Introduction
Sepsis, which reflects an uncontrolled systemic
inflammatory response, is among the most significant challenges in
critical care, with an estimated incidence of nearly 800,000
cases/year, and a mortality rate of 25%, and up to 60% when shock
is present (1). This syndrome
usually includes at least two of the four signs of a systemic
inflammatory response syndrome (SIRS): fever or hypothermia,
tachycardia, rapid breathing, hyperleukocytosis or leukopenia
(2). The pathophysiology of
sepsis involves dysfunctions of complex interactions of signal
pathways and impairment in host responses to pathogen-associated
molecular patterns, resulting in chaos in inflammatory cytokine
production, adhesion molecule expression, respiratory burst, and
production of reactive nitrogen species (3).
Endothelial cells, which are highly responsive to
their extracellular environment, are the primary targets of
sepsis-induced damage (4).
Exposure to septic serum, which contains lipopolysaccharide and
abnormal levels of cytokines, chemokines and irons, lead to
extensive endothelial dysfunction, manifested by a shift from the
hemostatic balance towards a procoagulant phenotype, elevated
leukocyte adhesion and abnormal regulation of vasomotor tone
(5). Notably, it is known that
progressive subcutaneous and body-cavity edema, usually leading to
impairment of organ function, typically develops in patients with
sepsis, suggesting wide-spread increases in vascular permeability
(6). Although increased
endothelial permeability has long been considered as a critical
step in sepsis-associated organ failure, the underlying molecular
mechanisms remain relatively unknown.
Endothelial-mesenchymal transition (EndMT), similar
to epithelial-mesenchymal transition, is characterized by loss of
endothelial cell markers, such as VE-cadherin and platelet
endothelial cell adhesion molecule-1 (PECAM-1, also known as CD31),
and gain of mesenchymal markers, such as α-smooth muscle actin
(α-SMA) and vimentin (7). During
EndMT, resident endothelial cells delaminate from a polarized cell
layer and migrate into the underlying tissue (8). Although EndMT is considered as an
essential mechanism of endocardial cushion formation during cardiac
and pulmonary development (9),
recently, accumulating evidence has linked EndMT to various
diseases. It is reported that loss of cell-cell junctions and
acquisition of invasive and migratory properties during EndMT have
also been linked to disruption of the endothelial barrier (10). Furthermore, a great portion of
fibroblasts in rodent models of renal diseases was found to
co-express the endothelial marker CD31 and the
fibroblast/myofibroblast markers, fibroblast-specific protein-1
and/or α-SMA (11). Furthermore,
a significant number of fibroblasts raised from bleomycin-induced
pulmonary fibrosis were of endothelial origin (12). However, whether EndMT occurs
during sepsis is relatively unknown.
Membrane-associated proteins, either embedded in the
lipid bilayers or anchored to the membrane, possess multiple
functions, including signal transduction, protein or iron
transport, cytoskeleton assembly and cell adhesion (13). 2-DE-based proteomic approaches
enable identification of alterations in membrane protein
expression, allowing proteome-wide profiling of cellular responses
upon stress (14). In this study,
barrier integrity of a HUVEC monolayer was rapidly decreased by
incubation with serum derived from sepsis patients. Notably,
exposure to septic serum induced clear EndMT in HUVECs.
Furthermore, proteomic analysis was introduced to screen for
cellular alterations underlying septic serum-mediated EndMT, and
caveolin-1 was identified as a key factor.
Materials and methods
Ethics statement
The study was approved by the Institutional Ethics
Committee of Sichuan Academy of Medical Sciences and Sichuan
Provincial People’s Hospital. All participants provided written
informed consent prior to sampling.
Clinical samples
The blood samples of sepsis patients and normal
healthy donors were collected from Sichuan Provincial People’s
Hospital. Sepsis was diagnosed according to the criteria of the
American College of Chest Physicians/Society of Critical Care
Medicine (25). The blood samples
were centrifuged at 900 × g for 10 min at room temperature, and the
cell-free supernatants were stored immediately in aliquots at −80°C
until use.
Cell culture and septic serum
treatment
Human umbilical vein endothelial cells (HUVECs) were
purchased from ATCC and were maintained in Dulbecco’s modified
Eagle’s medium (DMEM; Gibco, USA) containing 10% fetal calf serum
(Gibco, USA), penicillin (100 U/l) and streptomycin (10 mg/l).
Cells were incubated in a humidified atmosphere containing 5%
CO2 at 37°C and passaged every 5 days at a split ratio
of 1:4 using trypsin. For assessment of the effect of septic serum
on endothelial cells, confluent HUVECs were incubated with DMEM
medium containing 20% septic serum for the indicated time. Those
HUVECs treated with DMEM medium containing 20% healthy serum were
used as the control group.
Determination of endothelial barrier
dysfunction
HUVECs were seeded on 0.4-mm pore size culture
inserts and were cultured until confluence was reached. The cells
were treated with the medium containing septic serum for the
indicated time, and the medium was replaced with fresh medium
containing blue dextran dye (2.3 mg/ml). Fifty milliliters of
basolateral medium was sampled after 12 h of incubation, and was
measured using a microplate reader (Safire; Tecan, Morrisville, NC,
USA) at 610 nm.
Cell migration and invasion assays
In vitro cell migration and invasion assays
were performed using Transwell 24-well chambers (Corning). Cells
were seed in the upper chamber. For monitoring cell invasion, the
upper side of the filter was pre-covered with Matrigel
(Collaborative Research, Inc., Boston, MA, USA). After 12 h for the
migration assay or 24 h for the invasion assay, cells on the upper
side of the filter were removed and those cells on the underside of
the filter were stained with crystal violet, and counted with an
inverted microscope (Zeiss Axiovert).
Immunocytofluorescence
Cells were washed with phosphate-buffered saline
(PBS) (pH 7.4) three times and fixed in 4% paraformaldehyde at room
temperature for 10 min. After washing three times in PBS, the
slides were incubated with 3% bovine serum albumin for 30 min and
then incubated with the primary antibodies for 2 h at room
temperature. After washing for three times, the slides were
incubated with secondary antibody conjugated to TRITC for 30 min at
room temperature. The specimens were analyzed via a fluorescence
microscope.
Membrane protein purification and 2-DE
analyses
Cells (109) were harvested and
re-suspended in homogenization buffer (50 mM HEPES, pH 7.4, 1 mM
CaCl2, 1 mM EDTA, 1 mM vanadate, and 1 mM
phenylmethylsulfonyl fluoride). After cells were broken down using
a Dounce homogenizer (Wheaton), the cell samples were centrifuged
at 3,000 × g for 10 min at 4°C to remove nuclei. The supernatant
was then mixed with 80% (w/v) sucrose, and overlaid with sucrose at
gradient concentrations, including 35, 30, 25, 20, 15, 10 and 5%.
The gradients were centrifuged at 200,000 × g for 24 h at 4°C.
After centrifugation, two bands that appeared in the upper middle
region of the tube were collected, diluted in PBS, and centrifuged
for 166,000 × g for an additional 2 h at 4°C. The pellets were
suspended in lysis buffer (7 M urea, 2 M thiourea, 4% CHAPS)
containing protease inhibitor mixture 8340 (Sigma). Protein
concentrations were determined using the DC protein assay kit.
Protein samples were applied to IPG strips (all were from Bio-Rad)
for 16 h of rehydration. After rehydration, the strips were
submitted to IEF, and then equilibrated in equilibration buffer I
(containing 130 mM DTT) and II (containing 200 mM iodoacetamide)
for 15 min, respectively. The second dimension was performed using
12% SDS-PAGE, and the gels were stained using CBB R-250 (Merck).
The gels were scanned using the Bio-Rad GS-800 scanner (400–750
nm), and alterations in protein spots were analyzed using PDQuest
2-D analysis software (Bio-Rad). In-gel digestion of proteins was
carried out using mass spectrometry grade Trypsin Gold (Promega,
Madison, WI, USA) according to the manufacturer’s instructions.
Mass spectrum analysis was performed using a Q-TOF mass
spectrometer fit with an ESI source (Waters). The MS/MS data were
processed with MassLynx v. 4.1 software (both were from Micromass,
Manchester, UK) which converted MS/MS data into PKL files. The PKL
files were analyzed using the online Mascot search engine
(http://www.matrixscience.com).
Western blotting
Cell samples were lysed with RIPA buffer and
quantified by the DC protein assay kit (Bio-Rad). Protein samples
were separated by 12% SDS-PAGE and transferred to PVDF membranes
(Millipore). The membranes were incubated with 5% FBS overnight at
4°C, and subsequently probed with primary antibodies: mouse
anti-caveolin-1 (diluted 1:1,000; Santa Cruz Biotechnology, Inc.),
rabbit anti-galectin-1 (diluted 1:1,000), rabbit anti-α-enolase
(diluted 1:2,000), rabbit anti-S100A4 (diluted 1:2,000) (all were
from Abcam). After washing for three times, the membranes were then
incubated with secondary antibody conjugated to horseradish
peroxidase (diluted 1:10,000; Santa Cruz Biotechnology, Inc.) for 2
h at room temperature. Blots were visualized using enhanced
chemiluminescence reagents (Amersham Biosciences). β-actin was used
as an internal control.
Statistics analysis
Differences between two groups were assessed by the
Student’s t-test at P<0.05; P<0.01; or P<0.001.
Results
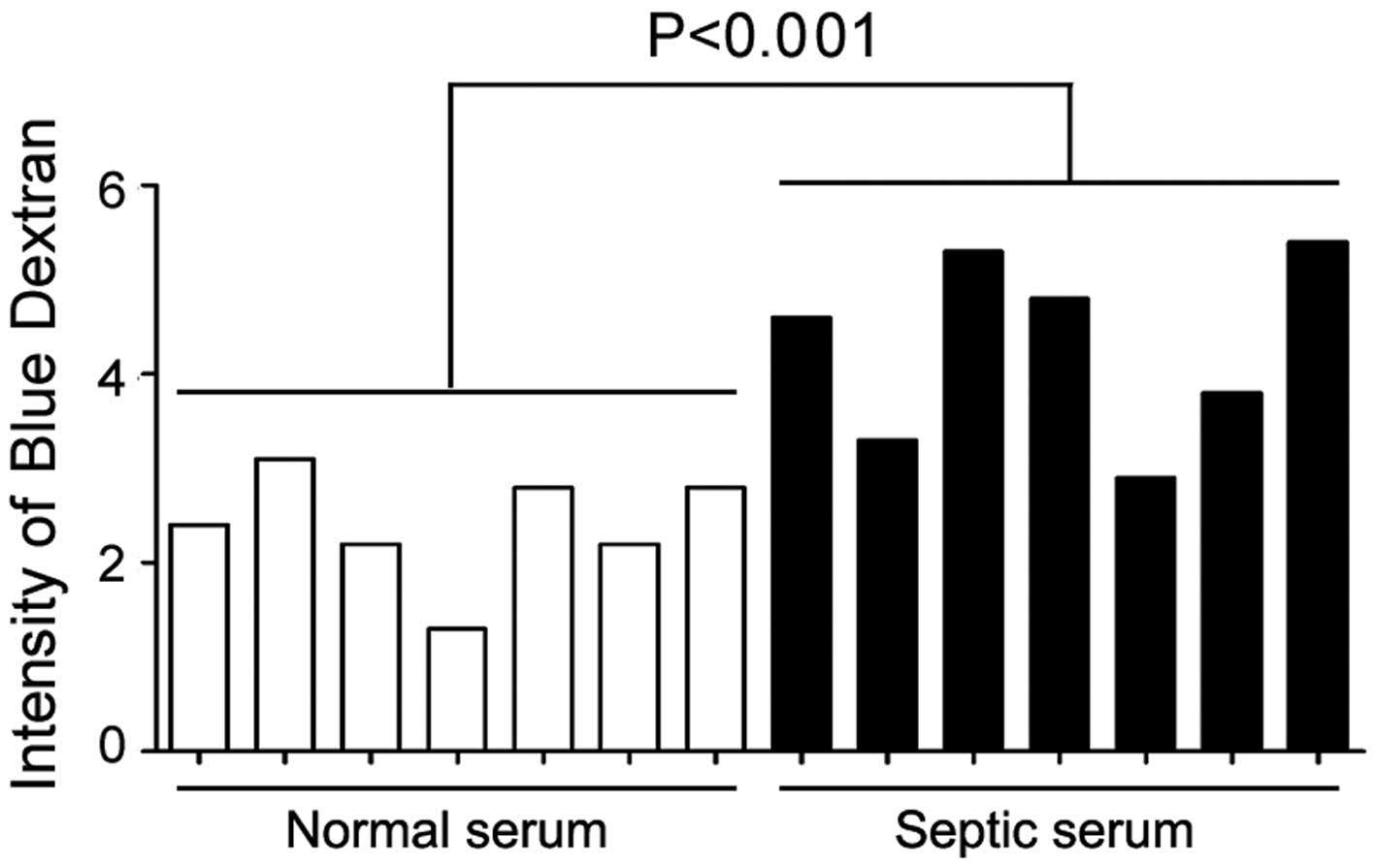
Septic serum decreases endothelial
barrier integrity
Previous studies have shed light on the loss of
endothelial barrier integrity under sepsis (15); however, the molecular mechanisms
underlying this process are relatively unknown. As an initial test,
we sought to determine whether incubation with the serum derived
from sepsis patients could alter endothelial barrier integrity. To
this end, HUVECs were used as an in vitro model, and septic
serum was obtained from 17 sepsis patients. The detailed
information of these patients is listed in Table I. As results, exposure to septic
serum for 12 h resulted in a significant decrease in HUVEC
monolayer integrity (Fig. 1).
 | Table IClinical parameters of sepsis
patients. |
Table I
Clinical parameters of sepsis
patients.
| Patient no. | Age (years) | APACHEII score | WBC
(109/l) | N | Cr (μmol/l) | PCT (ng/ml) | EM (years) | Prognosis |
|---|
| 1 | 32 | 22 | 18.34 | 78 | 233.6 | 18.1 | 13.9 | Death |
| 2 | 43 | 17 | 24.5 | 93 | 638.9 | 180.1 | 21.5 | Discharge |
| 3 | 57 | 32 | 1.05 | 82 | 294.7 | 49.5 | 72 | Death |
| 4 | 64 | 22 | 19.34 | 87 | 211.3 | 34.6 | 23.1 | Discharge |
| 5 | 63 | 13 | 6.92 | 89 | 238.2 | 23.7 | 21.8 | Death |
| 6 | 71 | 13 | 14.97 | 81 | 86.2 | 26.1 | 11 | Discharge |
| 7 | 67 | 39 | 5.72 | 85 | 165 | 21.1 | 23 | Death |
| 8 | 73 | 13 | 2.8 | 79 | 52.6 | 34 | 10 | Discharge |
| 9 | 79 | 15 | 5.88 | 83 | 63.7 | 32 | 15 | Death |
| 10 | 73 | 21 | 15.11 | 99 | 52.9 | 26.5 | 23 | Death |
| 11 | 72 | 21 | 9.52 | 90 | 160.9 | 2.14 | 22 | Discharge |
| 12 | 81 | 39 | 4.13 | 80 | 650.5 | >200 | 73 | Death |
| 13 | 64 | 19 | 18.12 | 92 | 213.4 | 43.3 | 21 | Discharge |
| 14 | 71 | 23 | 21.34 | 87 | 198.2 | 32.1 | 19 | Discharge |
| 15 | 63 | 21 | 25.42 | 89 | 122.4 | 33 | 16 | Discharge |
| 16 | 81 | 27 | 24.22 | 79 | 109.8 | 24 | 67 | Death |
| 17 | 71 | 35 | 22.11 | 92 | 215.4 | 22.5 | 78 | Death |
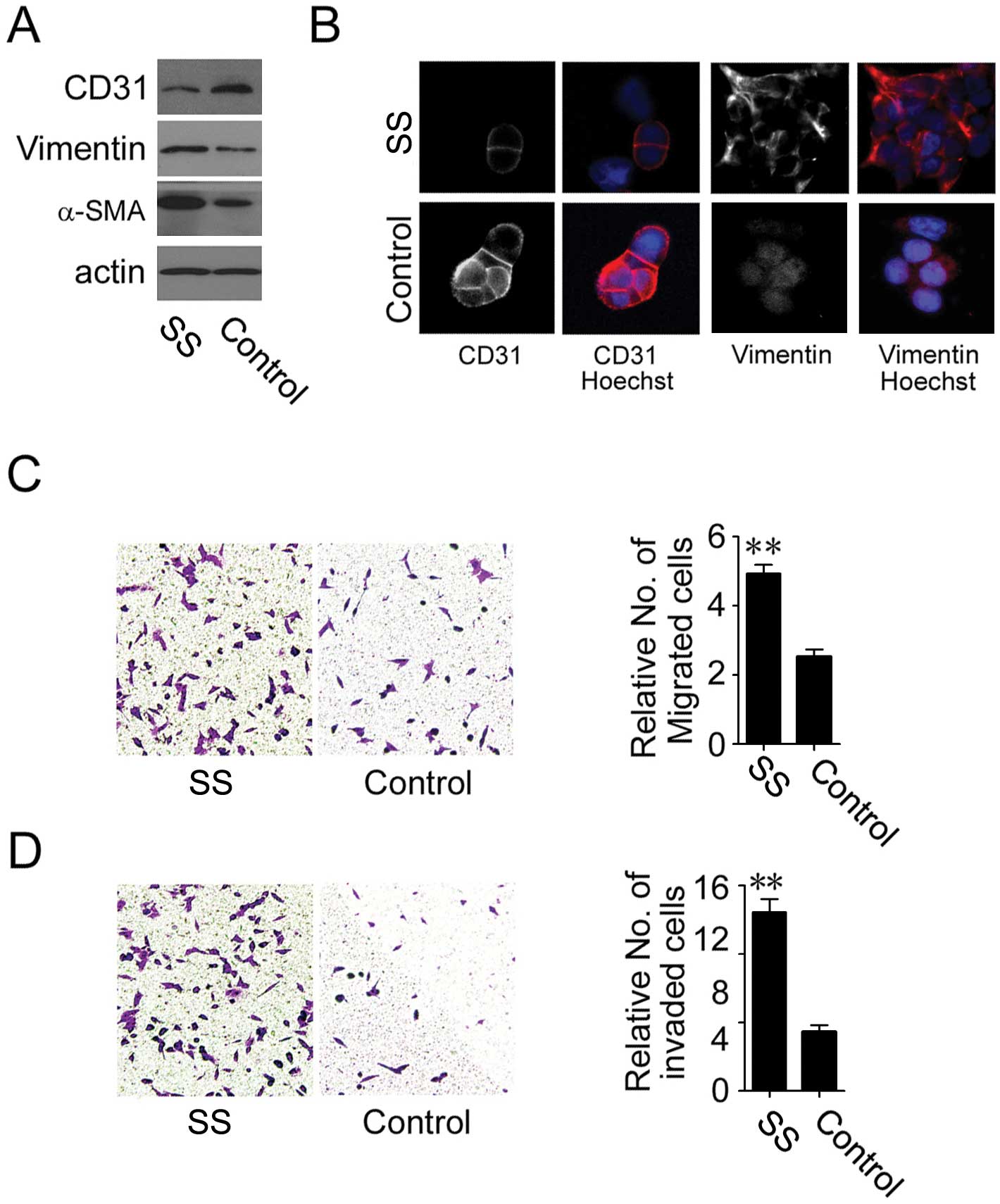
Septic serum induces EndMT in HUVECs
To elucidate septic serum-induced leakage in HUVEC
monolayer, we determined the vitality of HUVECs exposed to septic
serum. However, by TUNEL assay, no difference in the number of
apoptotic cells was found between the septic serum-treated cells
and untreated control after a 12-h incubation, although an
increased cell death was noted after 36 h of incubation (data not
shown). These observations suggest that endothelial barrier
dysfunction induced by a 12-h exposure to septic serum may not be
due to endothelial cell death.
Since tight junction proteins, which seal the
paracellular spaces between cells and contribute to endothelial
barrier function, are frequently repressed during the EndMT process
(10), we examined whether septic
serum induced EndMT in endothelial cells. Markedly reduced
expression of endothelial cell marker, CD31, and increased
expression of mesenchymal cell markers, vimentin and α-SMA, were
detected by immunoblot analysis in the septic serum-treated HUVECs
compared to the control cells (Fig.
2A). These observations were further supported by both loss of
CD31 on the cell membrane and an increase in vimentin in the
cytoplasm (Fig. 2B).
Additionally, both migratory and invasive capability of HUVECs were
elevated after treatment with septic serum, as shown by the
Transwell chamber migration (Fig.
2C) and Matrigel invasion assays, respectively (Fig. 2D). These observations suggest that
septic serum induces EndMT in endothelial cells.
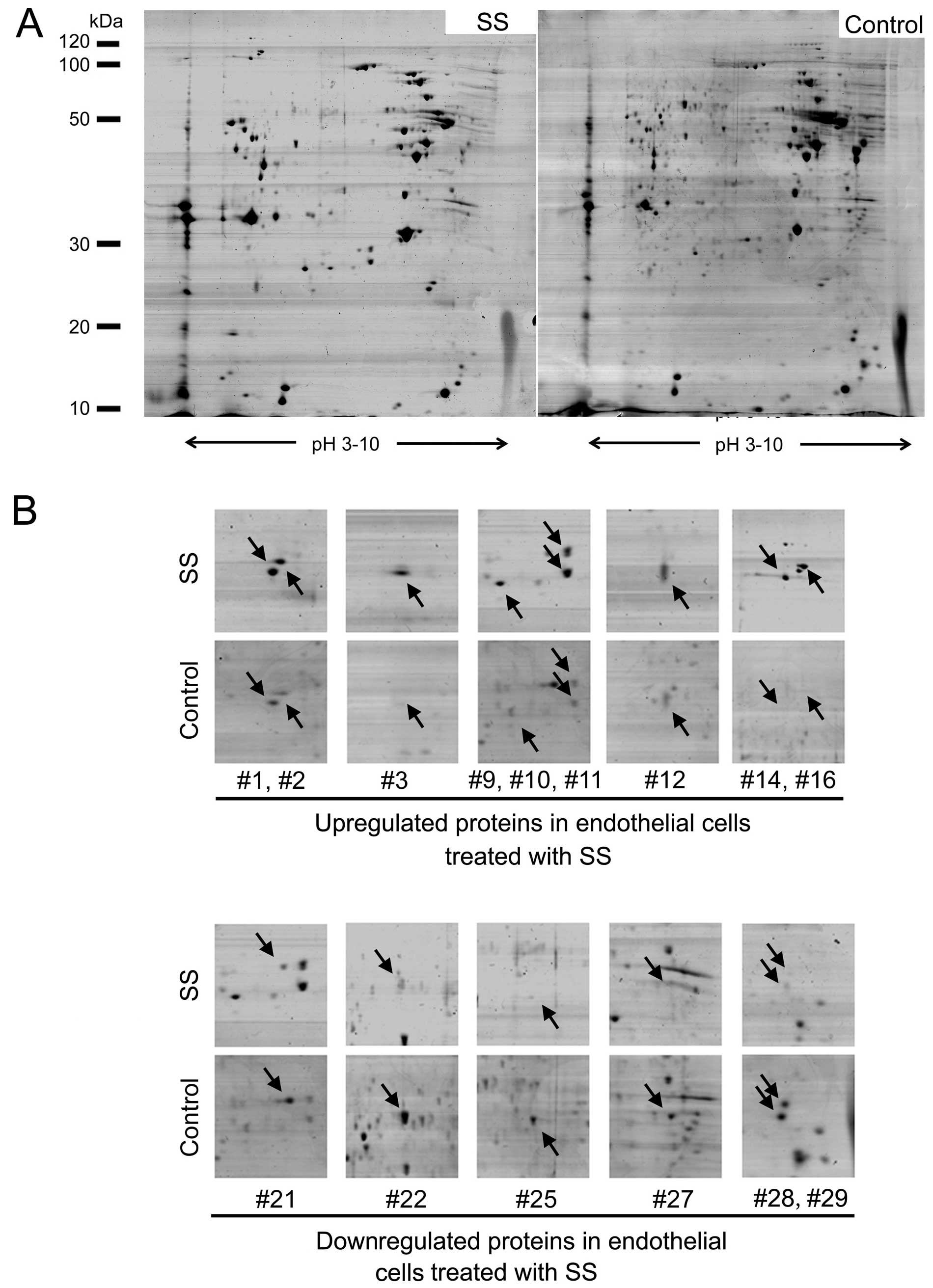
Proteomic profiling of endothelial cells
under septic stimuli
To further explore the mechanism of septic
serum-induced EndMT in endothelial cells, 2-DE-based proteomic
analysis was employed to profile the differentially expressed
membrane proteins between septic serum-treated and untreated
HUVECs. Representative 2-DE maps from seven parallel experiments
are shown in Fig. 3A. As a
result, a total of 29 protein spots were identified with an altered
expression level. Among them, 18 spots were downregulated in
response to septic serum, while 11 spots were upregulated. Fifteen
representative altered spots were boxed and enlarged with the
surrounding area (Fig. 3B).
The selected protein spots were then subjected to
and identified by MS/MS analysis. As a result, all of the 29 spots
were positively identified. The altered proteins were involved in
diverse biological processes, including signaling transduction,
cytoskeleton assembly, metabolism and adhesion, and the majority of
these proteins were reported as membrane proteins according to
ExPASy protein database. The detailed information of eight proteins
with the most significant alterations are listed in Table II.
| Table IIProteins demonstrating the most
significant alteration in endothelial cells treated or untreated
with septic serum. |
Table II
Proteins demonstrating the most
significant alteration in endothelial cells treated or untreated
with septic serum.
| Spot no. | Protein
description | Gene name | Accession no. | Function | Location | Theoretical
Mr/pI |
|---|
| 1 | Chloride
intracellular channel protein 1 | CLIC1 | O00299 | Chloride ion
channel | Cell membrane | 26,791.54/5.09 |
| 2 | Membrane-associated
progesterone receptor component 2 | PGRMC2 | O15173 | Signal
transduction | Cell membrane | 23,818.45/4.76 |
| 3 | S100A4 | S100A4 | P26447 | Calcium ion
binding | Cell membrane | 11,597.32/5.88 |
| 10 | Annexin A4 | ANXA4 | P09525 | Membrane fusion and
exocytosis | Cell membrane | 36,088.00/5.84 |
| 16 | Hephaestin | HEPH | Q9BQS7 | Iron ion
transport | Cell membrane |
127,792.48/5.64 |
| 21 | Caveolin-1 | CAV1 | Q2TNI1 | Lipid raft
regulation | Lipid raft | 20,471.62/5.64 |
| 22 | α-enolase | ENO1 | P06733 | Glycolysis | Cytoplasm, cell
membrane | 47,037.77/6.99 |
| 28 | Galectin-1 | LGALS1 | P09382 | ECM remodeling | Extracellular
space | 14,584.51/5.30 |
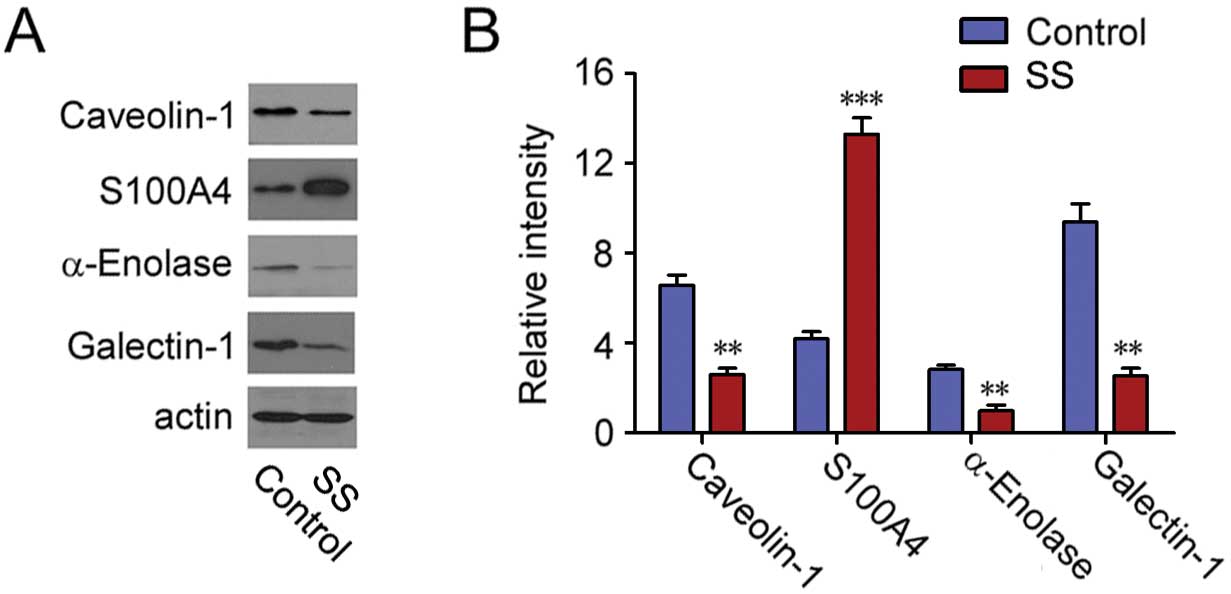
Validation of the altered proteins
To confirm the proteins exhibiting altered levels as
revealed by proteomic analysis, the expression levels of four
proteins with the most significant alterations, including
caveolin-1, S100A4, α-enolase and galectin-1, were examined by
immunoblot analysis. S100A4 was found to be upregulated, while the
level of caveolin-1, α-enolase and galectin-1 were downregulated
after septic serum treatment, which was in accordance with our
proteomic analysis (Fig. 4).
Caveolin-1 is a negative regulator of
EndMT during sepsis
Caveolin-1 was previously found to be involved in
EMT in either normal epithelial cells or cancer cell lines
(16–18). Therefore, we aimed to ascertain
whether caveolin-1 has a role in septic serum-induced EndMT in
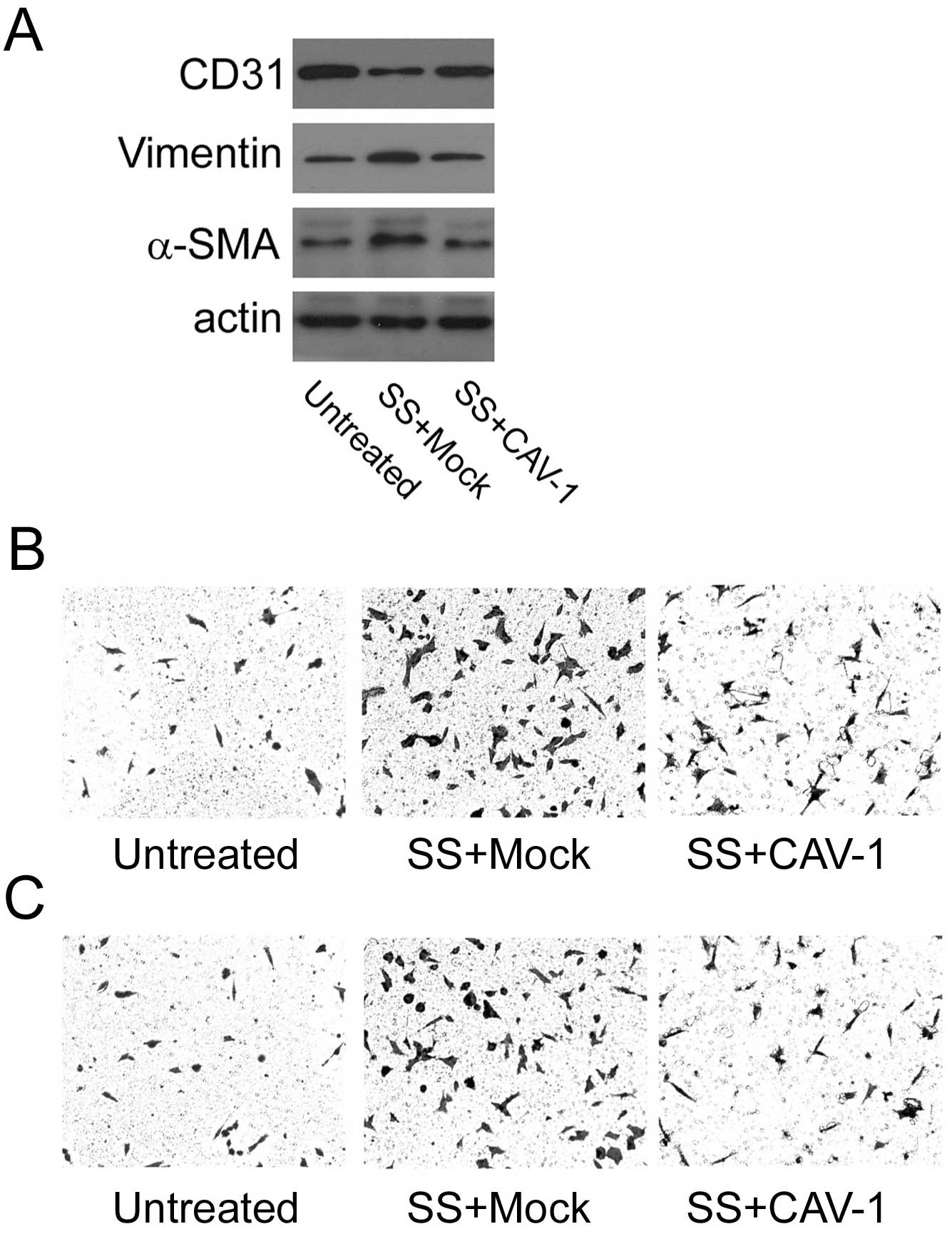
HUVECs. To this goal, HUVECs were transfected with a
caveolin-1-expressing vector prior to exposure to septic serum. As
shown, septic serum-induced overexpression of vimentin and α-SMA,
and reduction of CD31 was substantially abolished upon increased
caveolin-1 expression compared to mock control cells (Fig. 5A). These observations were further
accompanied by markedly attenuated migratory and invasive
capability of the HUVECs, as demonstrated by Transwell chamber
migration (Fig. 5B) and Matrigel
invasion assays (Fig. 5C),
respectively.
Discussion
Endothelial cells are involved in various aspects of
vascular biology, such as maintenance of endothelial barrier
integrity (4). Endothelial
dysfunction which results from direct exposure to various types of
stress, such as inflammatory mediators and microbial antigens, is
universal in sepsis (19). In the
present study, we evaluated endothelial dysfunction as a result of
septic serum stimuli. We demonstrated that treatment with the serum
derived from patients with sepsis induced a rapid decreased in the
barrier integrity of a HUVEC monolayer. Notably, endothelial
barrier dysfunction mediated by a 12-h exposure to septic serum was
not due to increased cell death, since a detectable increase in
HUVEC apoptosis was not observed until 36 h of incubation. We
further demonstrated that septic serum triggered EndMT in HUVECs,
as revealed by repression of CD31 and overexpression of α-SMA and
vimentin, which was accompanied by enhanced cell motility and
invasiveness. This is the first study to report that endothelial
cells undergo EndMT during sepsis. Furthermore, our data indicate
that EndMT may contribute to septic stimuli-induced endothelial
barrier dysfunction.
A small but increasing number of studies have shed
light on the regulatory signaling network of EndMT. It is reported
that Snail is required for TGF-β-induced EndMT of embryonic stem
cell-derived endothelial cells (MESECs) and that transient
expression of Snail induced the differentiation of MESECs into
mural cells, whereas knockdown of Snail expression abrogated
TGF-β-induced mural differentiation of MESECs (20). Inhibition of Smad3, a crucial node
protein in TGF-β signaling cascade, by a chemical antagonist,
blocked EndMT in pancreatic microvascular endothelial cells,
leading to a significant delay in the early development of
streptozotocin-induced diabetic nephropathy (21). Moreover, endothelin-1 has also
been suggested as an EndMT inducer in cardiac fibrosis in diabetic
hearts (22). In the present
study, 2-DE-based proteomic approaches were employed to analyze the
potential mediators of EndMT under sepsis, and 29 proteins with
significant alterations were identified. Four proteins, including
caveolon-1, S100A4, galectin-1 and α-enolase, were selected and
validated at the protein level.
Caveolin-1 is a 21- to 24-kDa cholesterol-binding
membrane protein and is the major scaffolding component of caveolae
(23). Abnormal expression of
caveolin-1 has long been implicated during EMT, which is a process
similar to EndMT. It is reported that downregulation of caveolin-1
by chronic exposure to EGF leads to the loss of E-cadherin, and
enhanced epidermoid carcinoma cell invasion (16). Consistently, it has been shown
that caveolin-1 promotes pancreatic cancer cell differentiation by
suppression of EMT via upregulation of E-cadherin expression
(17). Nevertheless, an enhanced
level of caveolin-1 was observed in both human embryonic carcinoma
cell line NT2/D1 and normal mouse mammary epithelial cell line
NMuMG following the induction of EMT. Upregulation of caveolin-1 in
these two cell models was probably mediated by activation of focal
adhesion kinase (FAK) and Src, two known tyrosine kinases involved
in EMT (18). Although both EMT
and EndMT give rise to cells that have a mesenchymal phenotype
(24), the role of caveolin-1 in
EndMT is still uncharacterized. Based on our data, exogenous
expression of caveolin-1 in HUVECs markedly blocked septic
serum-induced EndMT, by retrieving expression of CD31 and
repressing expression of both α-SMA and vimentin. In correlation,
increased migratory and invasive capability of HUVECs was markedly
attenuated upon caveolin-1 expression. Therefore, it is reasonable
to infer that caveolin-1 functions as a negative regulator of EndMT
in response to septic stimuli.
In summary, we reported that exposure to septic
serum induced EndMT and conferred a migratory and invasive
phenotype on endothelial cells. Furthermore, we profiled the
proteomic alterations in endothelial cells undergoing septic
stimuli, and 29 proteins with differentially expressed levels were
identified. Finally, we demonstrated that exogenous expression of
caveolin-1 attenuated septic serum-induced EndMT as well as
migratory and invasive capability in HUVECs. The present study may
lead to a better understanding of the biological role of
endothelial cells in the pathophysiology of sepsis.
Acknowledgements
This study was supported by the Department of Public
Health of Sichuan Province (grants 080326).
References
|
1
|
Cornell TT, Rodenhouse P, Cai Q, Sun L and
Shanley TP: Mitogen-activated protein kinase phosphatase 2
regulates the inflammatory response in sepsis. Infect Immun.
78:2868–2876. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Akrout N, Sharshar T and Annane D:
Mechanisms of brain signaling during sepsis. Curr Neuropharmacol.
7:296–301. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu SF and Malik AB: NF-kappa B activation
as a pathological mechanism of septic shock and inflammation. Am J
Physiol Lung Cell Mol Physiol. 290:L622–L645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shapiro NI, Schuetz P, Yano K, et al: The
association of endothelial cell signaling, severity of illness, and
organ dysfunction in sepsis. Crit Care. 14:R1822010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aird WC: The role of the endothelium in
severe sepsis and multiple organ dysfunction syndrome. Blood.
101:3765–3777. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee WL and Slutsky AS: Sepsis and
endothelial permeability. N Engl J Med. 363:689–691. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garcia J, Sandi MJ, Cordelier P, et al:
Tie1 deficiency induces endothelial-mesenchymal transition. EMBO
Rep. 13:431–439. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoshimatsu Y and Watabe T: Roles of TGF-β
signals in endothelial-mesenchymal transition during cardiac
fibrosis. Int J Inflam. 2011:7240802011.
|
|
9
|
van Meeteren LA and ten Dijke P:
Regulation of endothelial cell plasticity by TGF-β. Cell Tissue
Res. 347:177–186. 2012.
|
|
10
|
Nagasawa K, Chiba H, Fujita H, et al:
Possible involvement of gap junctions in the barrier function of
tight junctions of brain and lung endothelial cells. J Cell
Physiol. 208:123–132. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zeisberg EM, Potenta SE, Sugimoto H,
Zeisberg M and Kalluri R: Fibroblasts in kidney fibrosis emerge via
endothelial-to-mesenchymal transition. J Am Soc Nephrol.
19:2282–2287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tanjore H, Xu XC, Polosukhin VV, et al:
Contribution of epithelial-derived fibroblasts to bleomycin-induced
lung fibrosis. Am J Respir Crit Care Med. 180:657–665. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu CC and Yates JR III: The application of
mass spectrometry to membrane proteomics. Nat Biotechnol.
21:262–267. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiong P, Li Y, Tang Y and Chen H:
Proteomic analyses of Sirt1-mediated cisplatin resistance in OSCC
cell line. Protein J. 30:499–508. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tsao N, Hsu HP, Wu CM, Liu CC and Lei HY:
Tumour necrosis factor-alpha causes an increase in blood-brain
barrier permeability during sepsis. J Med Microbiol. 50:812–821.
2001.PubMed/NCBI
|
|
16
|
Lu Z, Ghosh S, Wang Z and Hunter T:
Downregulation of caveolin-1 function by EGF leads to the loss of
E-cadherin, increased transcriptional activity of β-catenin, and
enhanced tumor cell invasion. Cancer Cell. 4:499–515.
2003.PubMed/NCBI
|
|
17
|
Salem AF, Bonuccelli G, Bevilacqua G, et
al: Caveolin-1 promotes pancreatic cancer cell differentiation and
restores membranous E-cadherin via suppression of the
epithelial-mesenchymal transition. Cell Cycle. 10:3692–3700. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bailey KM and Liu J: Caveolin-1
up-regulation during epithelial to mesenchymal transition is
mediated by focal adhesion kinase. J Biol Chem. 283:13714–13724.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schuetz P, Jones AE, Aird WC and Shapiro
NI: Endothelial cell activation in emergency department patients
with sepsis-related and non-sepsis-related hypotension. Shock.
36:104–108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki
T, Watabe T and Miyazono K: Snail is required for TGFbeta-induced
endothelial-mesenchymal transition of embryonic stem cell-derived
endothelial cells. J Cell Sci. 121:3317–3324. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Qu X, Yao J, et al: Blockade of
endothelial-mesenchymal transition by a Smad3 inhibitor delays the
early development of streptozotocin-induced diabetic nephropathy.
Diabetes. 59:2612–2624. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Widyantoro B, Emoto N, Nakayama K, et al:
Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in
diabetic hearts through stimulation of endothelial-to-mesenchymal
transition. Circulation. 121:2407–2418. 2010. View Article : Google Scholar
|
|
23
|
Zhu Y, Liao HL, Wang N, et al: Lipoprotein
promotes caveolin-1 and ras translocation to caveolae: role of
cholesterol in endothelial signaling. Arterioscler Thromb Vasc
Biol. 20:2465–2470. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Potenta S, Zeisberg E and Kalluri R: The
role of endothelial-to-mesenchymal transition in cancer
progression. Br J Cancer. 99:1375–1379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bone RC, Balk RA, Cerra FB, et al:
Definitions for sepsis and organ failure and guidelines for the use
of innovative therapies in sepsis. The ACCP/SCCM Consensus
Conference Committee. American College of Chest Physicians/Society
of Critical Care Medicine. Chest. 101:1644–1655. 1992. View Article : Google Scholar
|