia-reperfusion injury in rats. Anesthesiology.
98:465–473. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shin IW, Jang IS, Lee SM, Park KE, Ok SH,
Sohn JT, Lee HK and Chung YK: Myocardial protective effect by
ulinastatin via an anti-inflammatory response after regional
ischemia/reperfusion injury in an in vivo rat heart model. Korean J
Anesthesiol. 61:499–505. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Molor-Erdene P, Okajima K, Isobe H, Uchiba
M, Harada N and Okabe H: Urinary trypsin inhibitor reduces
LPS-induced hypotension by suppressing tumor necrosis factor-α
production through inhibition of Egr-1 expression. Am J Physiol
Heart Circ Physiol. 288:H1265–H1271. 2005.PubMed/NCBI
|
|
19
|
Tanaka Y, Maehara S, Sumi H, Toki N,
Moriyama S and Sasaki K: Purification and partial characterization
of two forms of urinary trypsin inhibitor. Biochim Biophys Acta.
705:192–199. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanai T, Ishiwata T, Kobayashi T, Sato H,
Takizawa M, Kawamura Y, Tsujimoto H, Nakatani K, Ishibashi N,
Nishiyama M, Hatai Y, Asano Y, Kobayashi T, Takeshita S and
Nonoyama S: Ulinastatin, a urinary trypsin inhibitor, for the
initial treatment of patients with Kawasaki disease: a
retrospective study. Circulation. 124:2822–2828. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gao C, Li R and Wang S: Ulinastatin
protects pulmonary tissues from lipopolysaccharide-induced injury
as an immunomodulator. J Trauma Acute Care Surg. 72:169–176.
2012.PubMed/NCBI
|
|
22
|
Inoue K, Takano H, Shimada A, Yanagisawa
R, Sakurai M, Yoshino S, Sato H and Yoshikawa T: Urinary trypsin
inhibitor protects against systemic inflammation induced by
lipopolysaccharide. Mol Pharmacol. 67:673–680. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zaitsu M, Hamasaki Y, Tashiro K, Matsuo M,
Ichimaru T, Fujita I, Tasaki H and Miyazaki S: Ulinastatin, an
elastase inhibitor, inhibits the increased mRNA expression of
prostaglandin H2 synthase-type 2 in Kawasaki disease. J
Infect Dis. 181:1101–1109. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakamura H, Abe S, Shibata Y, Sata M, Kato
S, Saito H, Hino T, Takahashi H and Tomoike H: Inhibition of
neutrophil elastase-induced interleukin-8 gene expression by
urinary trypsin inhibitor in human bronchial epithelial cells. Int
Arch Allergy Immunol. 112:157–162. 1997. View Article : Google Scholar
|
|
25
|
Aosasa S, Ono S, Mochizuki H, Tsujimoto H,
Ueno C and Matsumoto A: Mechanism of the inhibitory effect of
protease inhibitor on tumor necrosis factor alpha production of
monocytes. Shock. 15:101–105. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qiu X, Ji S, Wang J, Li H, Xia T, Pan B,
Xiao S and Xia Z: The therapeutic efficacy of Ulinastatin for rats
with smoking inhalation injury. Int Immunopharmacol. 14:289–295.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu CE, Zhang MY, Zou CW and Guo L:
Evaluation of the pharmacological function of ulinastatin in
experimental animals. Molecules. 17:9070–9080. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Crofford LJ, Lipsky PE, Brooks P, Abramson
SB, Simon LS and van de Putte LB: Basic biology and clinical
application of specific cyclooxygenase-2 inhibitors. Arthritis
Rheum. 43:4–13. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hinz B, Brune K and Pahl A: Prostaglandin
E2 upregulates cyclooxygenase-2 expression in
lipopolysaccharide-stimulated raw 264.7 macrophages. Biochem
Biophys Res Commun. 272:744–748. 2000.
|
|
30
|
Ohshima H and Bartsch H: Chronic
infections and inflammatory processes as cancer risk factors:
possible role of nitric oxide in carcinogenesis. Mutat Res.
305:253–264. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Salerno L, Sorrenti V, Di Giacomo C, Romeo
G and Siracusa MA: Progress in the development of selective nitric
oxide synthase (NOS) inhibitors. Curr Pharm Des. 8:177–200. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Salvemini D, Misko TP, Masferrer JL,
Seibert K, Currie MG and Needleman P: Nitric oxide activates
cyclooxygenase enzymes. Proc Natl Acad Sci USA. 90:7240–7244. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jun CD, Choi BM, Kim HM and Chung HT:
Involvement of protein kinase C during taxol-induced activation of
murine peritoneal macrophages. J Immunol. 154:6541–6547.
1995.PubMed/NCBI
|
|
34
|
White KE, Ding Q, Moore BB, Peters-Golden
M, Ware LB, Matthay MA and Olman MA: Prostaglandin E2
mediates IL-1β-related fibroblast mitogenic effects in acute lung
injury through differential utilization of prostanoid receptors. J
Immunol. 180:637–646. 2008.PubMed/NCBI
|
|
35
|
Petrova TV, Akama KT and Van Eldik LJ:
Cyclopentenone prostaglandins suppress activation of microglia:
down-regulation of inducible nitric oxide synthase by
15-deoxy-Δ12,14-prostaglandin J2. Proc Natl
Acad Sci USA. 96:4668–4673. 1999.PubMed/NCBI
|
|
36
|
Ye SM and Johnson RW: Regulation of
interleukin-6 gene expression in brain of aged mice by nuclear
factor κB. J Neuroimmunol. 117:87–96. 2001.
|
|
37
|
Baeuerle PA and Baltimore D: NF-κB: ten
years after. Cell. 87:13–20. 1996.
|
|
38
|
Dijkstra G, Moshage H and Jansen PL:
Blockade of NF-κB activation and donation of nitric oxide: new
treatment options in inflammatory bowel disease? Scand J
Gastroenterol Suppl. 236:37–41. 2002.
|
|
39
|
Simmonds RE and Foxwell BM: Signalling,
inflammation and arthritis: NF-κB and its relevance to arthritis
and inflammation. Rheumatology. 47:584–590. 2008.
|
|
40
|
Wang H, Sun X, Gao F, Zhong B, Zhang YH
and Sun Z: Effect of ulinastatin on growth inhibition, apoptosis of
breast carcinoma cells is related to a decrease in signal
conduction of JNk-2 and NF-κB. J Exp Clin Cancer Res.
31:22012.PubMed/NCBI
|