Introduction
Retinoblastoma is the most common ocular tumor
classically initiated by the loss or mutation of both alleles of
the retinoblastoma gene (Rb1) during retinal development. It is a
pediatric tumor of the retina observed in approximately 1:15,000
live births (1). The current
treatment of retinoblastoma includes chemotherapy, radioactive
plaque, external beam radiotherapy, cryotherapy and surgery
(2–4). Although the current survival rates
of retinoblastoma exceed 90%, complications and side-effects exist,
such as severe visual impairment or the loss of one or both
eyes.
Previous studies on Rb1 have shown that the mutation
on chromosome 13q is often present in retinoblastoma tumors. Rb1, a
tumor suppressor gene, plays crucial role in the regulation of the
cell cycle, cell differentiation, cell aging, apoptosis and growth
suppression (5–7). Rb1 can be functionally inactivated
through a variety of mechanisms, including deregulated
phosphorylation and direct sequestration by oncoproteins. Evidence
supports the notion that the loss of Rb1 function leads to a
breakdown in genome integrity (8). According to previous studies,
SO-Rb50, an Rb1-deficient cell line, displays obvious chromosomal
instability. The chromosomal aberrations increase during long-term
culture in vitro (9,10).
The E2F family of transcription factors plays a
pivotal role in the regulation of cell cycle progression, DNA
repair and replication, apoptosis, differentiation and development
(11). E2F1, best known as the
founding member of the E2F transcription factor family, has been
implicated in the response to DNA damage in conjunction with
retinoblastoma family proteins. The Rb1-E2F1 complex is formed in
response to DNA damage and is recruited to the sites of DNA
double-strand breaks (DSBs). Certain studies have suggested that
E2F1 plays a crucial role in DNA DSB repair by promoting the
recruitment and/or retention of repair factors, such as XPA and
XPC, at the sites of DNA breaks (12,13). However, other studies have
indicated that the loss of Rb1 has no significant effects on DNA
DSBs, as shown by γ-H2AX foci intensity in the cells following
exposure to ionizing radiation (IR) (14). Therefore, the precise molecular
mechanism of action of Rb1 in chromosomal instability remains
unclear.
Two distinct pathways have been described which
ensure that DNA DSBs are repaired: DNA non-homologous end joining
(NHEJ) and homologous recombination (HR). During HR, the damaged
chromosome interacts via synapsis with an undamaged DNA molecule
with which it shares extensive sequence homology, usually its
sister chromatid (15,16). HR is most active in the late S and
G2 phase of the cell cycle. By contrast, NHEJ is active throughout
the cell cycle and requires little or no DNA homology during repair
(17,18). The NHEJ pathway plays a key role
in the repair of DNA DSBs caused by IR.
Based on the evidence of chromosomal instability in
Rb1-deficient cells, as well as no evidence of any significant
effects on DNA DSB repair in Rb1-deficient and wild-type (WT) cells
following exposure to IR, we hypothesized that Rb1 plays a
differential role in the sub-pathways of DNA DSB repair, NHEJ and
HR. In order to confirm this hypothesis, we evaluated the pathway
of Rb1-mediated DSB repair in retinoblastoma cells. We found that
Rb1 significantly promoted HR, had no effect on NHEJ in
retinoblastoma cells. This study provides new insight into the
mechanisms of action of the Rb1 gene in the chromosomal instability
of retinoblastoma cells.
Materials and methods
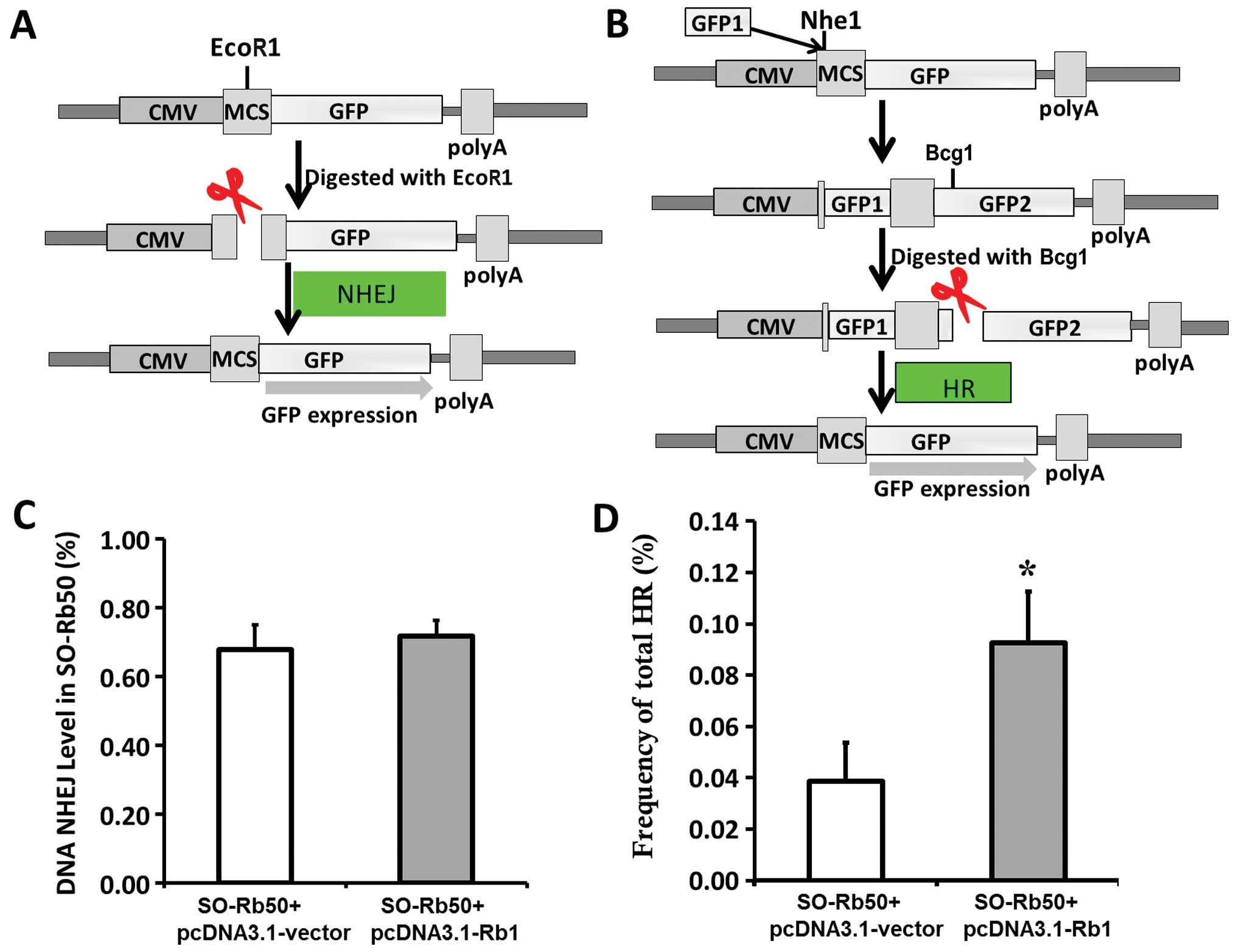
Plasmid construction
For pcDNA3.1-Rb1, WT Rb1 cDNA was inserted into the
pcDNA3.1-Myc-His vector between the KpnI and NotI
restriction sites. The pEGFP-HR plasmid as a substrate for
recombination was derived from pEGFP-N1 (Promega, Madison, WI,
USA). The structure of the HR substrate and the strategy to measure
HR is depicted in Fig. 3.
Briefly, GFP1 amplified by PCR was inserted into pEGFP-N1 at the
Nhe1 restriction site; GFP1 is 500 bp upstream from the
translation start site, ATG. There are 89-bp nucleotides before
GFP2.
Cell culture and transfection
HEK293 cells were grown in Dulbecco’s modified
Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum
(FBS), penicillin/streptomycin and glutamine. SO-Rb50
retinoblastoma cells, were established in 1991 in the State Key
Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun
Yat-sen University, Guangzhou, China. The cells were grown in DMEM
supplemented with 10% FBS, penicillin/streptomycin and glutamine.
SO-Rb50 cells were stably transfected with an expression plasmid
expressing WT Rb1 or an empty vector control (pcDNA3.1-Rb1 or
pcDNA3.1-vector) respectively, using
Lipofectamine®-Amine (Invitrogen, Carlsbad, CA, USA).
The positive clones were selected with G418 (500 μg/ml;
Sigma-Aldrich, St. Louis, MO, USA).
Reverse transcription-polymerase chain
reaction (RT-PCR)
HEK293 and SO-Rb50 cells in good condition were
harvested, and total RNA was isolated using TRIzol Reagent
(Invitrogen). RT-PCR was carried out using the one-step RT-PCR
system (Takara, Dalian, China). The following primer pairs were
used: for Rb1, 5′-TCTGTTTCAGGAAGAAGAACGA-3′ (sense) and
5′-TATGTGGCCATTACAACCTCAA-3′ (antisense); for β-actin,
5′-CACCACACCTTCTACAATGAG-3′ (sense) and 5′-TAGCACAGCCTGGATAGCAAC-3′
(antisense). For Rb1, RT-PCR was performed for 35 cycles each at
94°C for 30 sec, 60°C for 30 sec, 72°C for 1 min and a final
extension at 72°C for 10 min. RT-PCR for β-actin was performed for
20 cycles, each with the same temperature and time parameters as
for Rb1.
Immunofluorescence of cells in
suspension
Non-adherent SO-Rb50 cells, SO-Rb50 cells
transfected with pcDNA3.1-Rb1 and SO-Rb50 cells transfected with
the pcDNA3.1-vector were smeared across a gelatin-coated slide
forming a monolayer of cells. The cells were fixed in methanol and
then characterized by staining with mouse anti-rat Rb1 monoclonal
antibody (1:100; Wuhan Boster Biological Technology Ltd., Wuhan,
China). For the negative controls, the primary antibody was
replaced with PBS.
Cytogenetic techniques
For chromosome analysis, the retinoblastoma cells
(SO-Rb50) at the 825th passage were used. In brief, the cells in
the exponential growth phase were incubated with 40 mg/ml
colchicine at 37°C for 2 h and harvested by centrifugation (1,000
rpm, 5 min). The single cells were suspended in 8 ml hypotonic
solution of 0.075 mol/l KCl for 20 min at 37°C and then pre-fixed
for 5 min in 1 ml of cold Carnoy’s fixative (methanol:acetic acid,
3:1) by centrifugation (1,000 rpm, 5 min). The cell pellets were
fixed in 8 ml cold Carnoy’s fixative for 15 min. Following
centrifugation, the cells were resuspended in 8 ml Carnoy’s
fixative at 4°C overnight. Slides were prepared using the
conventional drop-splash technique [Lucas et al (19)] and then incubated at 70°C for 2 h.
Slides were incubated in the trypsin solution for 75 sec and then
stained with 5% Giemsa for 10 min. Fifty-six photographed cells at
metaphase on the slides were counted under an Olympus BX40
microscope, and the chromosome karyotype was analyzed according to
the ‘International System for Human Cytogenetic Nomenclature’ (ISCN
1978).
Assay of DNA repair efficiency in
vitro
To analyze the DNA repair efficiency of exogenous
Rb1, SO-Rb50 cells transfected with the pcDNA3.1-Rb1 or
pcDNA3.1-vector were exposed to IR [137Cs (dose rate,
0.67 Gy/min)]. Following incubation for 0, 2.5, 8 and 24 h, the
cells were smeared across a gelatin-coated slide forming a
monolayer of cells. The cells were then fixed with rabbit
monoclonal anti-phospho-H2AX ser-139 antibody (Millipore,
Billerica, MA, USA). The secondary antibody was anti-rabbit Alexa
Fluor 546-conjugated antibody (Invitrogen). Total cells were
counted under a fluorescent microscope (100 objective; Carl Zeiss,
Gottingen, Germany), and cells containing >10 foci were scored
as positive. At least 500 cells were counted. All experiments were
repeated up to four times. Error bars shown are standard errors of
the mean of at least three independent experiments.
Assay of NHEJ and HR by circularization
of linear plasmid substrate in SO-Rb50 cells
The experimental strategy for the NHEJ assay is
depicted in Fig. 3A and B. To
examine the efficiency of NHEJ or HR in the SO-Rb50 cells, the
cells were transfected with the linearized pEGFP-N1 or pEGFP-HR
plasmids digested with EcoR1 or Bcg1, respectively.
If NHEJ or HR occurred, the normal expression of GFP could be
ovserved. The intact pEGFP-N1 was used as the positive control and
treatment with PBS with no plasmid was used as the negative
control. Forty-eight hours later, the cells were harvested and
subjected to two-color fluorescence analysis. The green fluorescent
cells represented the repaired DSBs and the restoration of GFP
expression. The red fluorescent cells represented exogenous DNA
transfection efficiency. For each analysis, 200,000 cells were
processed. The relative NHEJ and HR rejoining activity was obtained
by the ratio of green to red fluorescent cells.
Cell viability assay (MTT)
The viability of the SO-Rb50 cells transfected with
the pcDNA3.1-Rb1 or pcDNA3.1-vector was determined using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(Sigma-Aldrich) assay at three weeks following G418 selection. A
total of 500 cells was seeded in 48-well culture plates. Cell
proliferation was determined using the MTT Cell Proliferation Assay
kit (American Type Culture Collection, Manassas, VA, USA) according
to the manufacturer’s instructions.
Cell cycle assay
The SO-Rb50 cells transfected with the pcDNA3.1-Rb1
or pcDNA3.1-vector were harvested, fixed with 75% ice-cold ethanol
in PBS and kept at 4°C. Prior to analysis, the cells were washed
twice with PBS and then incubated for 30 min in a propidium iodide
staining solution containing 0.05 mg/ml propidium iodide, 1 mM
ethylenediaminetetraacetic acid (EDTA), 0.1% Triton X-100™ and 1
mg/ml ribonuclease A (RNase A) (all from Sigma-Aldrich). The
staining fluorescence intensity was measured using a BD FACSort™
flow cytometer (BD Biosciences, San Jose, CA, USA) and used to
determine the G2/M ratio.
Statistical analysis
Data shown are representative of three independent
experiments with each experiment performed in triplicate. Data are
expressed as the means ± SD. Statistical analyses were performed
using the SPSS for Windows version 10.5 software package. The
differences between mean values were evaluated using the two-tailed
Student’s t-test (for two groups). A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
Rb1 gene mutation and DNA instability in
SO-Rb50 cells
As shown in previous studies, the Rb1 gene is
mutated in SO-Rb50 retinoblastoma cells (20). Our data confirmed that there was a
mutation with a primer pair located in exon 14 and 17 by RT-PCR
analysis (Fig. 1A). The results
of immunofluorescence staining also showed that the Rb1 protein is
not expressed in SO-Rb50 cells (Fig.
1C).
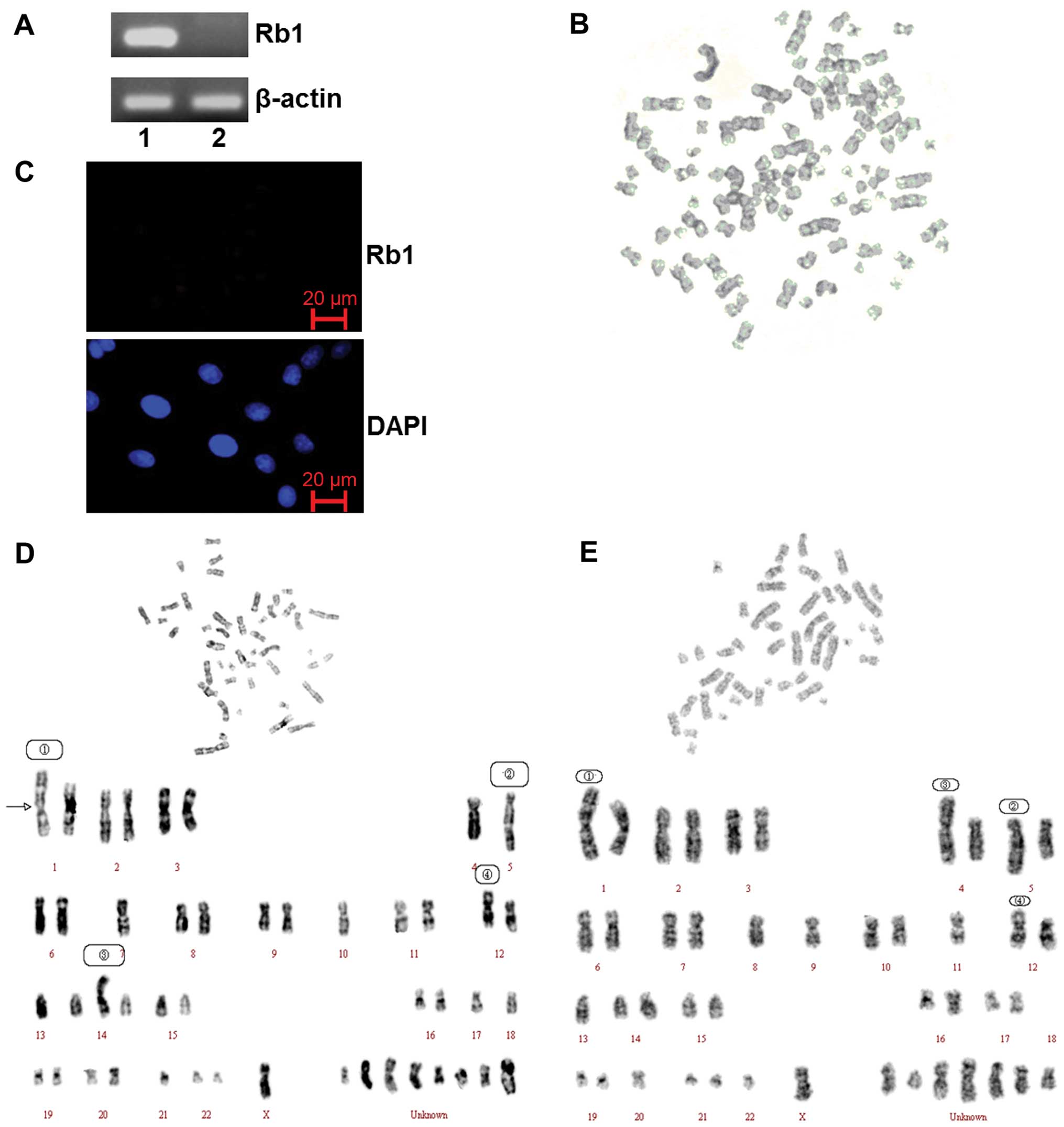 | Figure 1.Gene silencing induced by the mutation
of the Rb1 gene and the genomeicinstability of SO-Rb50 cells
without Rb1 gene function. (A) RT-PCR analysis of Rb1 mRNA in
HEK293 cells (lane 1) and SO-Rb50 cells (lane 2). (B) Karyotype of
a polyploidy cell. (C) Rb1 expression of SO-Rb50 cells was detected
by immunofluorescence staining. (D) Karyotype analysis of SO-Rb50
cells. The numerical chromosomal aberrations included: −1, −4,
−5×2, −7, −10, −11, −12, −13, −17, −18, −21, and 8 unknown
chromosomes. The structural aberrations included: 1, der(1)
t(1:?)(q33:?); 2, der(5) t(5:15)(q35:q11); 3, der(14)
t(14:?)(p11:?); 4, der(12) t(12:?)(p13:?). (E) Karyotype analysis
of SO-Rb50 cells. The numerical aberrations included: −1, −4, −5,
−8, −9, −11, −12, −13, −18×2, −20, −22, -X, and 7 unknown
chromosomes. The structural aberrations included: 1, der(1)
t(1:?)(q33:?); 2, der(5) t(5:15)(q35:q11); 3, der(4) t(4:?)(p14:?);
4, der(12) t(12:?)(p13:?). |
In order to confirm the data from previous studies
showing SO-Rb50 cells display obviously chromosomal instability
(8), we performed karyotype
analysis of 825th passage SO-Rb50 cells. Our data revealed both
numerical and structural chromosomal aberrations in the SO-Rb50
cells. The chromosome assay showed that the number of chromosomes
in the cells ranged from 22 to 93. The majority of the cells (47%)
had chromosome numbers of <46 and the metaphase spread with a
normal diploid number (2N=46) was approximately 30%. The cells with
chromosome numbers of >46 accounted for 23%. The type of
chromosomal aberrations included chromosome breakage, shift,
rearrangement, deletion, repeat, etc. The variation of several
chromosomes was so severe that they could not be recognized
(Fig. 1D and E). Polyploid cells
could also be observed occasionally (Fig. 1B).
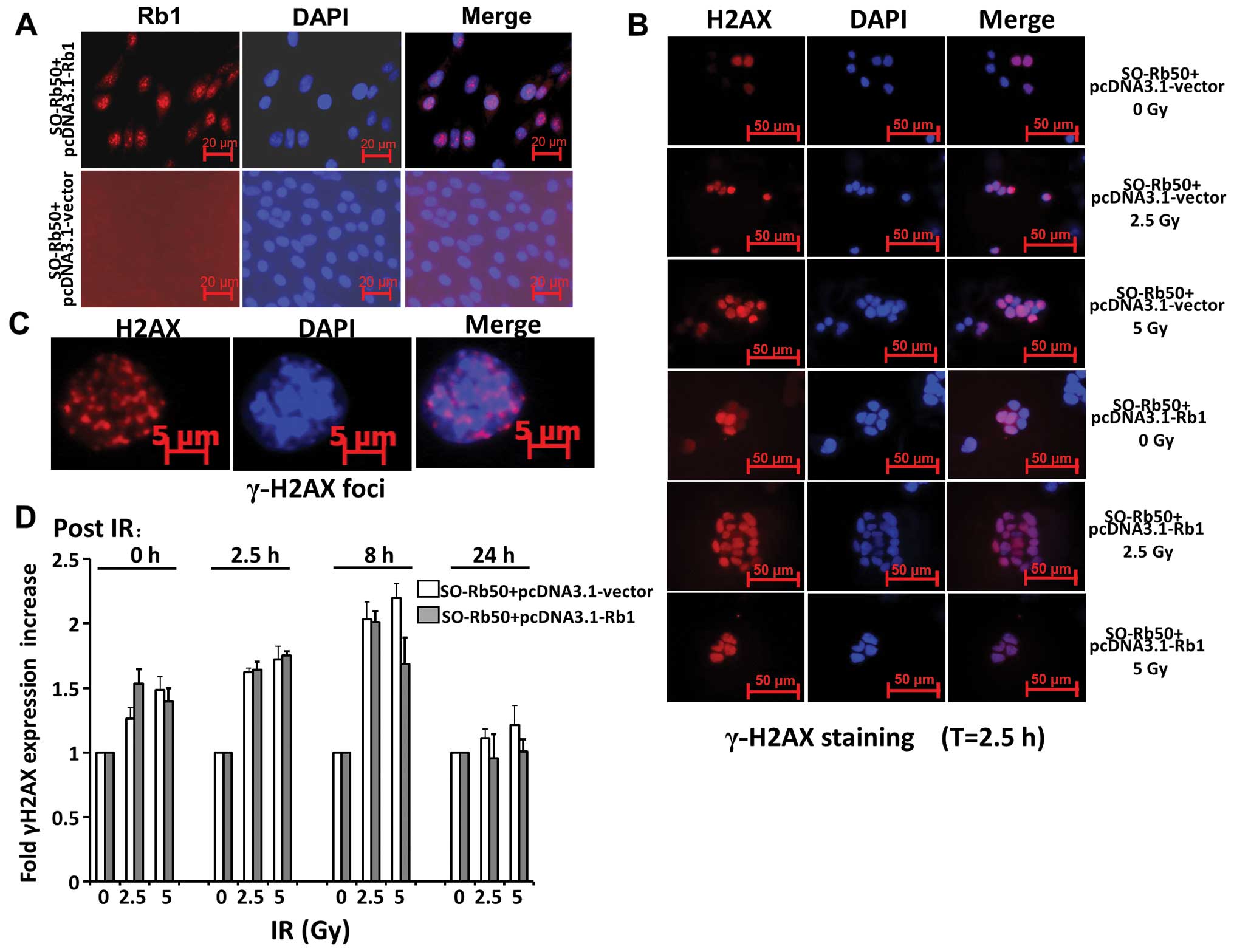
Rb1 does not affect the repair of DNA
DSBs following exposure to IR
In order to elucidate the mechanism behind the
chromosomal instability of retinoblastoma cells, we first
investigated the effect of Rb1 on the repair of DNA DSBs induced by
exposure to IR. The SO-Rb50 cells were transfected with the
pcDNA3.1-Rb1 plasmid, expressing WT Rb1 or the pcDNA3.1-vector.
Fig. 2A shows that exogenous Rb1
is highly expressed in the cytoplasm of SO-Rb50 cells following
transfection with the pcDNA3.1-Rb1 plasmid. Following exposure to
0, 2.5 or 5 Gy IR, the SO-Rb50 cells with and without exogenous Rb1
were fixed at different time points, 0, 2.5, 8 and 24 h
post-damage, and stained with γ-H2AX, a commonly used in
situ marker of DNA DSBs. Our data revealed that there was a
significant increase in γ-H2AX expression following exposure to 2.5
or 5 Gy IR compared with the controls (0 Gy) (set to 100%) in the
SO-Rb50 cells transfected with the pcDNA3.1-Rb1 or pcDNA3.1-vector
during the first 8 h. However, exogenous Rb1 did not affect γ-H2AX
expression, as shown by images of the cell population taken
following exposure to equal doses of IR (P>0.05) (Fig. 2D). These results are in agreement
with those from a previous study (14). These data suggest that Rb1 does
not affect the repair of DNA DSBs following exposure to IR.
Exogenous Rb1 does not affect NHEJ, but
promotes HR of SO-Rb50 cells
A previous study reported that NHEJ is a rapid
process, which can be completed in approximately 30 min, while HR
is much slower and takes 7 h or longer to complete (21). Moreover, the repair of IR-induced
DNA DSBs is catalyzed predominantly by the NHEJ pathway (22). Therefore, we further wished to
assess NHEJ and HR activity, separately, in SO-Rb50 cells, as
described in Materials and methods. For NHEJ assay, DNA substrate
with either complementary ends was prepared by linearizing pEGFP-N1
with EcoRI. The cleavage between the promoter and the GFP
reporter gene thereby prevents the expression of the reporter in
vivo (Fig. 3A). Intracellular
recircularization of the linearized DNA through NHEJ
repair-mediated end rejoining allows for the expression of GFP,
which was then assayed by flow cytometry analysis. The rejoining
levels in the cells revealed that exogenous Rb1 had no differential
effect on NHEJ (Fig. 3C).
For HR assay, we constructed the recombination
substrate, pEGFP-HR, which contains two GFP cDNA fragments after
the promoter, GFP1 in +1 to +400 bp; GFP2 covers the whole cDNA.
There is an inserter between GFP1 and GFP2, which results in the
abnormal expression of GFP. The DNA substrate with HR ends was
prepared by linearizing pEGFP-HR with Bcg1. If HR occurs,
GFP is expressesed by sharing extensive sequence homology (Fig. 3B). As shown in Fig. 3D, the level of HR in the cells
expressing exogenous Rb1 was significantly enhanced by 2.46-fold
compared with the control cells (P<0.01). These findings provide
direct evidence that Rb1 enhances HR, but does not affect NHEJ.
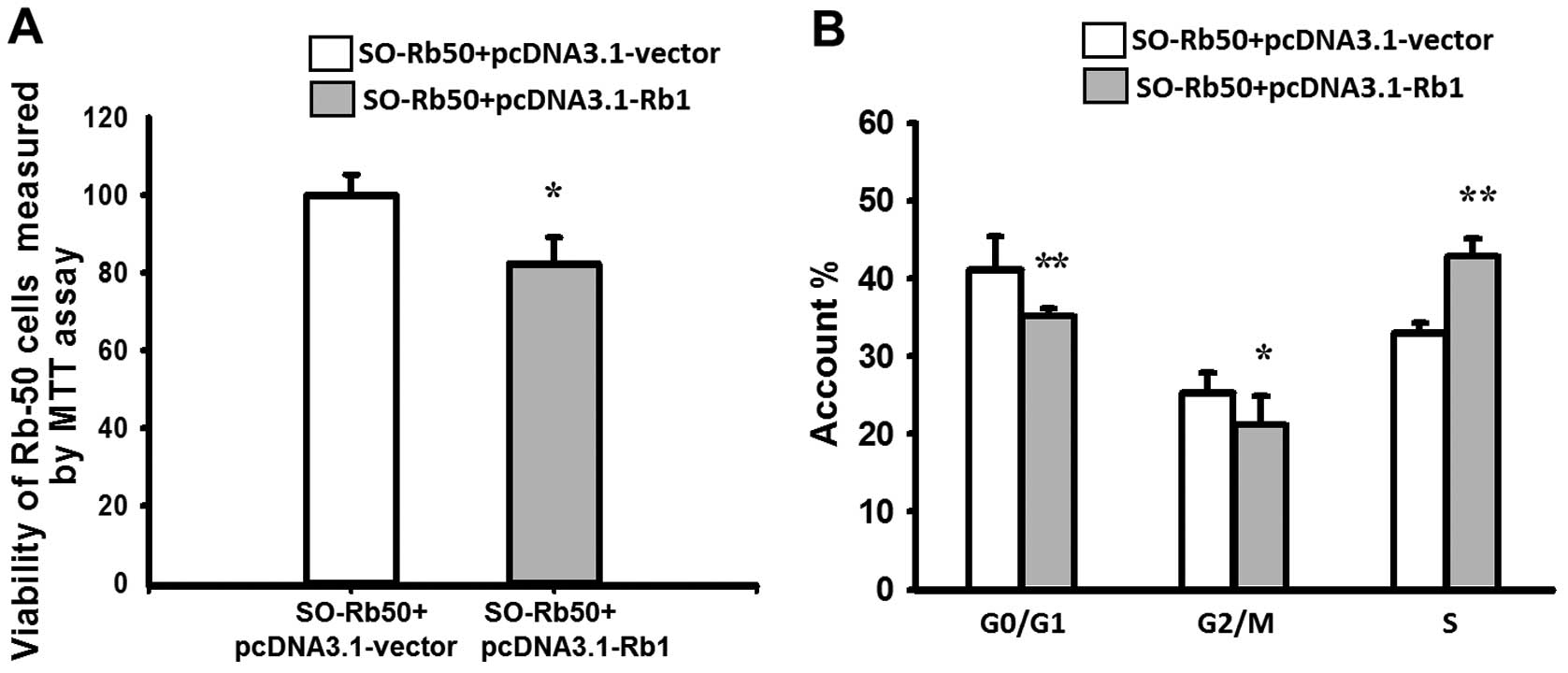
Exogenous Rb1 affects cell viability and
the cell cycle of SO-Rb50 cells in vitro
HR is most active during the late S and G2 phase of
the cell cycle. Therefore, we examined the cell cycle of SO-Rb50
cells with and without exogenous Rb1. The results of MTT assay
revealed that the viability of SO-Rb50 cells transfected with
pcDNA3.1-Rb1 was reduced by 17.46±2.66% compared with the control
group, which demonstrated that exogenous Rb1 significantly
inhibited the growth of SO-Rb50 cells (P<0.01) (Fig. 4A). Flow cytometric analysis
revealed that, compared with the control group, the percentage of
SO-Rb50 cells with exogenous Rb1 in the G0/G1 phase and G2/M phase
was decreased from 41.85±4.30 to 35.69±1.54% (t=3.665, P<0.01),
and from 25.23±2.77 to 21.23±4.00% (t=2.251, P<0.05)
respectively, and that in the S phase was increased from 32.92±1.48
to 43.08±2.23% (t=−10.396, P<0.001) (Fig. 4B). These results indicate that
exogenous Rb1 promotes the arrest of cells in the S phase of the
cell cycle, thereby inhibiting SO-Rb50 cell proliferation.
Discussion
A growing body of evidence suggests that the genomic
instability of retinoblastoma is observed both in vitro and
in vivo (9,10,23). However, the effect of Rb1 on the
DNA DSB repair process remains unclear. Whether the particular DNA
DSB repair sub-pathway is controlled by Rb1 is also unknown. To
address these issues, we first verified the loss of Rb1 in SO-Rb50
retinoblastoma cells. Our results indicated that there was a
mutation with a primer pair located in exon 14 and 17 by RT-PCR
(Fig. 1A). The staining of Rb1
confirmed that Rb1 was not expressed in the SO-Rb50 cells (Fig. 1C). These data are consistent with
those from a previous report (24).
Moreover, we further demonstrated the genomic
instability of SO-Rb50 cells by karyotype analysis. As shown in a
previous study, Feng et al (9) screened the promoter and 27 exons of
the Rb1 gene in SO-Rb50 cells using polymerase chain
reaction-single-strand conformation polymorphism (PCR-SSCP) and
southern blot analysis at different passages. They found new
mutation events that occurred in exons 23, 24 and 25 in consecutive
passages, compared to the 451st passage. G-banding and karyotype
analysis further proved that there were chromosomal aberrations,
which were observed in the same passage of different cell strains
of SO-Rb50 cells. We analyzed the SO-Rb50 cells at the 825th
passage. Our data showed that 47% of the SO-Rb50 cells displayed
both numerical and structural chromosomal aberrations. Only
approximately 30% of the cells had a normal diploid number (2N=46).
The heteromorphosis of several chromosomes was too severe to be
recognized (Fig. 1B, D and E).
These results strongly suggest that the mutation of Rb1 causes
dynamic chromosomal alterations during long-term culture in
vitro. Similarly, a study on sporadic unilateral retinoblastoma
tumors performed by Ganguly et al (23) indicated that tumors harbored novel
regions of amplification at 1q44, 3p25, 11q14, 11q25, 14q23, 15q21,
16p13, 17p11.2, 19q13 and 20q13, while regions of loss included
6q22, 7q21 and 21q2.
DNA DSB repair plays a key role in genomic
stability. IR-induced damage can result in DNA DSBs in cells.
Therefore, we first examined whether exogenous Rb1 affects the
repair of DNA DSBs induced by IR in SO-Rb50 cells. The stable cells
expressing WT Rb1 and the control cells were treated with 2.5 and 5
Gy radiation. At different time points, the cells were fixed and
stained with γ-H2AX. The formation of γ-H2AX foci are known to bind
at sites of DNA damage and more specifically at DNA DSBs (25,26). As shown by the foci formation in
cells, we found that IR treatment significantly induced DNA DSBs in
SO-Rb50 cells both with and without exogenous Rb1. However,
exogenous Rb1 had no significant effects upon γ-H2AX foci
intensity, as shown by images of the cell population taken at
following exposure to equal doses of IR. Both cells with WT Rb1 and
the empty vector displayed a similar increase in staining intensity
during the first 8 h post-damage and a similar kinetic decrease in
intensity from 8 to 24 h (Fig.
2D). These results are in agreement with those from a previous
study (14), which indicated that
Rb1 had no significant effects on direct DNA repair following
IR-induced damage in SO-Rb50 cells. Moreover, it is well
established that NHEJ is the main pathway for the repair of the
majority of IR-induced DNA DSBs throughout the cell cycle (27–30). Our results suggest that Rb1 does
not affect the NHEJ sub-pathway.
As described as above, DNA DSB repair involves NHEJ
and HR. The error-prone NHEJ pathway rapidly and promiscuously
rejoins the ends of broken chromosomes while HR repair uses a
homologous template in a sister chromatid or homologous chromosome
to perform error-free repair (31–33). Rb1 does not affect NHEJ.
Therefore, we hypothesized that the chromosomal aberration in
SO-Rb50 Rb1-deficient cells is induced by preventing error-free HR.
In order to demonstrate this hypothesis, we used recircularization
assay with the linearized pEGFP-N1 and pEGFP-HR substrate to
examine the role of Rb1 in NHEJ and HR. Consistent with our results
on IR-induced damage (Fig. 2D),
Rb1 did not affect NHEJ (Fig.
3C). Conversely, in the presence of WT Rb1, the level of HR in
the cells expressing exogenous Rb1 was significantly enhanced by
2.46-fold compared with the control cells (Fig. 3D). Therefore, the deficient HR may
result in chromosomal instability and chromosomal aberration in
SO-Rb50 cells.
HR is most active during the late S and G2 phases of
the cell cycle. Therefore, we investigated the effect of Rb1 on the
cell cycle and viability of SO-Rb50 cells. Our results revealed
that WT Rb1 reduced the viability of SO-Rb50 cells and
significantly promoted the arrest of cells in the S phase of the
cell cycle (Fig. 4), thereby
inhibiting SO-Rb50 cell proliferation. Our results are consistent
with those from previous studies, showing that Rb1 regulates the
cell cycle, differentiation, growth and apoptosis (5–7).
It is now clear that the complex of Rb1 binding transcript factor,
E2F, plays a crucial role in regulating the initiation of DNA
replication. A number of previous studies have demonstrated that
E2F regulates many downstream target genes that are involved in
cell cycle progression and DNA replication, such as cyclin A,
cyclin E, cdc2 and cdk2, proliferating cell nuclear antigen (PCNA),
mini-chromosome maintenance-7 (MCM-7), topoisomerase IIa and
thymidine kinase (34,35). It is possible that the loss of
Rb1, which occurs concomitantly with the vast target gene
deregulation, facilitates the bypass of the cell cycle checkpoint,
which is one of the mechanisms by which a tumor can occur.
In conclusion, this study provides evidence that
chromosomal aberrations occur in Rb1-deficient SO-Rb50
retinoblastoma cells. The assay of DNA DSB repair demonstrated that
Rb1 does not affect NHEJ and significantly promotes HR. To our
knowledge, this is the first study revealing the mechanism of
action of Rb1, namely the regulation of the sub-pathway of DNA DSB
repair. An in depth understanding of the mechanism by which the Rb1
tumor suppressor gene regulates the growth of tumor cells may
provide us with valuable information for the development of novel
methods of therapeutic intervention and treatment against
retinoblastoma tumors.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation (Project:
30970923).
References
|
1.
|
Tamboli A, Podgor MJ and Horm JW: The
incidence of retinoblastoma in the United States: 1974 through
1985. Arch Ophthalmol. 108:128–132. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Shields CL and Shields JA: Diagnosis and
management of retinoblastoma. Cancer Control. 11:317–327. 2004.
|
|
3.
|
Chintagumpala M, Chevez-Barrios P, Paysse
EA, Plon SE and Hurwitz R: Retinoblastoma: review of current
management. Oncologist. 12:1237–1246. 2007. View Article : Google Scholar
|
|
4.
|
Melamud A, Palekar R and Singh A:
Retinoblastoma. Am Fam Physician. 73:1039–1044. 2006.
|
|
5.
|
Lim Z and Quah BL: Unilateral
retinoblastoma in an eye with Peters anomaly. J AAPOS. 14:184–186.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kanber D, Berulava T, Ammerpohl O, Mitter
D, Richter J, Siebert R, et al: The human retinoblastoma gene is
imprinted. PLoS Genet. 5:e10007902009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Wilson PF, Nagasawa H, Fitzek MM, Little
JB and Bedford JS: G2-phase chromosomal radiosensitivity of primary
fibroblasts from hereditary retinoblastoma family members and some
apparently normal controls. Radiat Res. 173:62–70. 2010. View Article : Google Scholar
|
|
8.
|
Knudsen ES, Sexton CR and Mayhew CN: Role
of the retinoblastoma tumor suppressor in the maintenance of genome
integrity. Curr Mol Med. 6:749–757. 2006.PubMed/NCBI
|
|
9.
|
Feng G, Li H, Yi Y, Zheng J, Zhang Q, Wang
X and Du C: Study on the dynamic changes of retinoblastoma gene of
SO-Rb50 cell line. Yan Ke Xue Bao. 17:111–113. 2001.(In
Chinese).
|
|
10.
|
Li H, Feng G, Fang Y, Li Y, Zheng J and Yi
Y: An investigation on chromosome aberration of SO-Rb50 cloned cell
strains. Yan Ke Xue Bao. 14:220–223. 1998.(In Chinese).
|
|
11.
|
Attwooll C, Lazzerini DE and Helin K: The
E2F family: specific functions and overlapping interests. EMBO J.
23:4709–4716. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Chen J, Zhu F, Weaks RL, Biswas AK, Guo R,
Li Y and Johnson DG: E2F1 promotes the recruitment of DNA repair
factors to sites of DNA double-strand breaks. Cell Cycle.
10:1287–1294. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Guo R, Chen J, Zhu F, Biswas AK, Berton
TR, Mitchell DL and Johnson DG: E2F1 localizes to sites of
UV-induced DNA damage to enhance nucleotide excision repair. J Biol
Chem. 285:19308–19315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Bosco EE and Knudsen ES: Differential role
of RB in response to UV and IR damage. Nucleic Acids Res.
33:1581–1592. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Modesti M and Kanaar R: Homologous
recombination: from model organisms to human disease. Genome Biol.
2:reviews10142001. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
West SC: Molecular views of recombination
proteins and their control. Nat Rev Mol Cell Biol. 4:435–445. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lieber MR, Ma Y, Pannicke U and Schwarz K:
Mechanism and regulation of human non-homologous DNA end-joining.
Nat Rev Mol Cell Biol. 4:712–720. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Zhuang J, Jiang G, Willers H and Xia F:
Exonuclease function of human Mre11 promotes deletional
nonhomologous end joining. J Biol Chem. 284:30565–30573. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Lucas JM, Bryans M, Lo K, Wilkie NM,
Freshney M, Thornton D and Lang JC: The FGF-4 promoter is required
for transformation and is active in both embryonal and somatic
cells. Oncol Res. 6:139–149. 1994.PubMed/NCBI
|
|
20.
|
Qu B, Zhuo Z, Yi Y, Feng G, Zheng J, Liang
Q and Li Y: Dynamic investigation on chromosome aberration of a
human retinoblastoma cell line So-Rb50. Yan Ke Xue Bao. 9:38–39.
37:1993
|
|
21.
|
Mao Z, Bozzella M, Seluanov A and
Gorbunova V: Comparison of nonhomologous end joining and homologous
recombination in human cells. DNA Repair (Amst). 7:1765–1771. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Boeckman HJ, Trego KS and Turchi JJ:
Cisplatin sensitizes cancer cells to ionizing radiation via
inhibition of nonhomologous end joining. Mol Cancer Res. 3:277–285.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ganguly A, Nichols KE, Grant G, Rappaport
E and Shields C: Molecular karyotype of sporadic unilateral
retinoblastoma tumors. Retina. 29:1002–1012. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Wang X, Zeng F, Xu Z, Zheng Y and Wang L:
Detection of tumor suppressor gene and oncogene in So-Rb50 human
retinoblastoma cell line. Yan Ke Xue Bao. 9:34–37. 1993.PubMed/NCBI
|
|
25.
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Stucki M, Clapperton JA, Mohammad D, Yaffe
MB, Smerdon SJ and Jackson SP: MDC1 directly binds phosphorylated
histone H2AX to regulate cellular responses to DNA double-strand
breaks. Cell. 123:1213–1226. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Hinz JM, Yamada NA, Salazar EP, Tebbs RS
and Thompson LH: Influence of double-strand-break repair pathways
on radiosensitivity throughout the cell cycle in CHO cells. DNA
Repair (Amst). 4:782–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Branzei D and Foiani M: Regulation of DNA
repair throughout the cell cycle. Nat Rev Mol Cell Biol. 9:297–308.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Helleday T, Lo J, van Gent DC and
Engelward BP: DNA double-strand break repair: from mechanistic
understanding to cancer treatment. DNA Repair (Amst). 6:923–935.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Mahaney BL, Meek K and Lees-Miller SP:
Repair of ionizing radiation-induced DNA double-strand breaks by
non-homologous end-joining. Biochem J. 417:639–650. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Haber JE: Partners and pathwaysrepairing a
double-strand break. Trends Genet. 16:259–264. 2000.PubMed/NCBI
|
|
32.
|
Karran P: DNA double strand break repair
in mammalian cells. Curr Opin Genet Dev. 10:144–150. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Thompson LH and Schild D: Recombinational
DNA repair and human disease. Mutat Res. 509:49–78. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Chellappan SP, Hiebert S, Mudryj M,
Horowitz JM and Nevins JR: The E2F transcription factor is a
cellular target for the RB protein. Cell. 65:1053–1061. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Stevaux O and Dyson NJ: A revised picture
of the E2F transcriptional network and RB function. Curr Opin Cell
Biol. 14:684–691. 2002. View Article : Google Scholar : PubMed/NCBI
|