Introduction
Atrial fibrillation (AF) represents the most common
form of sustained cardiac arrhythmia with an estimated prevalence
of 1% in the general population (1). The incidence of AF increases
markedly with advancing age, ranging from less than 1% in
individuals under 60 years of age to approximately 10% of those
over 80 years (2). According to
the Framingham Heart Study, the lifetime risk for development of AF
is approximately 25% for individuals who have reached the age of 40
years (3). AF accounts for
substantially increased cardiovascular morbidity and mortality. It
is associated with an approximately 5-fold increase in the risk of
stroke, and more than 15% of all strokes are ascribed to this
disordered heart rhythm (4). The
risk of AF-related thromboembolism also increases strikingly with
age, rising from 1.5% at age 50–59 years to 23.5% at age 80–89
years (4). The total death rate
is roughly doubled among patients with AF compared with people in
normal sinus rhythm (5). AF is
also responsible for compromised exercise performance, degraded
quality of life, impaired cognitive function or dementia,
tachycardia-induced cardiomyopathy, and left ventricular
dysfunction or even congestive heart failure, conferring a large
economic burden on national healthcare system worldwide (6). Despite the significant clinical
importance, the molecular mechanisms involved in the pathogenesis
of AF remain poorly understood.
Traditionally, AF has been regarded as a
complication attributed to miscellaneous adverse cardiac or
systemic conditions, including hypertension, coronary artery
disease, rheumatic heart disease, valvular heart disease, pulmonary
heart disease, cardiomyopathy, cardiac sur gery, diabetes mellitus
type 2, obstructive sleep apnea, hyperthyroidism, and electrolyte
imbalance (1). However, in 30–45%
of AF patients, an underlying cause cannot be identified by routine
procedures, and such AF is termed ‘idiopathic’ or ‘lone’ (1), of which at least 15% have a positive
family history, a condition classified as familial AF (7). Increasing evidence has demonstrated
the familial aggregation of AF and enhanced susceptibility to AF in
the close relatives of patients with AF, suggesting that genetic
risk factors play a pivotal role in the initiation and maintenance
of AF in a subset of cases (8–14).
Genome-wide linkage analyses with microsatellite markers mapped
susceptibility loci for AF on human chromosomes 10q22, 6q14–16,
11p15.5, 5p13 and 5p15, of which AF-causative mutations in 2 genes,
including KCNQ1 on chromosome 11p15.5 and NUP155 on
chromosome 5p13, were identified and functionally characterized
(15–20). The genetic screening of candidate
genes has revealed a great number of AF-associated genes, including
KCNE2, KCNE3, KCNE5, KCNH2, KCNJ2, KCNA5, SCN5A, SCN1B, SCN2B,
SCN3B, NPPA, GJA1, GJA5, GATA4, GATA5 and GATA6
(21–44). Nevertheless, AF is of substantial
genetic heterogeneity and the genetic basis for AF in an
overwhelming majority of patients remains unclear.
Recent studies have highlighted the essential role
of the cardiac sodium channel complex in the generation and
propagation of the cardiac action potential. The complex comprises
multiple protein factors, including the pore-forming α-subunit
encoded by SCN5A, auxiliary β-subunits, and other accessory
proteins, such as MOG1, ankyrin-G, FHF1B, Fyn and PTPH1 (45). In humans, 4 sodium channel
β-subunits (β1 to β4, encoded by SCN1B to SCN4B),
which are expressed in both atrial and ventricular cardiomyocytes,
have been identified thus far. They share a common protein topology
with an extracellular immunoglobulin-like domain, a single
transmembrane spanning segment, and an intracellular C-terminal
domain, and are implicated in the trafficking of sodium channels to
plasma membranes, the modulation of channel gating and voltage
dependence, and play a role in cell adhesion and recruitment of
cytosolic proteins such as ankyrin-G (45). Mutations in SCN5A, SCN1B,
SCN2B and SCN3B have been implicated in AF (28–33), which prompts us to hypothesize
that SCN4B is another gene contributing to AF.
To examine this hypothesis, the coding exons and
splice sites of SCN4B were sequenced in patients with
familial AF in contrast to ethnically matched control individuals,
and the functional effect of the mutated SCN4B gene was
analyzed in silico by using the online program,
MutationTaster.
Materials and methods
Study subjects
A cohort of 170 unrelated index patients with
familial AF identified among the Han Chinese population was
recruited. The available relatives of the probands harboring the
identified mutations were also enrolled in this study. A total of
200 unrelated ethnically matched healthy individuals used as the
controls were enlisted. All the participants underwent evaluation
by medical history, physical examination, electrocardiography and
echocardiography. Peripheral venous blood specimens were prepared
and clinical data including medical records, electrocardiogram and
echocardiography reports were collected. The study subjects were
clinically classified using a consistently applied set of
definitions (7,38). In brief, AF was diagnosed by a
standard 12-lead electrocardiogram demonstrating no P waves and
irregular R-R intervals irrespective of clinical symptoms. Lone AF
was defined as AF occurring in individuals under the age of 60
without other cardiac or systemic diseases by physical examination,
electrocardiogram, transthoracic echocardiogram and extensive
laboratory tests. Familial AF was defined as that present in a
family with more than one first- or second-degree relative affected
with AF. Relatives were classified as ‘unaffected’ if they were
asymptomatic and had a normal electrocardiogram. Paroxysmal AF was
defined as AF lasting more than 30 sec that terminated
spontaneously. Persistent AF was defined as AF lasting more than 7
days and requiring either pharmacological therapy or electrical
cardioversion for termination. AF that was refractory to
cardioversion or that was allowed to continue was classified as
permanent. The study protocol was reviewed and approved by the
local Institutional Ethics Committee and written informed consent
was obtained from all participants prior to investigation.
Genetic analysis
Genomic DNA from all participants was extracted from
blood lymphocytes with the Wizard Genomic DNA Purification kit
(Promega, Madison, WI, USA). Initially, the coding exons and
intron/exon boundaries of the SCN4B gene were sequenced in
170 unrelated index patients with familial AF. Subsequently,
genotyping for SCN4B in the available relatives of the
probands carrying the identified mutations and 200 ethnically
matched unrelated healthy individuals used as the controls was
performed. The reference genomic DNA sequence of SCN4B was
derived from GenBank (accession no. NG_011710). With the aid of
online Primer3 software (http://frodo.wi.mit.edu), the primer pairs used to
amplify the coding regions and splice junctions of SCN4B by
polymerase chain reaction (PCR) were designed as shown in Table I. PCR was carried out using
HotStar Taq DNA Polymerase (Qiagen, Hilden, Germany) on a PE 9700
Thermal Cycler (Applied Biosystems, Foster, CA, USA) with standard
conditions and concentrations of reagents. Amplified products were
purified with the QIAquick Gel Extraction kit (Qiagen). Both
strands of each PCR product were sequenced with a
BigDye® Terminator version 3.1 Cycle Sequencing kit
(Applied Biosystems) under an ABI PRISM 3130XL DNA Analyzer
(Applied Biosystems). The sequencing primers were those designed
previously for specific region amplifications. DNA sequences were
viewed and analyzed with the DNA Sequencing Analysis Software
version 5.1 (Applied Biosystems). The variant was validated by
resequencing of an independent PCR-generated amplicon from the same
subject and met the quality control threshold with a call rate
>99%. Additionally, an identified variant was searched in the
single nucleotide polymorphism (SNP) database from the National
Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/SNP) to confirm the
novelty.
 | Table I.The intronic primers used to amplify
the coding exons and exon-intron boundaries of SCN4B. |
Table I.
The intronic primers used to amplify
the coding exons and exon-intron boundaries of SCN4B.
| Exon | Forward primer
(5′→3′) | Reverse primer
(5′→3′) | Size (bp) |
|---|
| 1 | CTC, TCT, GCC, CGC,
TAA, CTT, TC | CTA, TGA, ACC, AGG,
CAG, GAA, CC | 371 |
| 2 | TTG, GCA, CTG, AGG,
GTG, ATA, GA | CAG, AAG, GGA, CCA,
GAG, CGT, AG | 372 |
| 3 | GAG, GAC, CCC, GAT,
TCT, TTC, TC | AAA, CAC, CAA, CAC,
GGT, CCA, TT | 387 |
| 4 | TGA, TAG, ATG, CCA,
TGC, TCT, GC | GGG, GTA, GAT, GAG,
AGG, GTG, GT | 382 |
| 5 | TCT, GTA, GAA, GGC,
CAG, GGA, GA | GGC, AGG, ACT, CTG,
GTT, TCT, TG | 361 |
Alignment of multiple SCN4B protein
sequences across species
The multiple SCN4B protein sequences across various
species were aligned using the online program, MUSCLE version 3.6
(http://www.ncbi.nlm.nih.gov/homologene?cmd=retrieve&dopt=multipleAlignment&list_uids=18384).
Prediction of the causative potential of
a SCN4B sequence variation
The disease-causing potential of a SCN4B
sequence variation was predicted using MutationTaster (http://www.mutationtaster.org), which automatically
gave a probability for the variation to be either a pathogenic
mutation or a benign polymorphism. Notably, the P-value is the
probability of the prediction rather than the probability of error
as used in t-test statistics, i.e., a value close to 1 indicates a
high ‘security’ of the prediction.
Statistical analysis
Data are expressed as the means ± SD. Continuous
variables were examined for normality of distribution and the
unpaired Student’s t-test was used for the comparison of numeric
variables between 2 groups. Comparison of the categorical variables
between 2 groups was performed using Pearson’s χ2 or
Fisher’s exact tests when appropriate. A two-tailed P-value
<0.05 was considered to indicate a statistically significant
difference.
Results
Characteristics of the study
population
A cohort of 170 unrelated patients with familial AF
and a total of 200 ethnically matched unrelated healthy individuals
used as the controls were registered and clinically evaluated. None
of them had apparent traditional risk factors for AF. There were no
significant differences between the patient and control groups in
baseline characteristics including age, gender, body mass index,
blood pressure, fasting blood glucose levels, serum lipid levels,
left atrial dimension, left ventricular ejection fraction, heart
rate at rest, as well as life style (data not shown). In the
present study, 12 patients were also diagnosed with hyper-tension
in accordance to the criterion that the average systolic or
diastolic blood pressure (2 readings performed after 5 min of rest
in the sitting position) was ≥140 or 90 mmHg, respectively, but at
the time of initial diagnosis of AF, their blood pressures were
normal. The baseline clinical characteristics of the 170 patients
with familial AF are summarized in Table II.
| Table II.The baseline clinical characteristics
of the 170 pro-bands with familial atrial fibrillation. |
Table II.
The baseline clinical characteristics
of the 170 pro-bands with familial atrial fibrillation.
| Parameter | Statistic |
|---|
| Age at initial
diagnosis of atrial fibrillation (years) | 44±9 |
| Age at present
study (years) | 49±8 |
| Male (n, %) | 112 (66) |
| Body mass index
(kg/m2) | 23±3 |
| Left ventricular
ejection fraction (%) | 61±5 |
| Left atrial
diameter (mm) | 37±4 |
| Paroxysmal atrial
fibrillation (n, %) | 98 (58) |
| Persistent atrial
fibrillation (n, %) | 51 (30) |
| Permanent atrial
fibrillation (n, %) | 21 (12) |
| Positive family
history of atrial fibrillation (n, %) | 170 (100) |
| History of
cardioversion (n, %) | 88 (52) |
| Catheter-based
ablation for atrial fibrillation (n, %) | 76 (45) |
| History of
thromboembolic stroke (n, %) | 24 (14) |
| History of
pacemaker (n, %) | 9 (5) |
| Systolic blood
pressure (mmHg) | 128±10 |
| Diastolic blood
pressure (mmHg) | 80±5 |
| Fasting blood
glucose (mmol/l) | 6±1 |
| Total cholesterol
(mmol/l) | 5±1 |
| Aspirin (n, %) | 37 (22) |
| Warfarin (n,
%) | 78 (46) |
| Amiodarone (n,
%) | 92 (54) |
| β-blocker (n,
%) | 33 (19) |
| Calcium channel
blocker (n, %) | 27 (16) |
| Digitalis
(n, %) | 43 (25) |
SCN4B mutations
Direct sequencing of the entire coding sequences and
flanking intronic sequences of the SCN4B gene was performed
following PCR amplification of genomic DNA from each of the 170
unrelated patients with familial AF. Two heterozygous SCN4B
mutations were identified in 2 out of the 170 patients, with a
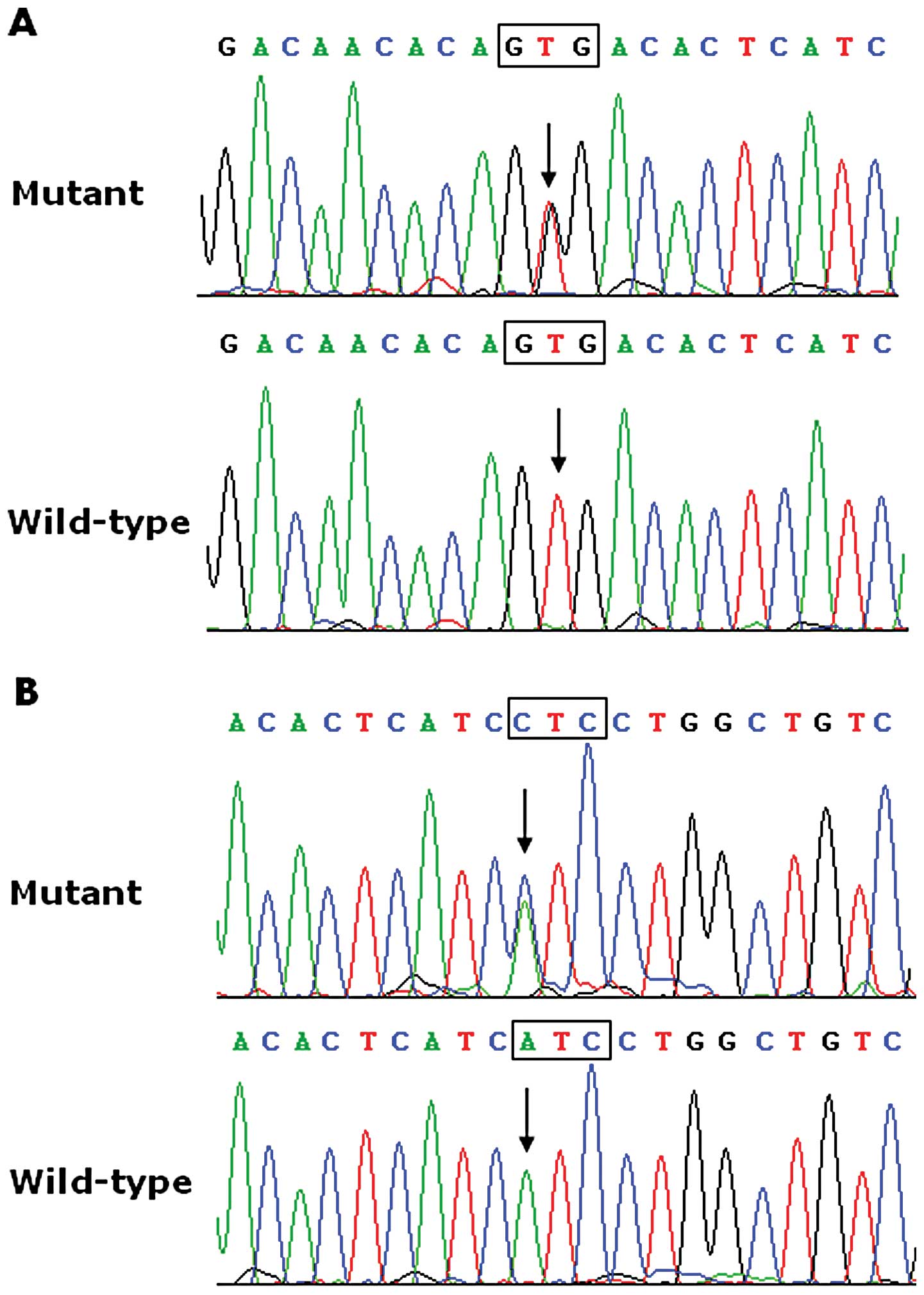
mutational prevalence of approximately 1.18%. Specifically, a
substitution of guanine (G) for thymine (T) in the second
nucleotide of codon 162 (c.485T>G), predicting the transition of
valine (V) into glycine (G) at amino acid 162 (p.V162G), was
identified in the proband from family 1. A replacement of adenine
(A) by cytosine (C) in the first nucleotide of codon 166
(c.496A>C), equivalent to a transversion of isoleucine (I) into
leucine (L) at amino acid 166 (p.I166L), was identified in the
proband from family 2. The sequence chromatograms showing the
detected heterozygous SCN4B mutations in contrast to the
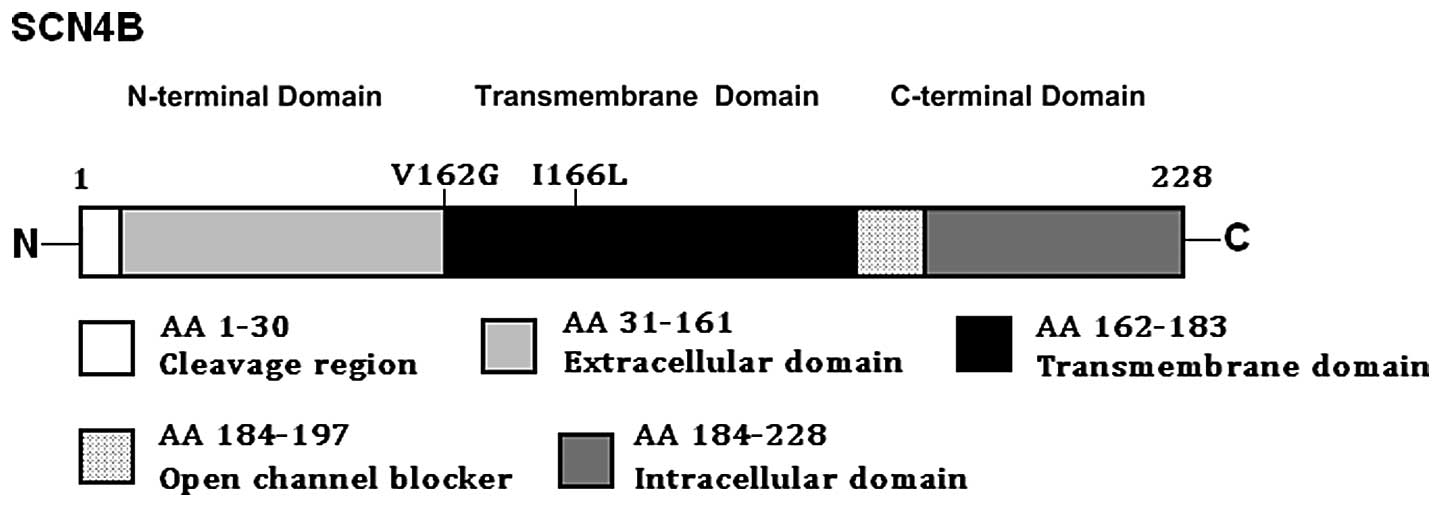
corresponding control sequences are shown in (Fig. 1). A schematic linear topology of
the SCN4B-encoded β4 subunit indicating the locations of the
mutations identified in AF patients is presented in (Fig. 2). The missense mutations were not
found in the 400 control alleles nor were they reported in the SNP
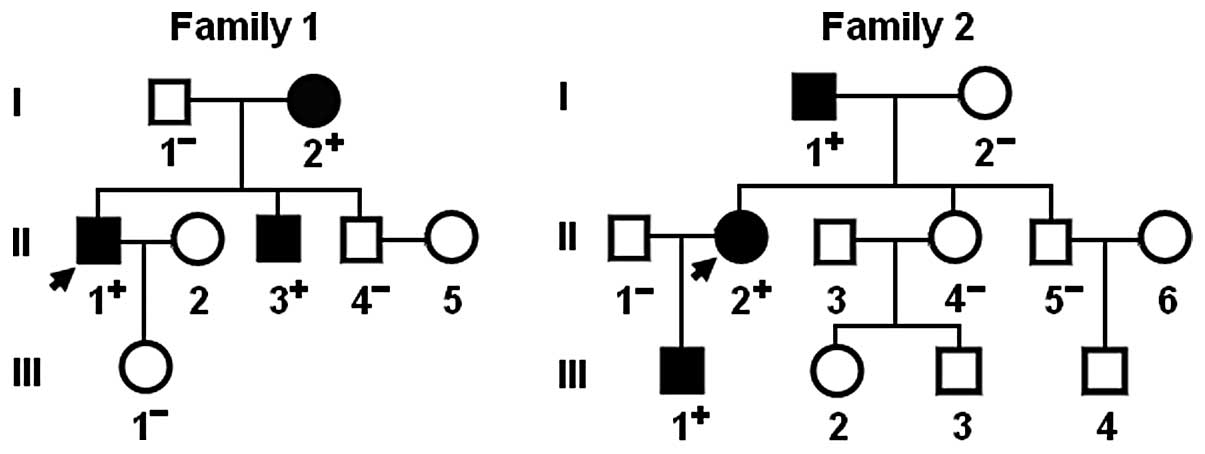
database. A genetic scan of the families of the 2 mutation carriers
showed that in each family the mutation was present in all affected
living family members, but absent in the unaffected family members
examined. Analysis of the pedigrees revealed that each mutation
co-segregated with AF transmitted in an autosomal dominant pattern
in the family with a complete penetrance. The pedigree structures
of the 2 families are illustrated in (Fig. 3). The phenotypic characteristics
and genotypic status of the affected family members are listed in
Table III.
| Table III.The phenotypic characteristics and
status of SCN4B mutations of the affected pedigree members. |
Table III.
The phenotypic characteristics and
status of SCN4B mutations of the affected pedigree members.
Subject information
| Phenotype
| Electrocardiogram
| Cardiac
echocardiogram
| Genotype
|
|---|
| Identity | Gender | Age at time of
study (years) | Age at initial
diagnosis of AF (years) | AF
(classification) | Heart rate
(beats/min) | QRS interval
(msec) | QTc | LAD (mm) | LVEF (%) | SCN4B
mutations |
|---|
| Family 1 | | | | | | | | | | V162G |
| I-2 | F | 62 | 32 | Permanent | 71 | 98 | 542 | 38 | 62 | +/- |
| II-1 | M | 40 | 36 | Paroxysmal | 73 | 106 | 445 | 35 | 68 | +/- |
| II-3 | M | 38 | 30 | Paroxysmal | 79 | 90 | 444 | 32 | 60 | +/- |
| Family 2 | | | | | | | | | | I166L |
| I-1 | M | 69 | 41 | Permanent | 68 | 108 | 426 | 37 | 58 | +/- |
| II-2 | F | 46 | 38 | Persistent | 66 | 100 | 404 | 34 | 67 | +/- |
| III-1 | M | 22 | 22 | Paroxysmal | 128 | 86 | 397 | 30 | 64 | +/- |
According to a commonly used criterion to diagnose
long QT syndrome (46), the
corrected QT interval was defined as normal range (≤440 msec) or
prolonged (>440 msec). Using this definition, all 3 AF patients
from family 1 had long QT (Table
III). The mother of the proband experienced recurrent syncopal
episodes that began when she was 26 years old. Since that time, she
had experienced >20 syncopal episodes, the majority of which
were preceded by emotional or physical stress. The
electrocardiogram revealed a markedly prolonged QT interval
(corrected QT interval was 542 msec), and the echocardiogram
documented a structurally normal heart. Therefore, she was
diagnosed with long QT syndrome.
Multiple alignments of SCN4B protein
sequences
A cross-species alignment of SCN4B protein sequences
displayed that the altered amino acids were evolutionarily highly
conserved, as presented in (Fig.
4), suggesting that the amino acids are functionally
important.
Causative potential of SCN4B sequence
variations
The SCN4B sequence variations of c.485T>G
and c.496A>C were both automatically predicted to be
disease-causing mutations by MutationTaster, with P-values of
0.745474 for c.485T>G and 0.996496 for c.496A>C. No SNPs in
the altered regions were found in the MutationTaster database.
Discussion
In the present study, 2 novel heterozygous SCN4B
mutations, p.V162G and p.I166L, were identified in 2 families with
AF, respectively. In each family, the missense mutation was present
in all the affected family members examined but was absent in the
unaffected family members available. These 2 mutations were not
detected in the 400 normal chromosomes from an ethnically-matched
control population. A cross-species alignment of multiple SCN4B
protein sequences exhibited that the altered amino acids were
evolutionarily highly conserved. Functional analysis in
silico demonstrated that the mutations were both
disease-causing. Therefore, it is highly likely that mutated
SCN4B gene contributes to the pathogenesis of AF in these
families.
The SCN4B gene maps on human chromosome
11q23.3, and is composed of 5 exons, encoding a type 1 membrane
protein of 228 amino acids, which forms an auxiliary β4 subunit of
voltage-gated sodium channel (47). Quantitative analysis of the tissue
distribution of the sodium channel β4 subunit showed that β4 was
expressed primarily in excitable tissues, including neuronal,
muscular and cardiac tissues from mice, rats and humans (47,48). Similar to the β1–β3 subunits, β4
contains an N-terminal cleaved signal sequence, an extracellular
V-type immunoglobulin-like fold, a single transmembrane α helix,
and a short intracellular C-terminal tail that may participate in
protein-protein interactions. In the immunoglobulin-like fold of
the predicted mature β4 protein, there are 3 cysteines, of which
the cysteines at positions 23 and 101 are completely conserved
across all other β subunits, as well as other V-type
immunoglobulin-like folds, and have been proposed to form an
intramolecular disulfide bond that stabilizes the structure of the
extracellular domain. The β4 subunit is covalently associated with
sodium channel α subunit via a disulfide bond to constitute a
functional ion channel complexity and functions to increase the
expression of sodium channel at the cell surface and modulate its
gating kinetics and voltage dependence, which suggests an important
role of the β4 subunit in cardiac electrophysiology (45,47).
The findings that the mutated SCN4B gene
predisposes to AF may be partially attributed to dysfunctional
sodium channels. Sodium channels play a pivotal role not only in
the initiation of the action potential but also in the maintenance
of the action potential dome, and the loss of sodium channel
function can result in shortened refractoriness and slowed
conduction, which creates an important electrophysiological
substrate for reentry in favor of AF (49,50). Additionally, the gain of sodium
channel function may give rise to enhanced cellular excitability,
increased spontaneous action potential depolarization and reduced
threshold for action potential firing, forming an arrhythmogenic
matrix prone to AF (51–53). The SCN4B mutations, p.V162G and
p.I166L identified in this study, were both located in the
transmembrane domain, and thus may be expected to exert a critical
effect on the conduction of sodium ions across the membrane and
voltage-dependent gating of sodium channel. Therefore, it can by
hypothesized that SCN4B is an integral structural component of the
cardiac sodium channel complex required for the sodium channel to
function adequately, and the mutations, p.V162G and p.I166L, may
alter sodium current density and the voltage dependence of sodium
channel activation or inactivation. However, the detailed
electrophysiological mechanisms by which the mutated SCN4B
gene confers susceptibility to AF remain to be elucidated.
Of note, all 3 AF patients from family 1, who
harbored the SCN4B mutation p.V162G, had a prolonged QT interval,
and the mother of the proband had been diagnosed as having long QT
syndrome. Since 10–15% of patients with long QT syndrome have a
normal QT interval (54), it
could not be ruled out that other family members carrying a SCN4B
mutation had long QT syndrome. Consistent our results,
Medeiros-Domingo et al (55) performed a genetic analysis of
SCN4B in 263 patients with congenital long QT syndrome and
found the heterozygous missense mutation, p.L179F, with a
mutational prevalence of approximately 0.38%. This mutation was not
observed in 800 reference alleles and led to an increase in late
sodium current. Tan et al (56) genotyped SCN4B in 292 cases
with sudden infant death syndrome and discovered the heterozygous
mutation, p.S206L, with a mutational prevalence of approximately
0.34%. Functional analysis revealed that this mutation accentuated
the late sodium current and increased the ventricular action
potential duration. These findings indicate that AF may share a
common genetic origin with long QT syndrome as well as sudden
infant death. Considering that congenital long QT syndrome is
potentially lethal secondary to malignant ventricular arrhythmias
and that the mutated SCN4B gene has been linked to sudden
infant death, the present study is of significant clinical
importance.
In conclusion, to our knowledge, this is the first
study presenting SCN4B as a novel AF-susceptibility gene and
suggests a common genetic basis for AF and congenital long QT
syndrome, as well as sudden infant death. The findings provide
significant insight into the molecular mechanisms underlying
arrhythmias and provide potential therapeutic strategies for the
early prophylaxis and personalized therapy of arrhythmias.
Acknowledgements
We would like to thank the
participants for their devotion to the study. This study was
supported by grants from the National Natural Science Fund of China
(81070153, 81270161 and 30570768), the Natural Science Fund of
Shanghai, China (10ZR1428000), the Personnel Development Foundation
of Shanghai, China (2010019) and the Key Program of Basic Research
of Shanghai, China (10JC1414000, 10JC1414001 and 10JC1414002).
References
|
1.
|
Fuster V, Rydén LE, Cannom DS, Crijns HJ,
Curtis AB, Ellenbogen KA, Halperin JL, Kay GN, Le Huezey JY, Lowe
JE, Olsson SB, Prystowsky EN, Tamargo JL, Wann LS, Smith SC Jr,
Priori SG, Estes NA III, Ezekowitz MD, Jackman WM, January CT, Lowe
JE, Page RL, Slotwiner DJ, Stevenson WG, Tracy CM, Jacobs AK,
Anderson JL, Albert N, Buller CE, Creager MA, Ettinger SM, Guyton
RA, Halperin JL, Hochman JS, Kushner FG, Ohman EM, Stevenson WG,
Tarkington LG and Yancy CW; American College of Cardiology
Foundation/American Heart Association Task Force: 2011 ACCF/AHA/HRS
focused updates incorporated into the ACC/AHA/ESC 2006 guidelines
for the management of patients with atrial fibrillation: a report
of the American College of Cardiology Foundation/American Heart
Association Task Force on practice guidelines. Circulation.
123:e269–e367. 2011.
|
|
2.
|
Go AS, Hylek EM, Phillips KA, Chang Y,
Henault LE, Selby JV and Singer DE: Prevalence of diagnosed atrial
fibrillation in adults: national implications for rhythm management
and stroke prevention: the AnTicoagulation and Risk Factors in
Atrial Fibrillation (ATRIA) Study. JAMA. 285:2370–2375. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Lloyd-Jones DM, Wang TJ, Leip EP, Larson
MG, Levy D, Vasan RS, D’Agostino RB, Massaro JM, Beiser A, Wolf PA
and Benjamin EJ: Lifetime risk for development of atrial
fibrillation: the Framingham Heart Study. Circulation.
110:1042–1046. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Wolf PA, Abbott RD and Kannel WB: Atrial
fibrillation as an independent risk factor for stroke: the
Framingham Study. Stroke. 22:983–988. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Benjamin EJ, Wolf PA, D’Agostino RB,
Silbershatz H, Kannel WB and Levy D: Impact of atrial fibrillation
on the risk of death: the Framingham Heart Study. Circulation.
98:946–952. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Magnani JW, Rienstra M, Lin H, Sinner MF,
Lubitz SA, McManus DD, Dupuis J, Ellinor PT and Benjamin EJ: Atrial
fibrillation: current knowledge and future directions in
epidemiology and genomics. Circulation. 124:1982–1993. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Darbar D, Herron KJ, Ballew JD, Jahangir
A, Gersh BJ, Shen WK, Hammill SC, Packer DL and Olson TM: Familial
atrial fibrillation is a genetically heterogeneous disorder. J Am
Coll Cardiol. 41:2185–2192. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Ellinor PT, Yoerger DM, Ruskin JN and
MacRae CA: Familial aggregation in lone atrial fibrillation. Hum
Genet. 118:179–184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Arnar DO, Thorvaldsson S, Manolio TA,
Thorgeirsson G, Kristjansson K, Hakonarson H and Stefansson K:
Familial aggregation of atrial fibrillation in Iceland. Eur Heart
J. 27:708–712. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Junttila MJ, Raatikainen MJ, Perkiömäki
JS, Hong K, Brugada R and Huikuri HV: Familial clustering of lone
atrial fibrillation in patients with saddleback-type ST-segment
elevation in right precordial leads. Eur Heart J. 28:463–468. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Christophersen IE, Ravn LS,
Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH and
Christensen K: Familial aggregation of atrial fibrillation: a study
in Danish twins. Circ Arrhythm Electrophysiol. 2:378–383. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Yang YQ, Zhang XL, Wang XH, Tan HW, Shi
HF, Fang WY and Liu X: Familial aggregation of lone atrial
fibrillation in the Chinese population. Intern Med. 49:2385–2391.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Lubitz SA, Yin X, Fontes JD, Magnani JW,
Rienstra M, Pai M, Villalon ML, Vasan RS, Pencina MJ, Levy D,
Larson MG, Ellinor PT and Benjamin EJ: Association between familial
atrial fibrillation and risk of new-onset atrial fibrillation.
JAMA. 304:2263–2269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Fox CS, Parise H, D’Agostino RB Sr,
Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA and Benjamin EJ:
Parental atrial fibrillation as a risk factor for atrial
fibrillation in offspring. JAMA. 291:2851–2855. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Brugada R, Tapscott T, Czernuszewicz GZ,
Marian AJ, Iglesias A, Mont L, Brugada J, Girona J, Domingo A,
Bachinski LL and Roberts R: Identification of a genetic locus for
familial atrial fibrillation. N Engl J Med. 336:905–911. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Ellinor PT, Shin JT, Moore RK, Yoerger DM
and MacRae CA: Locus for atrial fibrillation maps to chromosome
6q14–16. Circulation. 107:2880–2883. 2003.PubMed/NCBI
|
|
17.
|
Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang
Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y,
Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J and Huang W: KCNQ1
gain-of-function mutation in familial atrial fibrillation. Science.
299:251–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Oberti C, Wang L, Li L, Dong J, Rao S, Du
W and Wang Q: Genome-wide linkage scan identifies a novel genetic
locus on chromosome 5p13 for neonatal atrial fibrillation
associated with sudden death and variable cardiomyopathy.
Circulation. 110:3753–3759. 2004. View Article : Google Scholar
|
|
19.
|
Darbar D, Hardy A, Haines JL and Roden DM:
Prolonged signal-averaged P-wave duration as an intermediate
phenotype for familial atrial fibrillation. J Am Coll Cardiol.
51:1083–1089. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Zhang X, Chen S, Yoo S, Chakrabarti S,
Zhang T, Ke T, Oberti C, Yong SL, Fang F, Li L, de la Fuente R,
Wang L, Chen Q and Wang QK: Mutation in nuclear pore component
NUP155 leads to atrial fibrillation and early sudden cardiac death.
Cell. 135:1017–1027. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Yang Y, Xia M, Jin Q, Bendahhou S, Shi J
and Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R,
Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J
and Chen Y: Identification of a KCNE2 gain-of-function mutation in
patients with familial atrial fibrillation. Am J Hum Genet.
75:899–905. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Lundby A, Ravn LS, Svendsen JH, Hauns S,
Olesen SP and Schmitt N: KCNE3 mutation V17M identified in a
patient with lone atrial fibrillation. Cell Physiol Biochem.
21:47–54. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ravn LS, Aizawa Y, Pollevick GD,
Hofman-Bang J, Cordeiro JM, Dixen U, Jensen G, Wu Y, Burashnikov E,
Haunso S, Guerchicoff A, Hu D, Svendsen JH, Christiansen M and
Antzelevitch C: Gain of function in IKs secondary to a mutation in
KCNE5 associated with atrial fibrillation. Heart Rhythm. 5:427–435.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Hong K, Bjerregaard P, Gussak I and
Brugada R: Short QT syndrome and atrial fibrillation caused by
mutation in KCNH2. J Cardiovasc Electrophysiol. 16:394–396. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Xia M, Jin Q, Bendahhou S, He Y, Larroque
MM and Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J,
Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings
P, Barhanin J and Chen Y: A Kir2.1 gain-of-function mutation
underlies familial atrial fibrillation. Biochem Biophys Res Commun.
332:1012–1019. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Olson TM, Alekseev AE, Liu XK, Park S,
Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A and
Terzic A: Kv1.5 channelopathy due to KCNA5 loss-of-function
mutation causes human atrial fibrillation. Hum Mol Genet.
15:2185–2191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Yang Y, Li J, Lin X, Yang Y, Hong K, Wang
L, Liu J, Li L, Yan D, Liang D, Xiao J, Jin H, Wu J, Zhang Y and
Chen YH: Novel KCNA5 loss-of-function mutations responsible for
atrial fibrillation. J Hum Genet. 54:277–283. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Olson TM, Michels VV, Ballew JD, Reyna SP,
Karst ML, Herron KJ, Horton SC, Rodeheffer RJ and Anderson JL:
Sodium channel mutations and susceptibility to heart failure and
atrial fibrillation. JAMA. 293:447–454. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Darbar D, Kannankeril PJ, Donahue BS,
Kucera G, Stubblefield T, Haines JL, George AL Jr and Roden DM:
Cardiac sodium channel (SCN5A) variants associated with atrial
fibrillation. Circulation. 117:1927–1935. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Watanabe H, Darbar D, Kaiser DW,
Jiramongkolchai K, Chopra S, Donahue BS, Kannankeril PJ and Roden
DM: Mutations in sodium channel beta1- and beta2-subunits
associated with atrial fibrillation. Circ Arrhythm Electrophysiol.
2:268–275. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Olesen MS, Holst AG, Svendsen JH, Haunsø S
and Tfelt-Hansen J: SCN1Bb R214Q found in 3 patients: 1 with
Brugada syndrome and 2 with lone atrial fibrillation. Heart Rhythm.
9:770–773. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Wang P, Yang Q, Wu X, Yang Y, Shi L, Wang
C, Wu G, Xia Y, Yang B, Zhang R, Xu C, Cheng X, Li S, Zhao Y, Fu F,
Liao Y, Fang F, Chen Q, Tu X and Wang QK: Functional
dominant-negative mutation of sodium channel subunit gene SCN3B
associated with atrial fibrillation in a Chinese GeneID population.
Biochem Biophys Res Commun. 398:98–104. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Olesen MS, Jespersen T, Nielsen JB, Liang
B, Møller DV, Hedley P, Christiansen M, Varró A, Olesen SP, Haunsø
S, Schmitt N and Svendsen JH: Mutations in sodium channel β-subunit
SCN3B are associated with early-onset lone atrial fibrillation.
Cardiovasc Res. 89:786–793. 2011.
|
|
34.
|
Hodgson-Zingman DM, Karst ML, Zingman LV,
Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett
JC Jr and Olson TM: Atrial natriuretic peptide frameshift mutation
in familial atrial fibrillation. N Engl J Med. 359:158–165. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Thibodeau IL, Xu J, Li Q, Liu G, Lam K,
Veinot JP, Birnie DH, Jones DL, Krahn AD, Lemery R, Nicholson BJ
and Gollob MH: Paradigm of genetic mosaicism and lone atrial
fibrillation: physiological characterization of a connexin
43-deletion mutant identified from atrial tissue. Circulation.
122:236–244. 2010. View Article : Google Scholar
|
|
36.
|
Gollob MH, Jones DL, Krahn AD, Danis L,
Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F,
Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L and Bai D:
Somatic mutations in the connexin 40 gene (GJA5) in atrial
fibrillation. N Engl J Med. 354:2677–2688. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Yang YQ, Zhang XL, Wang XH, Tan HW, Shi
HF, Jiang WF, Fang WY and Liu X: Connexin40 nonsense mutation in
familial atrial fibrillation. Int J Mol Med. 26:605–610.
2010.PubMed/NCBI
|
|
38.
|
Jiang JQ, Shen FF, Fang WY, Liu X and Yang
YQ: Novel GATA4 mutations in lone atrial fibrillation. Int J Mol
Med. 28:1025–1032. 2011.PubMed/NCBI
|
|
39.
|
Yang YQ, Wang MY, Zhang XL, Tan HW, Shi
HF, Jiang WF, Wang XH, Fang WY and Liu X: GATA4 loss-of-function
mutations in familial atrial fibrillation. Clin Chim Acta.
412:1825–1830. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Wang J, Sun YM and Yang YQ: Mutation
spectrum of the GATA4 gene in patients with idiopathic atrial
fibrillation. Mol Biol Rep. 39:8127–8135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Yang YQ, Wang J, Wang XH, Wang Q, Tan HW,
Zhang M, Shen FF, Jiang JQ, Fang WY and Liu X: Mutational spectrum
of the GATA5 gene associated with familial atrial fibrillation. Int
J Cardiol. 157:305–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Yang YQ, Wang XH, Tan HW, Jiang WF, Fang
WY and Liu X: Prevalence and spectrum of GATA6 mutations associated
with familial atrial fibrillation. Int J Cardiol. 155:494–496.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Yang YQ, Li L, Wang J, Zhang XL, Li RG, Xu
YJ, Tan HW, Wang XH, Jiang JQ, Fang WY and Liu X: GATA6
loss-of-function mutation in atrial fibrillation. Eur J Med Genet.
55:520–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Li J, Liu WD, Yang ZL and Yang YQ: Novel
GATA6 loss-of-function mutation responsible for familial atrial
fibrillation. Int J Mol Med. 30:783–790. 2012.PubMed/NCBI
|
|
45.
|
Meadows LS and Isom LL: Sodium channels as
macromolecular complexes: implications for inherited arrhythmia
syndromes. Cardiovasc Res. 67:448–458. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Schwartz PJ, Moss AJ, Vincent GM and
Crampton RS: Diagnostic criteria for the long QT syndrome: an
update. Circulation. 88:782–784. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Yu FH, Westenbroek RE, Silos-Santiago I,
McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS,
Catterall WA, Scheuer T and Curtis R: Sodium channel beta4, a new
disulfide-linked auxiliary subunit with similarity to beta2. J
Neurosci. 23:7577–7585. 2003.PubMed/NCBI
|
|
48.
|
Maier SK, Westenbroek RE, McCormick KA,
Curtis R, Scheuer T and Catterall WA: Distinct subcellular
localization of different sodium channel alpha and beta subunits in
single ventricular myocytes from mouse heart. Circulation.
109:1421–1427. 2004. View Article : Google Scholar
|
|
49.
|
Terrenoire C, Simhaee D and Kass RS: Role
of sodium channels in propagation in heart muscle: how subtle
genetic alterations result in major arrhythmic disorders. J
Cardiovasc Electrophysiol. 18:900–905. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Nattel S: New ideas about atrial
fibrillation 50 years on. Nature. 415:219–226. 2002.PubMed/NCBI
|
|
51.
|
Makiyama T, Akao M, Shizuta S, Doi T,
Nishiyama K, Oka Y, Ohno S, Nishio Y, Tsuji K, Itoh H, Kimura T,
Kita T and Horie M: A novel SCN5A gain-of-function mutation M1875T
associated with familial atrial fibrillation. J Am Coll Cardiol.
52:1326–1334. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Li Q, Huang H, Liu G, Lam K, Rutberg J,
Green MS, Birnie DH, Lemery R, Chahine M and Gollob MH:
Gain-of-function mutation of Nav1.5 in atrial fibrillation enhances
cellular excitability and lowers the threshold for action potential
firing. Biochem Biophys Res Commun. 380:132–137. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Blana A, Kaese S, Fortmüller L, Laakmann
S, Damke D, van Bragt K, Eckstein J, Piccini I, Kirchhefer U,
Nattel S, Breithardt G, Carmeliet P, Carmeliet E, Schotten U,
Verheule S, Kirchhof P and Fabritz L: Knock-in gain-of-function
sodium channel mutation prolongs atrial action potentials and
alters atrial vulnerability. Heart Rhythm. 7:1862–1869. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Moric-Janiszewska E, Markiewicz-Łoskot G,
Loskot M, Weglarz L, Hollek A and Szydlowski L: Challenges of
diagnosis of long-QT syndrome in children. Pacing Clin
Electrophysiol. 30:1168–1170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Medeiros-Domingo A, Kaku T, Tester DJ,
Iturralde-Torres P, Itty A, Ye B, Valdivia C, Ueda K,
Canizales-Quinteros S, Tusié-Luna MT, Makielski JC and Ackerman MJ:
SCN4B-encoded sodium channel beta4 subunit in congenital long-QT
syndrome. Circulation. 116:134–142. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Tan BH, Pundi KN, Van Norstrand DW,
Valdivia CR, Tester DJ, Medeiros-Domingo A, Makielski JC and
Ackerman MJ: Sudden infant death syndrome-associated mutations in
the sodium channel beta subunits. Heart Rhythm. 7:771–778. 2010.
View Article : Google Scholar : PubMed/NCBI
|