Introduction
Prostate cancer (PCa) is one of the most commonly
diagnosed types of cancer in the older male population (1). It accounts for 15 and 4% of male
cancer cases in developed and developing countries, respectively
(2). The androgen receptor (AR)
plays a pivotal role in the onset and malignant progression of PCa
(3). It has been reported that
Cdc6, a replication licensing protein, is involved in AR signal
transduction and plays a role in the malignant progression of PCa.
AR targets the human Cdc6 gene for transcriptional regulation and
Cdc6 is also modulated at the transcription level by androgen or
anti-androgen treatment in androgen-sensitive PCa cells (4,5).
AR binds at a distinct androgen-response element (ARE) in the Cdc6
promoter that is functionally required for androgen-dependent Cdc6
transcription (6–9). AR silencing in PCa cells has been
shown to markedly decrease Cdc6 expression and androgen-dependent
cellular proliferation (10).
In all eukaryotes, there are several initiation
sites of DNA replication, and each DNA segment replicates only once
during each cell cycle (11).
Prior to DNA replication, pre-replication complexes (pre-RCs) must
be assembled at the replication sites in cells in the G1 phase
(12). Cdc6 plays a crucial role
in the assembly of pre-RCs by linking origin recognition complex
(Orc) with minichromosome maintenance (Mcm) proteins to form
pre-RCs at the sites of DNA replication (13). During the early G1 phase, Cdc6 is
firstly recruited to bind to Orc, which is a indispensable step for
the subsequent loading of Mcms onto the chromatin (14). Cdc6 is also involved in regulating
the progression of the cell cycle at the G2/M phase by interacting
with ATR (15). If Cdc6 is
inhibited, the ATR-dependent checkpoint pathway cannot be activated
in the case of DNA replication stress (15), which may lead to aberrant mitosis,
a condition that is lethal to cancer cells as a consequence of the
lack of supervision of mitotic entry. Therefore, Cdc6 may be a
potential anticancer target.
Cantharidin is the main ingredient for the
anticarcinogenic effect of Mylabris, which has been used for
antitumor treatment in Traditional Chinese Medicine for centuries.
However, its nephrotoxic and phlogogenic side-effects limit its
clinical application (16).
Norcantharidin (NCTD), a demethylated analog of cantharidin, has
been shown to exert a strong antitumor effect on many types of
cancer, such as primary hepatic carcinoma (17), lung (18), colorectal (19), breast (20) and oral cancer (21). Unlike the majority of anticancer
drugs, the significant advantages of NCTD are that it induces
little myelosuppression and induces leucocytosis (22). In this study, we report that NCTD
inhibits androgen-insensitive PCa cell growth, induces mitotic
catastrophe and enhances the anticancer effects of paclitaxel in
vitro. As shown by our results, NCTD degraded Cdc6 and Mcm6 in
DU145 cells, and disturbed the chromatin binding of Cdc6, Mcm2 and
Mcm6. Although DNA synthesis was inhibited by NCTD, the ATR, which
participates in the checkpoint pathway in the case of replication
stress, was inhibited. As a result, aberrant mitosis was observed.
In addition, combination treatment with NCTD and paclitaxel
displayed strong synergistic anticancer effects.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM) was
purchased from Gibco/Life Technologies (Carlsbad, CA, USA). Fetal
bovine serum (FBS), penicillin, streptomycin, and all other tissue
culture reagents were obtained from Thermo Scientific HyClone
(Logan, UT, USA). Paclitaxel and propidium iodide (PI) were
purchased from Sigma Chemical Co. (St. Louis, MO, USA).
Cell culture and synchronization
DU145 cells were cultured in DMEM with 10% FBS (v/v)
and penicillin (100 U/ml)/streptomycin (100 μg/ml). Cultures
were maintained in a humidified incubator at 37°C in a 5%
CO2 incubator. When the cells reached 30% confluence,
they were synchronized at the late G1 phase if necessary by the
addition of mimosine (0.5 mM) for 20 h and then released into fresh
medium.
Cell proliferation assay
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT; Sigma) tests were performed to evaluate the cytotoxic
effects in vitro. Briefly, DU145 cells were plated in
96-well plates (8×103 cells/well). Following treatment
with NCTD for 48 h at the indicated concentrations, MTT was added
to each well (100 μg/well) followed by incubation for an
additional 4 h. The produced insoluble formazan was dissolved with
200 μl DMSO and the optical density (OD) was measured using
an ELISA reader (Thermo Labsystems, Helsinki, Finland) at
wavelengths of 570 and 630 nm. The experiments were carried out in
triplicate.
BrdU incorporation assay
The DU145 cells were seeded onto 22-mm diameter
coverglasses placed in 6-well plates (3×105
cells/coverglass). One hour prior to fixing the cells, 10 μM
BrdU (Sigma Chemicals) were added to the cultures. The cells were
rinsed and fixed in 4% phosphate-buffered paraformaldehyde for 10
min. Following aspiration, the cells were rinsed 3 times in PBS for
5 min and 0.2% Triton X-100 was added to the specimens for 10 min.
The specimens were then incubated in 4 M HCl after being rinsed 3
times in PBS for 5 min. After neutralization using PBS, the
specimens were blocked in goat serum for 60 min. The blocking
solution was aspirated and the specimens were incubated in diluted
primary mouse-monoclonal antibody to BrdU (1:1,000, Cell Signaling
Technology Inc., Beverly, MA, USA) overnight at 4°C. After rinsing
3 times in PBS for 5 min, the specimens were incubated in
fluorochrome-conjugated secondary antibody diluted in PBS at room
temperature in the dark and observed under fluorescent microscope.
At least 1,000 cells/treatment using at least 2
cover-glasses/treatment were counted, and the number of positive
cells was recorded. Labeling indexes were calculated as the number
of positively stained cells divided by the number of total
cells.
Western blot analysis
The cells were harvested and suspended in ice-cold
lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM
PMSF and 10 U/ml aprotinin) for 20 min, then centrifuged at 12,000
rpm for 10 min at 4°C. Total proteins were separated by 10%
SDS-PAGE and transferred onto polyvinylidene fluoride membranes.
The membranes were blocked with TBS containing 0.1% Triton X-100
and 5% non-fat milk for 1 h at room temperature, then incubated
with rabbit-anti-human monoclonal antibody against Cdc6 (1:1,000;
Cell Signaling Technology Inc., Danvers, MA, USA),
rabbit-anti-human monoclonal against Mcm2 (1:1,000, Cell Signaling
Technology), goat polyclonal antibody against Mcm3 (1:500, Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA), mouse-anti-human
monoclonal antibody against Mcm6 (1:500, Santa Cruz Biotechnology,
Inc.) and rabbit-anti-human monoclonal against GAPDH (1:2,000, Cell
Signaling Technology) at 4°C overnight. After washing, the
membranes were incubated with HRP-conjugated Ig at room temperature
for 1 h. Signal detection was carried out with an ECL system
(Millipore, Billerica, MA, USA).
Chromatin binding assay
The cells were harvested and resuspended in tubes
with extraction buffer (100 mM KCl, 50 mM HEPES-KOH pH 7.5, 2.5 mM
MgCl2, 50 mM Na4P2O7,
0.1 mM NaVO3, 0.5% Triton X-100) containing protease
inhibitors, then set on ice for 5 to 10 min for incubation. The
tubes were flicked to mix the solution every 2–3 min during
incubation. Subsequently, 30% ice-cold sucrose containing protease
inhibitors was added to the bottom of the tubes. The tubes were
then spinned in a microfuge, 12–15 krpm, 10 min, 4°C and the
supernatants were transferred to new tubes. The pellets were washed
with EB buffer and flicked to dislodge the pellets from the wall of
the tubes and vortexed briefly for resuspension, followed by
spinning in a microfuge, 12–15 krpm, 5 min, 4°C. The supernatants
were saved and combined (this is the nonchromosomal fraction) from
this and the previous step. The pellets were resuspended with EB
buffer (the pellets are the chromatin-binding fraction). The
supernatants and the pellets were then used for western blot
analysis.
Nuclear staining
For nuclear staining, the cells were treated with or
without 50 or 100 μM NCTD for 48 h, washed twice with cold
PBS, and fixed with 4% paraformaldehyde for 15 min. The fixed cells
were permeabilized with 0.2% Triton X-100 and then incubated with
4, 6-diamidino-2-phenylindole (DAPI) at the concentration of 5
μg/ml for 5 min. A fluorescent microscope was used for
imaging. At least 1,000 cells/treatment were counted, and the
number of cells with signs of mitotic catastrophe was recorded.
Labeling indexes were calculated as the number of cells with signs
of mitotic catastrophe divided by the number of total cells.
Flow cytometric assay
Following treatment with or without hydroxyurea (HU)
or NCTD/HU for 24 h, the cells in each group were collected by
Trypsin digestion. The cells were fixed with 70% alcohol and then
centrifuged again for 10 min at 8,000 rpm. The supernatant was
discarded and 500 μl PI staining solution (5 μg/ml
RNase, 0.1% Triton X-100, 0.1 mM EDTA, 50 μg/ml PI) was
added. The cells were then incubated away from light for 30 min at
4°C. The cell cycle and apoptosis were then measured by flow
cytometry (Becton-Dickinson, San Jose, CA, USA).
In vitro combination treatment of DU145
cells with NCTD and paclitaxel
DU145 cells were treated with NCTD in combination
with paclitaxel at various concentrations for 48 h. MTT assays were
performed to detect the cell viability in vitro. Briefly,
the cells were seeded on 96-well plates in 10% FBS-containing
medium at a density of 4,000 cells/well and incubated at 37°C for
24 h prior to exposure to the drugs. The cells were then treated
with NCTD, paclitaxel, or a combination of both for 48 h at the
indicated concentrations. The combination ratio of NCTD (μM)
and paclitaxel (nM) was 1:1 and the concentration range is
presented in Table I. Cell
viability was determined following treatment with the drugs for 48
h by MTT assay. Each experiment was performed in quintuplicate
wells for each drug concentration and independently carried out 3
times. By combining both agents at graded concentrations, the
combined effects of growth inhibition were obtained and analyzed
using Calcusyn1 software. The combination index (CI) was calculated
as follows: CI = (D)1/(Dx)1 + (D)2/(Dx)2 + (D)1(D)2/(Dx)1(Dx)2;
where (Dx)1 is the dose of drug 1 required to produce an X% effect
alone, (D)1 is the dose of drug 1 required to produce the same X%
effect in combination with drug 2, (Dx)2 is the dose of drug 2
required to produce an X% effect alone, and (D)2 is the dose of
drug 2 required to produce the same X% effect in combination with
drug 1.
 | Table I.ED50 and concentration
range for NCTD and paclitaxel. |
Table I.
ED50 and concentration
range for NCTD and paclitaxel.
| Drug |
ED50 | Concentration
range |
|---|
| Paclitaxel
(nM) | 772.5±316.8 | 6.25–400 |
| NCTD
(μM) | 206.6±12.0 | 6.25–400 |
The effects of the combination treatment were then
transformed and displayed as FA-CI plots. The combined effects were
determined as follows: CI <1, synergy; CI = 1, zero interaction
(strictly additive effects); and CI >1, antagonism.
Statistical analysis
Average values are expressed as the means ± standard
deviation (SD). Statistical significance between different groups
was determined using the Student’s t-test. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
Effect of NCTD on tumor cell
proliferation
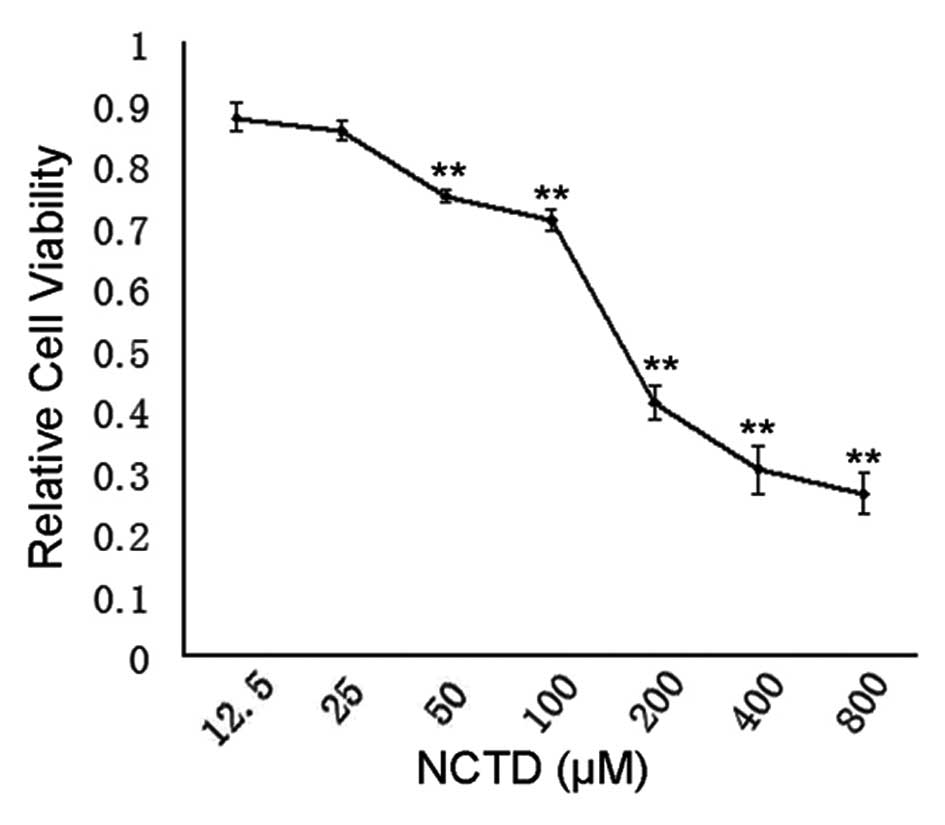
To evaluate the effects of NCTD inhibition on the
proliferation of DU145 PCa cells, MTT assay was carried out. In the
NCTD-treated group, a dose-dependent suppression of cell
proliferation was observed. NCTD effectively inhibited the
proliferation of DU145 cells, with an IC50 of ~200
μM (Fig. 1).
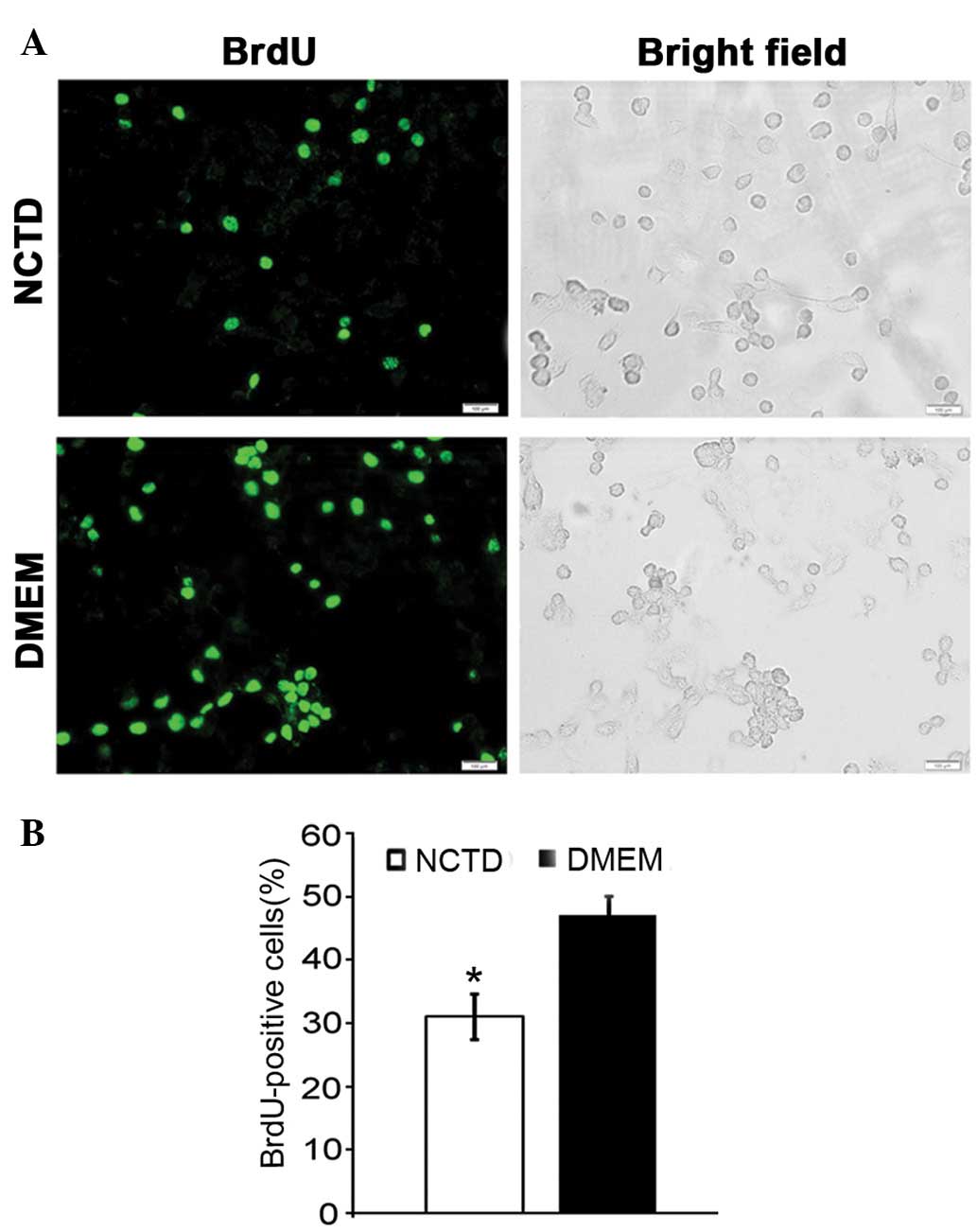
BrdU incorporation assay
We investigated whether NCTD inhibits the
proliferation of DU145 cells by suppressing DNA synthesis. The BrdU
incorporation assays revealed that ~47% of the PCa cells in the
control group were able to incorporate BrdU, while in the
NCTD-treated group, the number of BrdU-positive cells decreased to
31% (Fig. 2A and B). NCTD
significantly inhibited DNA replication (P<0.05).
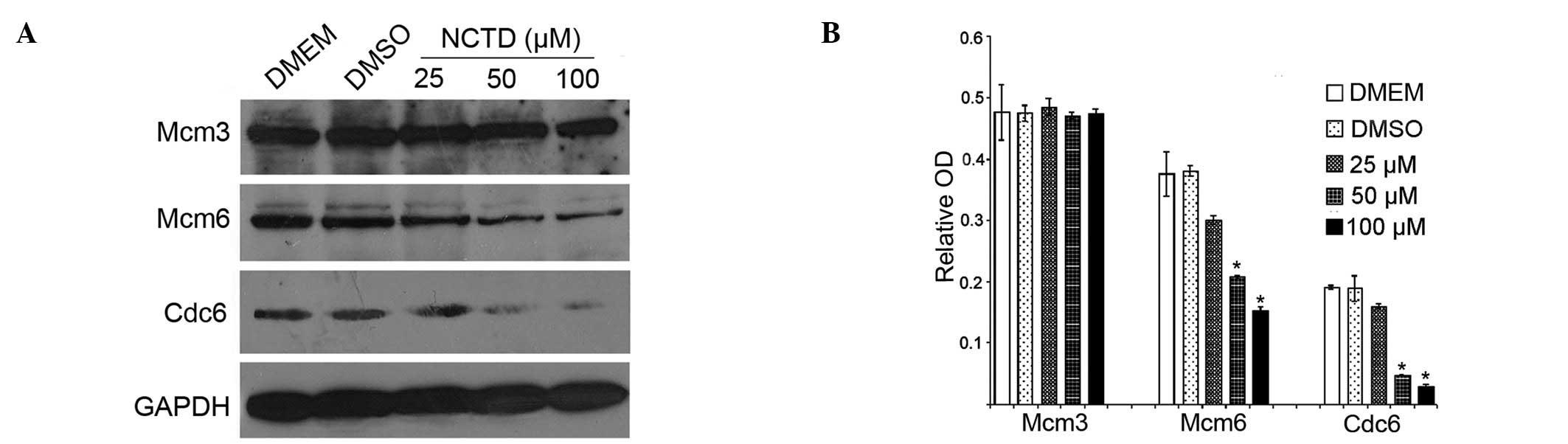
NCTD induces the degradation of pre-RC
proteins
To further characterize the inhibitory effect of
NCTD on DNA replication, we examined the effects of NCTD on pre-RC
proteins. Total proteins from NCTD-treated DU145 cells were
analyzed by western blot analysis. Of the pre-RC components, Cdc6,
Mcm3 and Mcm6 are the most frequently reported proteins that are
overexpressed in tumors. Our results revealed that Cdc6 and Mcm6
were significantly degraded in a dose-dependent manner following
treatment with NCTD, as compared with the control group (Fig. 3A and B).
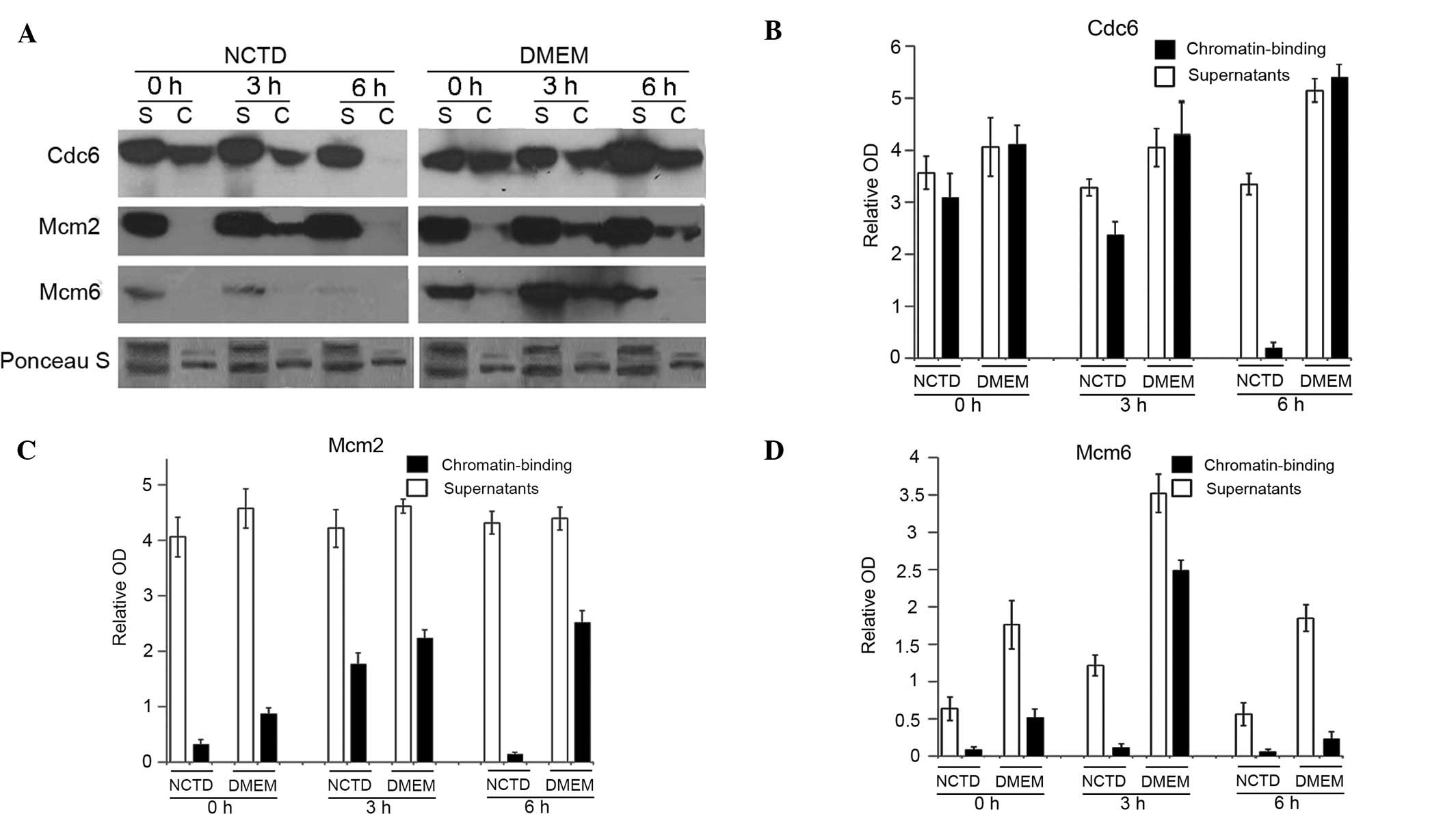
NCTD disturbs the assembly of pre-RC in
G1 cells
In G1 phase cells, Cdc6 resides in the nucleus and
is essential for pre-RC formation and replication initiation
(23). The end result of pre-RC
formation is the loading of Mcms onto origin DNA (24). To examine the inhibitory effect of
NCTD on the assembly of pre-RC, the cells were incubated with
mimosine for 20 h and then released into fresh medium with or
without NCTD (NCTD was added 8 h prior to the release of mimosine).
The cells were collected at 0, 3 and 6 h after the release of
mimosine. The chromatin binding of Cdc6, Mcm2 and Mcm6 was examined
by chromatin-binding assay. Our results indicated that NCTD
significantly reduced the chromatin binding of Cdc6 and Mcm2 in the
cells at the 6 h time point (Fig.
4A–C). The chromatin binding of Mcm6 was significantly
decreased in cells at all time points (Fig. 4A and D). The results revealed that
NCTD disturbed the assembly of Cdc6, Mcm2 and Mcm6 onto
chromatin.
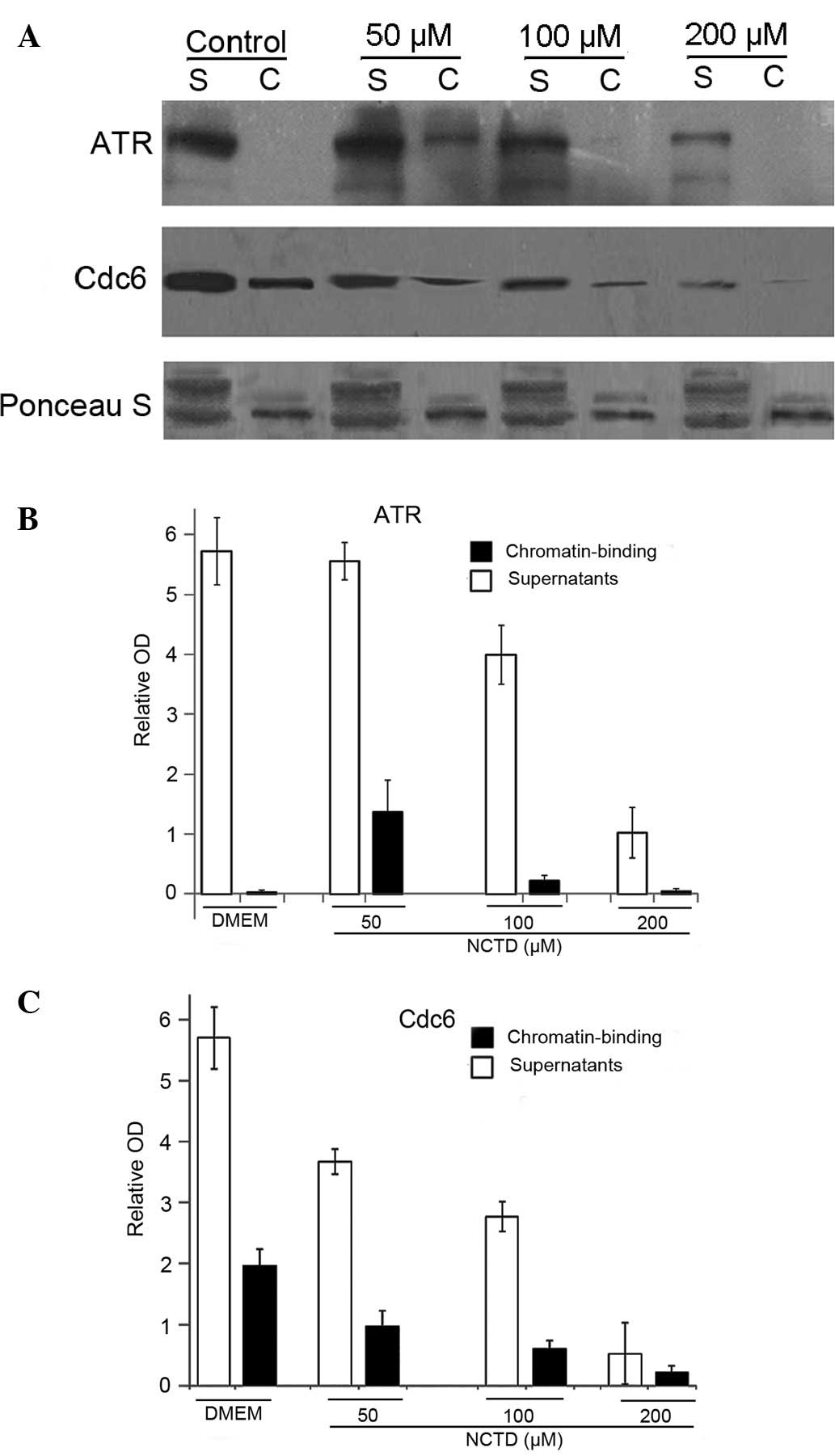
NCTD disturbs ATR binding to chromatin
and disables the S-M checkpoint
Previous studies have shown that Cdc6 is not only
required for G1 origin licensing, but is also crucial for proper S
phase DNA replication and participates in the S-M checkpoints
(25). As shown in a previous
study, the loss of Cdc6 did not activate the ATR-dependent DNA
damage response and induced aberrant mitosis (25). It has been reported that human
Cdc6 physically interacts with ATR, in a manner that is stimulated
by phosphorylation by Cdk, and the presence of Cdc6 during the S
phase is essential for ATR to bind to chromatin in response to
replication inhibition (15). In
our study, NCTD effectively degraded Cdc6 in DU145 cells (Figs. 3 and 4). Thus, there is a possibility that
NCTD can inhibit ATR binding to chromatin by the degradation of
Cdc6. Our results revealed that few ATR chromatin-binding fractions
were detected in the control group, suggesting that the
ATR-dependent checkpoint pathway was not activated without DNA
replication stress (Fig. 5A and
B). In the low-dose NCTD-treated groups (50 μM), ATR was
detected in chromatin, suggesting that DNA replication was
inhibited and ATR was activated. However, under higher doses of
NCTD (100 and 200 μM), ATR disappeared from chromatin. In
addition, the chromatin binding of Cdc6 signficantly decreased in a
dose-dependent manner following treatment with NCTD (Fig. 5A and C). These results suggested
that NCTD (lower dose) activates the ATR-dependent checkpoint
pathway by inducing DNA replication stress. However, at the same
time, NCTD (higher dose) also attenuates the ATR-dependent
checkpoint activity by inhibiting ATR binding to chromatin, which
may be due to the Cdc6 degradation.
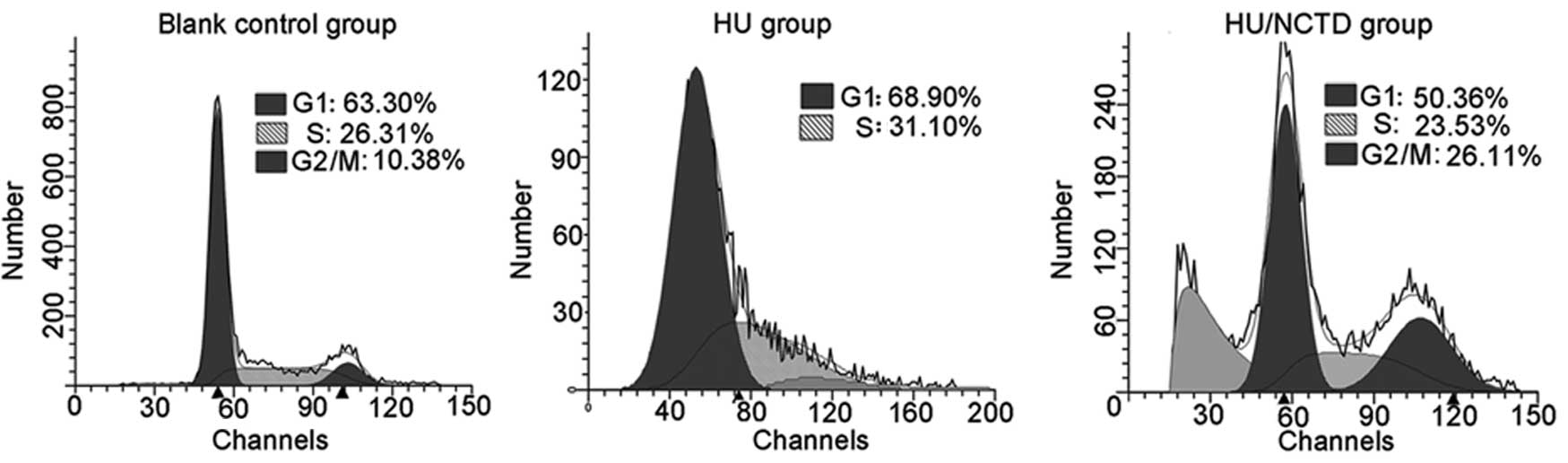
ATR plays a crucial role in the S-M checkpoint
pathway by signaling to Chk1 and conferring G2/M arrest when the
cells are under replication stress (26). This surveillance warrants cells
will not transit into mitosis before the completion of the S phase
(15,27). To further characterize the
inhibitory effect of NCTD on the S-M checkpoint, we examined the
influence of NCTD on HU-induced S phase arrest. The results
revealed that HU effectively induced S phase arrest (31%) and the
cells did not progress into the G2/M phase (0%). In the NCTD group,
26% of the cells still progressed into the G2/M phase under HU
stress. Of note, there was a significant accumulation of cells in
the sub-G1 phase in the NCTD/HU-treated group, suggesting that the
aberrant mitosis led to apoptosis (Fig. 6).
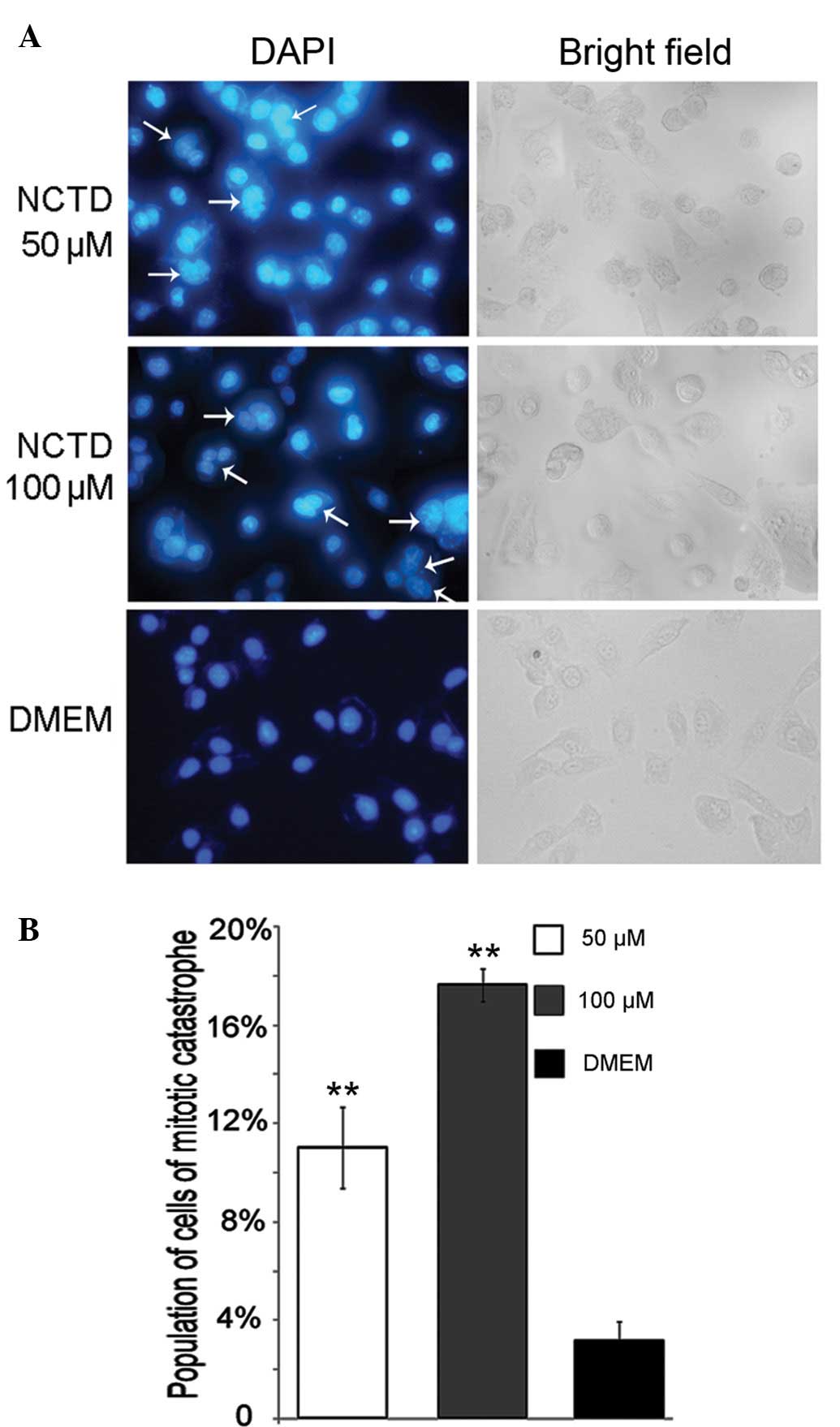
NCTD induces mitotic catastrophe in DU145
cells
In our experiments, DNA replication was inhibited by
NCTD. Simultaneously, the chromatin binding of ATR and hence, the
S-M checkpoint was also prevented. Thus, it is rational to suggest
that treatment with NCTD may induce aberrant mitosis by
simultaneously disturbing DNA replication and disabling the S-M
checkpoint. The results from DAPI nuclear staining revealed that
following treatment with NCTD, the cells underwent mitotic
catastrophe, indicated by the presence of cells with micronuclei
and giant multinucleated cells (28). The percentages of DU145 cells
exposed to 50 or 100 μM NCTD which underwent mitotic
catastrophe were ~11 and 17%, respectively, significantly higher
than those of the control group (Fig.
7A and B) (P<0.05).
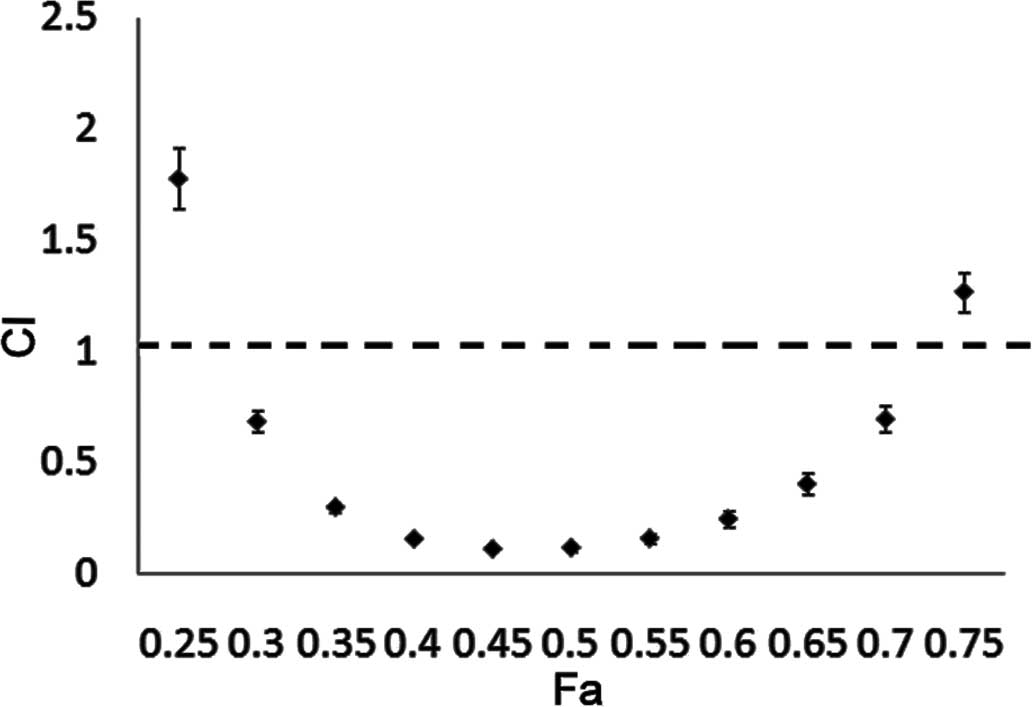
Synergism of NCTD with paclitaxel in PCa
DU145 cells
As a microtubule-poisoning drug, paclitaxel can
induce mitotic catastrophe and is effective for PCa therapy. As
opposed to the NCTD-induced mitotic catastrophe, paclitaxel
interferes with the normal breakdown of microtubules during cell
division (29,30). Therefore, a combination of these 2
drugs, may produce synergistic effects. In this study, we
investigated whether NCTD and paclitaxel exert synergistic effects.
As shown by our results, a strong synergistic anticancer effect on
DU145 cells was observed with the combination treatment of NCTD and
paclitaxel at a low concentration range (CI <1) (Fig. 8). Our findings provide
justification for the further development of the combined treatment
with NCTD and paclitaxel for the treatment of PCa.
Discussion
As is well known, cancer cells have an infinite
proliferation ability. The ability of DNA replication in cancer
cells is promoted in order to achieve cell division. The assembly
of pre-RCs on chromatin is indispensable to initiate DNA
replication (12). Cdc6 plays an
important role in the formation and maintainence of pre-RCs
(12). In our study, NCTD was
verified to have the ability of inhibiting PCa cell growth with an
IC50 of approximately 200 μM (Fig. 1). The BrdU labeling assay
demonstrated that NCTD effectively inhibited DNA replication in
cultured DU145 cells (Fig. 2).
The initiation proteins, Cdc6 and Mcm6, were degraded (Fig. 3), and were prevented from binding
to chromatin following treatment with NCTD (Fig. 4). These results demonstrate that
NCTD suppresses DNA replication by the degradation of Cdc6 and Mcm6
and as a result, blocks the formation of pre-RCs.
Cell cycle dysregulation is a hallmark of tumor
cells. The regulation of proteins that mediate critical events in
the cell cycle can be a useful method for the treatment of tumors
(31). In our study, a larger
proportion of PCa cells with aberrant mitosis following treatment
with NCTD was observed (Fig. 7).
Previous studies have indicated that human Cdc6 physically
interacts with ATR in a Cdk-phosphorylation-stimulated manner and
that Cdc6 is required for the ATR-dependent replication-checkpoint
response activated by modest replication stress (15). In this study, we found that the
chromatin binding fraction of ATR emerged and was reduced following
treatment with NCTD in a dose-dependent manner (Fig. 5). The blocking of ATR binding to
chromatin leads to premature mitosis before replication has been
completed; mitotic catastrophe is lethal to cells (28). In this study, we compared the cell
cycle distribution of HU-treated and HU/NCTD-treated DU145 cells
and found that a large proportion of cells progressed to the G2/M
phase; we also observed cells in the sub-G1 phase in the
HU/NCTD-treated group. The HU-treated cells were blocked in the S
phase and were prevented from entering the G2/M phase in the
presence of the activation of the checkpoint pathway (Fig. 6). Cell cycle analysis revealed
that NCTD induced mitotic catastrophe, possibly by disturbing the
interaction of Cdc6 and ATR. However, the exact correlation between
Cdc6, ATR and mitotic catastrophe requires further investigation.
Therefore, NCTD kills DU145 cells not only by suppressing DNA
replication in the G1 phase but also by inducing mitotic
catastrophe during the S/G2M transition.
In this study, we investigated the synergistic
anti-neoplastic effect of paclitaxel combined with NCTD in DU145
PCa cells. Our results revealed that the combination of NCTD and
paclitaxel was highly synergistic at low concentrations (Fig. 8). Firstly, NCTD can induce
apoptosis through multiple pathways, including inhibiting the
initiation of DNA replication (32,33). Secondly, NCTD can inhibit the
assembly of Cdc6 and ATR on chromatin, which is essential in the
ATR-dependent checkpoint pathway. The blocking of the activation of
the ATR-dependent checkpoint pathway results in premature entry
into mitosis before the completion of DNA replication and mitotic
catastrophe, a lethal event for cells (28). When the ATR-dependent checkpoint
pathway is obstructed, more cells, which should have been blocked
in the S phase, will abnormally enter into mitosis. Therefore, a
larger proportion of vulnerable cells in aberrant mitosis may
favorably contribute to the anticancer effects of paclitaxel. A
lower dose of the combination of the 2 drugs may achieve a stronger
effect with fewer side-effects. Unlike the majority of anticancer
drugs, NCTD has significant advantages of inducing
myelo-suppression and inducing leucocytosis, making it a promising
candidate for use in combination treatments.
In conclusion, NCTD exerts anticancer effects by
inhibiting the formation and maintenance of pre-RCs, and inducing
mitotic catastrophe in DU145 cells. Its multistage and multipath
antitumor effects make it a promising candidate for use in
combination treatments. In addition, Cdc6 may be a promising
anticancer target in PCa.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (nos.
30901822, 81272482 and 81072113).
References
|
1.
|
Greenlee RT, Murray T, Bolden S and Wingo
PA: Cancer statistics. CA Cancer J Clin. 50:7–33. 2000.
|
|
2.
|
Quinn M and Babb P: Patterns and trends in
prostate cancer incidence, survival, prevalence and mortality. Part
I: international comparisons. BJU Int. 90:162–173. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Cude KJ, Dixon SC, Guo Y, Lisella J and
Figg WD: The androgen receptor: genetic considerations in the
development and treatment of prostate cancer. J Mol Med (Berl).
77:419–426. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Robles LD, Frost AR, Davila M, Hutson AD,
Grizzle WE and Chakrabarti R: Down-regulation of Cdc6, a cell cycle
regulatory gene, in prostate cancer. J Biol Chem. 277:25431–25438.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Mallik I, Davila M, Tapia T, Schanen B and
Chakrabarti R: Androgen regulates Cdc6 transcription through
interactions between androgen receptor and E2F transcription factor
in prostate cancer cells. Biochim Biophys Acta. 1783:1737–1744.
2008. View Article : Google Scholar
|
|
6.
|
Claessens F, Verrijdt G, Schoenmakers E,
et al: Selective DNA binding by the androgen receptor as a
mechanism for hormone-specific gene regulation. J Steroid Biochem
Mol Biol. 76:23–30. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Wang Q, Li W, Liu XS, et al: A
hierarchical network of transcription factors governs androgen
receptor-dependent prostate cancer growth. Mol Cell. 27:380–392.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bolton EC, So AY, Chaivorapol C, Haqq CM,
Li H and Yamamoto KR: Cell- and gene-specific regulation of primary
target genes by the androgen receptor. Genes Dev. 21:2005–2017.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Massie CE, Adryan B, Barbosa-Morais NL, et
al: New androgen receptor genomic targets show an interaction with
the ETS1 transcription factor. EMBO Rep. 8:871–878. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Jin F and Fondell JD: A novel androgen
receptor-binding element modulates Cdc6 transcription in prostate
cancer cells during cell-cycle progression. Nucleic Acids Res.
37:4826–4838. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Cook JG: Replication licensing and the DNA
damage checkpoint. Front Biosci. 14:5013–5030. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Stillman B: Cell cycle control of DNA
replication. Science. 274:1659–1664. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Saxena S and Dutta A: Geminin-Cdt1 balance
is critical for genetic stability. Mutat Res. 569:111–121. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Lei M and Tye BK: Initiating DNA
synthesis: from recruiting to activating the MCM complex. J Cell
Sci. 114:1447–1454. 2001.PubMed/NCBI
|
|
15.
|
Yoshida K, Sugimoto N, Iwahori S, et al:
CDC6 interaction with ATR regulates activation of a replication
checkpoint in higher eukaryotic cells. J Cell Sci. 123:225–235.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Wang GS: Medical uses of Mylabris
in ancient China and recent studies. J Ethnopharmacol. 26:147–162.
1989.
|
|
17.
|
Chang C, Zhu Y, Tang X and Tao W: The
anti-proliferative effects of norcantharidin on human HepG2 cells
in cell culture. Mol Biol Rep. 38:163–169. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Luan J, Duan H, Liu Q, Yagasaki K and
Zhang G: Inhibitory effects of norcantharidin against human lung
cancer cell growth and migration. Cytotechnology. 62:349–355. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Chen YJ, Chang WM, Liu YW, et al: A
small-molecule metastasis inhibitor, norcantharidin, downregulates
matrix metalloproteinase-9 expression by inhibiting Sp1
transcriptional activity in colorectal cancer cells. Chem Biol
Interact. 181:440–446. 2009. View Article : Google Scholar
|
|
20.
|
Huang Y, Liu Q, Liu K, Yagasaki K and
Zhang G: Suppression of growth of highly-metastatic human breast
cancer cells by norcantharidin and its mechanisms of action.
Cytotechnology. 59:2092009. View Article : Google Scholar
|
|
21.
|
Kok SH, Cheng SJ, Hong CY, et al:
Norcantharidin-induced apoptosis in oral cancer cells is associated
with an increase of proapoptotic to antiapoptotic protein ratio.
Cancer Lett. 217:43–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Liu XH, Blazsek I, Comisso M, et al:
Effects of norcantharidin, a protein phosphatase type-2A inhibitor,
on the growth of normal and malignant haemopoietic cells. Eur J
Cancer. 31A:953–963. 1995.PubMed/NCBI
|
|
23.
|
Ofir Y, Sagee S, Guttmann-Raviv N, Pnueli
L and Kassir Y: The role and regulation of the preRC component Cdc6
in the initiation of premeiotic DNA replication. Mol Biol Cell.
15:2230–2242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Forsburg SL: Eukaryotic MCM proteins:
beyond replication initiation. Microbiol Mol Biol Rev. 68:109–131.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Lau E, Zhu C, Abraham RT and Jiang W: The
functional role of Cdc6 in S-G2/M in mammalian cells. EMBO Rep.
7:425–430. 2006.PubMed/NCBI
|
|
26.
|
Liu Q, Guntuku S, Cui XS, et al: Chk1 is
an essential kinase that is regulated by Atr and required for the
G(2)/M DNA damage checkpoint. Genes Dev. 14:1448–1459.
2000.PubMed/NCBI
|
|
27.
|
Clay-Farrace L, Pelizon C, Santamaria D,
Pines J and Laskey RA: Human replication protein Cdc6 prevents
mitosis through a checkpoint mechanism that implicates Chk1. EMBO
J. 22:704–712. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Vakifahmetoglu H, Olsson M and Zhivotovsky
B: Death through a tragedy: mitotic catastrophe. Cell Death Differ.
15:1153–1162. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
McGuire WP, Rowinsky EK, Rosenshein NB, et
al: Taxol: a unique antineoplastic agent with significant activity
in advanced ovarian epithelial neoplasms. Ann Intern Med.
111:273–279. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Horwitz SB: Taxol (paclitaxel): mechanisms
of action. Ann Oncol. 5(Suppl 6): S3–S6. 1994.PubMed/NCBI
|
|
31.
|
Stewart ZA, Westfall MD and Pietenpol JA:
Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol
Sci. 24:139–145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Chen YN, Chen JC, Yin SC, et al: Effector
mechanisms of norcantharidin-induced mitotic arrest and apoptosis
in human hepatoma cells. Int J Cancer. 100:158–165. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Chen YN, Cheng CC, Chen JC, Tsauer W and
Hsu SL: Norcantharidin-induced apoptosis is via the extracellular
signal-regulated kinase and c-Jun-NH2-terminal kinase signaling
pathways in human hepatoma HepG2 cells. Br J Pharmacol.
140:461–470. 2003. View Article : Google Scholar : PubMed/NCBI
|