Introduction
Hearing loss (HL) affects 1–3 of every 1,000
newborns; thus, it is one of the most common sensory disorders in
humans (1). This condition is
caused by environmental factors, such as noise or treatment with
ototoxic drugs (e.g., aminoglycoside antibiotics) or genomic
alterations. Hereditary HL occurs in the presence of defects either
in the nuclear genome, as the 35delG mutation in the gene encoding
connexin 26 (GJB2), or in mitochondrial DNA (mtDNA).
Mutations in mtDNA have been shown to be responsible for both
maternally inherited syndromic and non-syndromic HL (NSHL) and play
a role in the predisposition to aminoglycoside-induced ototoxicity.
Jacobs et al(2)
demonstrated that in Italy, at least 5% of cases of post-lingual,
non-syndromic hearing impairment may be attributed to mtDNA
mutations. Furthermore, it has been estimated that up to 67% of
patients with mtDNA disorders also manifest sensorineural HL (SNHL)
(3). This may be explained by the
fact that cells of the cochlea have high oxidative phosphorylation
demands, and are thus affected to a greater extent than other cells
by a mitochondrial decrease in the protein synthesis rate provoked
by mutations in mtDNA.
Non-syndromic SNHL associated with mtDNA mutations
is generally progressive (4,5),
involving mainly higher frequencies (6–8)
and is generally symmetric HL. The onset of HL usually occurs in
childhood, is predominantly post-lingual and may be accompanied
with vertigo (9) and tinnitus
(10,11). There is a high variability in
severity ranging from normal hearing to profound deafness, even
within families presenting similar genotypes (12–14); this may be due to the fact that
the phenotypic effects are a result of several factors and can
develop gradually. Some mtDNA variants, in particular in the
MT-RNR1 and tRNASer(UCN)
genes, have been identified in several cases as the main cause of
SNHL, suggesting that these two loci in particular are hotspots for
deafness-associated mutations.
The most commonly reported mutations known to cause
HL are A1555G (15), 961delT
(16–18), C1494T (19), A7445G (20,21), 7472insC (22,23) and A3243G (24,25). These variants together with the
use of aminoglycosides or in association with other mutations,
either mitochondrial or nuclear, can aggravate the condition of
hearing impairment.
In particular, it has been documented that, even
though the presence of the mutation, A1555G, itself may induce HL
(15), this effect may be
worsened in combination with aminoglycoside therapy, as this
variant produces a modification in 12 rRNA, making its secondary
structure more similar to the corresponding region of E.
coli 16S rRNA, thus much more vulnerable to the effects of this
class of antibiotics (16).
mtDNA variants, as mutations, deletions or
insertions, at position 961 in the same MT-RNR1 gene,
have been found in patients with SNHL either with or without a
history of aminoglycoside therapy (26,27). The T>G substitution in position
961 in particular, has been observed more frequently in
hearing-impaired patients compared with controls; thus, it has been
suggested to correlate with SNHL (28).
Taking into consideration that thus far, several
mutations have been examined and many are yet to be discovered, in
our study, we aimed to identify novel potentially pathogenic mtDNA
variants and establish the frequency of the known mutations in our
cohort of deaf patients.
Patients and methods
Patients
In collaboration with the Audiology Clinic at the
Hospital of Ferrara, Ferrara, Italy we retrieved data on 169
patients suffering from hearing impairment without known aetiology
and some of their close relatives. The present study was composed
of 102 females and 67 males, with an average age of 20 years
(ranging from 0 to 67 years). Their only clinical feature was HL
and they did not present any syndromic sign or other clinical
abnormalities, including muscular diseases, diabetes, visual
dysfunction or neurological disorders. The analysis referred to the
audiological tests data. In the audiometric tests, the severity of
hearing impairment was defined by pure-tone threshold average (PTA)
in frequencies: 500, 1,000, 2,000 and 4,000 Hz. HL of <20 dB was
considered as normal hearing, 21–40 dB mild HL, 41–70 dB moderate
HL, 71–90 dB severe HL and >90 dB profound HL. Written informed
consent was provided from all study participants prior to
enrollment. Any research involving human subjects was conducted in
accordance with the ethical standards of all applicable national
and institutional committees and with the World Medical
Association’s Helsinki Declaration.
Sequence analysis of mtDNA, secondary
structure analysis and sequence conservation
Total DNA was extracted from peripheral blood using
the Wizard Genomic DNA Purification kit from Promega (Madison, WI,
USA). The analysis and search for the mutations in the genes coding
for connexin 26 (GJB2), connexin 30 (GJB6) and
pendrin (SLC26A4) were carried out by the Department of
Medical Genetics at the Hospital of Ferrara.
From each subject, four regions corresponding to the
mitochondrial genomes coding for 12S RNA (MT-RNR1),
tRNA serine 1 (UCN) (MT-TS1), tRNA valine (MT-TV),
tRNA leucine 1 (MT-TL1), tRNA aspartic acid (MT-TD)
and part of 16S rRNA (MT-RNR2), NADH dehydrogenase subunit I
(MT-ND1), cytochrome c oxidase subunit I
(MT-CO1), cytochrome c oxidase subunit II
(MT-CO2) were PCR-amplified. The PCR products were analysed
by direct sequencing in the ABI 3730XL or ABI 3100 sequencing
machines at BMR Genomics (Padova, Italy). The sequence data were
compared to the revised Cambridge Sequence (rCRS), GenBank
accession no. NC_012920 (http://www.ncbi.nlm.nih.gov/nuccore/NC_012920).
The presence and the nature of all identified
nucleotide changes (polymorphisms, putative pathogenic variants,
mutations) were confirmed through mitomap (http://mitomap.org/MITOMAP) and the Human
Mitochondrial Genome Database (http://www.genpat.uu.se/mtDB/) which report published
and unpublished data on human mtDNA variations and contain a
comprehensive database of the complete human mitochondrial genomes,
including sequences from GenBank (16,411 sequences with size >14
kbp) and other sources.
In the subjects harbouring the mutations, A1555G,
A3213G, C7792T and T961G, homo/heteroplasmy was determined by
electrophoresis on a 1.2% agarose gel following enzymatic digestion
as previously described (28).
The RNAfold software (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) was
used to predict the RNA secondary structure based on minimum energy
requirements and base pair probability. The folding of sequences
containing novel mutations was compared to the wild-type
prediction.
The rCRS and the mitochondrial sequence of 18
different mammals [Gorilla gorilla, Cavia porcellus, Capra
hircus, Bos Taurus, Macaca (fascicularis, sylvanus, mulatta,
thibetana), Canis lupus familiaris, Felis catus, Equus asinus, Sus
scrofa, Mus musculus, Rattus norvegicus, Pongo abelii, Pongo
pygmaeus, Pan paniscus, Pan troglodytes] were aligned using the
ClustalW2 sequence alignment program (http://www.ebi.ac.uk/Tools/msa/clustalw2/) to analyse
the conservation of the positions of the new sequence variants
identified in our patients. We considered the variants conserved
with a conservation rate >50%.
Results
The 169 subjects presented with idiopathic SNHL and
no other symptoms. We performed a mutation analysis of four mtDNA
fragments corresponding to the hot spots for HL. We detected
mutations in GJB2 in 43 patients and excluded 18 of them
from our analysis as they did not show any association with mtDNA
variants.
Comparing the mitochondrial genomes to the rCRS, in
our cohort of patients, we found 81 different sequence alterations
(Table I), including
HL-associated A1555G, putatively pathogenic T961G and five other
mutations that have never been reported to date. Among the five
novel mutations, we hypothesised that one in particular (G786A) may
play a role in the onset of aminoglycoside-induced HL.
 | Table ImtDNA alterations detected and
conservation. |
Table I
mtDNA alterations detected and
conservation.
| Hypoacusic
(n=168) | % hypoacusia with
no GJB2 mutation (n=150) | Cons. | mtDB % on 2704 | Mitomap % on
GenBank 16411 |
|---|
| Polymorphism |
| G709A | 18 | 12 | Yes | 16.4 | 13.79 |
| T710C | 1 | 0.67 | No | 0.89 | 1.16 |
| A750G | 166 | 100 | Yes | 99.18 | 97.64 |
| G930A | 1 | 0.67 | No | 2.25 | 2.42 |
| G951A | 3 | 2 | No | 0.29 | 0.61 |
| T1189C | 5 | 3.33 | Yes | 3.85 | 4.42 |
| T1243C | 6 | 4 | Yes | 2.11 | 1.44 |
| A1438G | 163 | 100 | Yes | 96.89 | 94.99 |
| T1700C | 4 | 2.67 | No | 0.18 | 0.74 |
| G1719A | 15 | 10 | No | 4.10 | 4.41 |
| A1811G | 12 | 8 | No | 7.54 | 8.49 |
| G1888A | 9 | 6 | No | 5.32 | 6.19 |
| T3336C | 1 | 0.67 | Yes | 0.33 | 0.64 |
| A3348G | 1 | 0.67 | Yes | 1.74 | 0.80 |
| T3394C | 2 | 1.33 | Yes | 1.44 | 1.64 |
| T3396C | 1 | 0.67 | No | 0.22 | 0.83 |
| A3447G | 1 | 0.67 | Yes | 0.44 | 0.52 |
| A3480G | 4 | 2.67 | Yes | 4.85 | 5.26 |
| A3505G | 3 | 2 | No | 2.07 | 1.25 |
| G3591A | 2 | 1.33 | No | 0.74 | 0.53 |
| T3644C | 1 | 0.67 | Yes | 0.48 | 0.67 |
| G3666A | 1 | 0.67 | No | 2.15 | 2.14 |
| G3705A | 1 | 0.67 | No | 1.15 | 1.21 |
| A3720G | 1 | 0.67 | Yes | 0.70 | 0.65 |
| T3847C | 1 | 0.67 | No | 0.26 | 0.74 |
| G3915A | 7 | 4.67 | Yes | 0.81 | 1.41 |
| G7337A | 2 | 1.33 | No | 0.55 | 0.97 |
| G7521A | 3 | 2 | No | 5.62 | 5.45 |
| A7768G | 4 | 2.67 | Yes | 2.22 | 2.16 |
| G7805A | 1 | 0.67 | No | 1.37 | 0.86 |
| G7853A | 1 | 0.67 | No | 1.66 | 1.15 |
| T7961C | 2 | 1.33 | No | 0.18 | 0.72 |
| G8027A | 1 | 0.67 | No | 2.14 | 3.22 |
| Possible
HL-associated mutations |
| T961G | 6 | 4 | No | 0.18 | 0.37 |
| HL-associated
mutations |
| A1555G | 3 | 2 | Yes | 0.44 | nd |
| Novel
mutations |
| C712A | 1 | 0.67 | Yes | nd | nd |
| G786A | 1 | 0.67 | Yes | nd | nd |
| A3213G | 1 | 0.67 | Yes | nd | nd |
| C7534T | 1 | 0.67 | No | nd | nd |
| A7746G | 1 | 0.67 | No | nd | nd |
| Rare mutations |
| A644G | 1 | 0.67 | Yes | 0.04 | 0.07 |
| T721C | 2 | 1.33 | No | 0.18 | 0.24 |
| T742C | 1 | 0.67 | No | 0.07 | 0.06 |
| A813G | 1 | 0.67 | No | 1.63 | 0.49 |
| C867T | 1 | 0.67 | No | 0.04 | 0.03 |
| A942G | 2 | 1.33 | No | 0.11 | 0.09 |
| C959T | 1 | 0.67 | No | nd | 0.13 |
| T980C | 2 | 1.33 | No | 0.51 | 0.46 |
| A1118G | 1 | 0.67 | Yes | 0.04 | nd |
| T1119C | 1 | 0.67 | Yes | 0.96 | 0.45 |
| T1193C | 1 | 0.67 | Yes | 0.29 | 0.26 |
| C1405T | 2 | 1.33 | Yes | 0.04 | nd |
| T1406C | 2 | 1.33 | Yes | 0.37 | 0.32 |
| A1618G | 1 | 0.67 | Yes | 0.04 | 0.03 |
| A1708T | 1 | 0.67 | Yes | 0.04 | 0.01 |
| T3308C | 2 | 1.33 | Yes | 0.81 | 0.01 |
| C3342T | 2 | 1.33 | No | 0.04 | 0.06 |
| C3388A | 1 | 0.67 | Yes | 0.07 | 0.07 |
| T3504C | 1 | 0.67 | No | nd | 0.12 |
| A3511G | 1 | 0.67 | No | 0.04 | 0.14 |
| C3546A | 1 | 0.67 | Yes | 0.11 | 0.05 |
| T3645C | 1 | 0.67 | No | 0.18 | 0.15 |
| A3672G | 1 | 0.67 | Yes | 0.07 | 0.14 |
| A3714G | 2 | 1.33 | Yes | 0.15 | 0.17 |
| C3741T | 1 | 0.67 | No | 0.18 | 0.20 |
| C3792T | 1 | 0.67 | No | nd | nd |
| A3808G | 2 | 1.33 | Yes | 0.04 | 0.07 |
| C3903T | 1 | 0.67 | Yes | 0.04 | nd |
| C3936T | 1 | 0.67 | Yes | 0.07 | 0.04 |
| A7385G | 1 | 0.67 | Yes | 0.63 | 0.40 |
| T7440G | 1 | 0.67 | No | nd | nd |
| C7471T | 1 | 0.67 | No | nd | 0.04 |
| G7642A | 1 | 0.67 | No | 0.30 | 0.25 |
| T7645C | 2 | 1.33 | No | 0.22 | 0.29 |
| T7705C | 2 | 1.33 | No | 0.18 | 0.40 |
| A7717G | 1 | 0.67 | Yes | nd | nd |
| A7720G | 1 | 0.67 | Yes | nd | 0.01 |
| C7792T | 1 | 0.67 | Yes | nd | 0.04 |
| G7830A | 1 | 0.67 | No | 0.15 | 0.10 |
| C7873T | 1 | 0.67 | No | 0.15 | 0.12 |
| G7984A | 1 | 0.67 | No | 0.07 | 0.07 |
| A8014T | 1 | 0.67 | No | 0.15 | 0.32 |
A1555G
Three genetically unrelated subjects harboured the
homoplasmic A1555G mutation in the MT-RNR1 gene, a
mtDNA variant that has been associated with deafness. The subjects
were two females and one male with an average age of 47 years
suffering from SNHL. The enzymatic digestion of the fragment showed
homoplasmy in all cases. The phenotypes were different as one was
congenitally deaf, and the other two had the onset of the symptoms
at 5 and at 19 years, respectively; unfortunately, none of them
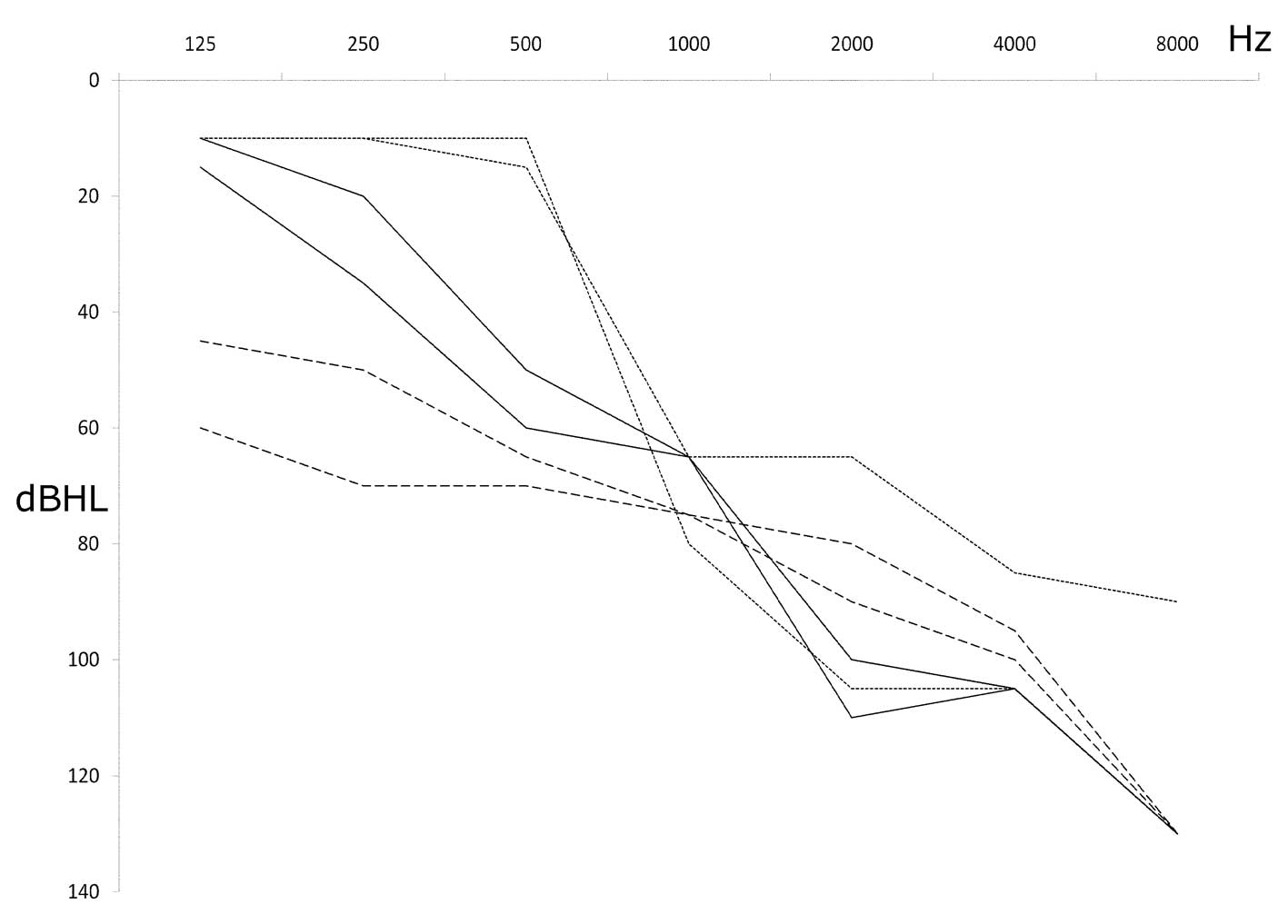
could recall any previous exposure to aminoglycosides (Table II).
| Table IIPatients harbouring the hearing
loss-associated A1555G mutation. |
Table II
Patients harbouring the hearing
loss-associated A1555G mutation.
| Patient | Gender | Age (years) |
Homo/heteroplasmy | 2d | GJB2 | GJB6 | SLC26A4 | Age of onset
(years) | PTA dx | PTA sn | Family history of
HL | Other mtDNA
mutations | Type of line in
Fig. 1 |
|---|
| Mit76 | F | 47 | Homo | Yes | wt | wt | wt | 5 | 80 | 81.2 | No, sporadic | C7471T; T3504C | Continuous |
| Mit114 | F | 44 | Homo | Yes | wt | wt | wt | 19 | 56.2 | 75 | No, sporadic | No | Dotted |
| Mit140 | M | 50 | Homo | Yes | wt | wt | wt | At birth | 80 | 82.5 | nd | No | Dashed |
Audiometric examination in all the affected
individuals showed a downsloping curve confirming the typical
pattern of mitochondrial SNHL, which implicates the loss of high
hearing frequencies (Fig. 1). One
of these patients with severe progressive hearing impairment
harboured two additional mutations whose pathogenicity has yet to
be defined: T3504C, a rare variant in the gene coding for
MT-ND1; and C7471T, a very rare mtDNA variant located in the
extraloop of tRNASer(UCN).
T961G
Six patients harboured the mutation, T961G, in
MT-RNR1. The phenotypes, as well as the audiometric
tests in our T961G cases, were quite disparate as we found two
young sisters (mit26 and mit29) with a moderate hearing impairment,
whose father and mother were normoacusic even though the latter had
the same mtDNA variant. Mit51 showed post-lingual asymmetric
progressive HL and in addition to T961G, harboured an additional
mutation close to it (C959T) with a low frequency in the databases.
Mit116 presented with profound familial congenital SNHL. As for the
last two patients, mit178 had hypoplasia of the cochlea and mit186
presented with progressive bilateral HL, which was later diagnosed
as partial trisomy of chromosome 6p.
We defined the homoplasmy in all of the cases, with
the exception of mit51 whose state could not be determined as the
presence of the other mutation in position 959 prevented the
AciI restriction enzyme digestion. The comparison of the RNA
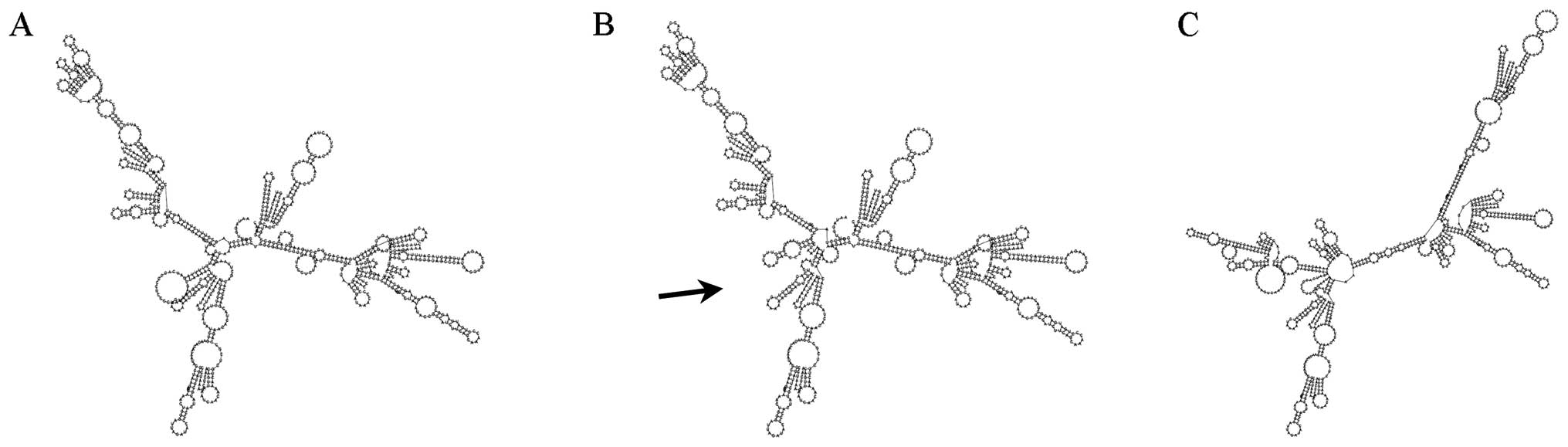
secondary structure determined by this mutation shows a clear
difference with the wild-type one (Fig. 2).
Novel mutations
We detected novel sequence variants not present in
the literature or in mitochondrial databases (Table III), including C712A and the
heteroplasmic G786A in MT-RNR1, A3213G in
MT-RNR2, C7534T in the D-loop of TRND
(tRNAAsp) and in the MT-CO2 gene, A7746G, which
produces an aminoacidic change in translation. All the mutations
were recorded in mitomap (http://www.mitomap.org/bin/view.pl/MITOMAP/VariantSubmissionList)
and numbered from 20111230001 onwards. In a phylogenetic analysis,
we compared the human nucleotide variants with other 18 different
mammals and found a conservation rate of >50% for variants 712,
786 and 3213.
| Table IIINovel mutations. |
Table III
Novel mutations.
| Mutation |
Homo/heteroplasmy | Gene | GJB2 | GJB6 | SLC26A4 | Age of onset
(years) | PTA dx | PTA sn | Family history of
HL | Other mtDNA
mutations | Conserved | Patient | Age (years) | Gender | Notes | Origin | 2d |
|---|
| C712A | Homo | RNR1 | wt/wt | wt/wt | nd | 0 | 35 | 40 | Uncle? | A1811G | Yes | Mit184 | 13 | M | SNHL
Perinatal asphyxia?
Normoacusic brother | Italy
Sardinia | Yes |
| G786A | Hetero | RNR1 | wt/wt | wt/wt | nd | Post-lingual | 85 | 72.5 | No, sporadic | No | Yes | Mit7 | 39 | F | SNHL
progressive | Italy | Yes |
| A3213G | nd | RNR3 | wt/wt | wt/wt | 6,7, 8, 10,19
wt | Congenital | 95 | 95 | No, sporadic | A3348G; G3591A;
A3714G; G7642A; G7805A | Yes | Mit70 | 4 | F | SNHL | Morocco | No |
A7746G
Asn→Ser | Homo | CO2 | wt/wt | wt/wt | wt/wt | 0 | 61 | 59 | No, sporadic | T980C | No | Mit100 | 4 | M | SNHL otitis CT
ok | Italy | Yes |
Among the novel mutations, we particularly
considered the heteroplasmic G786A in mit7, a 39-year-old female.
Her parents and sister were normoacusic and did not harbour any
mutations either in mtDNA or in HL-associated genes. She suffered
from asymmetric progressive SNHL and had been treated with
streptomycin in her childhood. In our alignment analysis, position
786 in the MT-RNR1 gene was quite conserved (14/18);
moreover, the mutated secondary structure prediction showed to be
different compared to the wild-type one (Fig. 2).
In MT-RNR1 we also found the mutation,
C712A, which may have an effect on HL as the site shows a 100%
conservation even if no differences in the RNA structure of the
gene are detectable. The patient harbouring this variant, a
13-year-old subject with mild SNHL (mit184), harboured two
additional mutations of A1811G: a polymorphism and a quite rare
C>A mutation in the evolutionarily conserved position 3546 in
the MT-ND1 gene.
A3213G in MT-RNR2 was detected in a young
girl from Morocco harbouring several other variants (A3348G,
G3591A, A3714G, G7642A and G7805A) with congenital profound SNHL.
This was conserved and had a different RNA structure.
Mit145 harboured C7534T in the D-loop of
tRNAAsp together with G709A and the rare A8014T mutation in
MT-CO2. The other novel variant, A7746G, detected in the
MT-CO2 gene, not conserved, was found in a 5-year-old boy
also harbouring the T980C variant in MT-RNR1. A7746G
presents a missense mutation with the aminoacidic change Asn>Ser
in the subunit of cytochrome c oxidase (complex IV).
Low frequency mutations
We identified several other variants that may be
associated with hearing impairment, presenting a low frequency in
mtDB and mitomap (Table IV).
Among these, we preferably considered the mutations in subjects
presenting audiograms compatible to a mitochondrial mutation HL
diagnosis, in conserved positions and with a frequency <0.05%,
such as: i) the mutation A>G in the conserved position 644
(0,04% in mtDB) in MT-TF, located in the acceptor stem of
tRNAPhe. 644A>G found in a 13-year-old girl with SNHL,
harbouring the polymorphisms, G709A, G1888A and C7873T; ii) T721C
in MT-RNR1. This 36-year-old female had progressive HL which
began at age 22; the RNA structure though did not seem to differ
from the wild-type one; in fact, we eventually found the same
mutation in a 34-year-old male heterozygotic for connexin 26 35delG
who was normoacusic; iii) T1119C in MT-RNR1 found in
mit110: a 36-year-old patient with progressive post-lingual
bilateral SNHL which began at age 33; the RNA showed a different
structure; iv) C3342T in the ND1 gene in two deaf sisters
harbouring both the additional mutation T7961C; v) A3808G a
mutation in a conserved site found in two sisters with audiograms
compatible to mitochondrial deafness; vi) A3847G in a case of a
37-year-old female whose mild sporadic hearing impairment began in
her thirties; vii) A7720G in MT-CO2 in a 3-year-old subject
presenting with mild progressive hearing impairment; viii) C7792T
in MT-CO2, observed in a 42-year-old male with progressive
hearing impairment which began in his twenties, presenting with
moderate to severe symmetric impairment confirmed by a downward
overlapping audiogram; ix) G7830A G7984A together with G709A and
G1888A in a 45-year-old female with moderate HL at high
frequencies.
| Table IVRare mutations. |
Table IV
Rare mutations.
| Mutation | Gene | GJB2 | GJB6 | SLC26A4 | Age of onset
(years) | PTA dx | PTA sn | Family history of
HL | Other mtDNA
mutations | Conserved | Patient | Age (years) | Gender | Notes |
|---|
| A644G | RNR1 | wt/wt | wt/wt | nd | nd | 40 | 43.75 | No | G709A; G1888A;
C7873T | Yes | Mit22 | 13 | F | Italy, SNHL,
microhematuria, close to 642 (T>C = HL) |
| T721C | RNR1 | wt/wt | wt/wt | wt/wt | 22 | 89 | 81 | No, sporadic | No | No | Mit109 | 36 | F | Italy, SNHL |
| T721C | RNR1 | 35delG/wt | wt/wt | nd | No | 0 | 0 | No, sporadic | No | No | Mit162 | 34 | M | Italy,
normoacusic |
| A813G | RNR1 | nd | nd | nd | nd | 70 | 70 | No, sporadic | No | No | Mit58 | 49 | M | Italy, SNHL, MRI
ok |
| T1119C | RNR1 | wt/wt | wt/wt | wt/wt | 33 | 28 | 25 | Maybe mother | G709A; T1243C | Yes | Mit110 | 36 | F | Italy, progressive
SNHL |
| C3342T | ND1 | wt/wt | wt/wt | wt/wt | nd | 61 | 60 | nd | T7961C | No | Mit158–159 | 45–49 | M-F | Italy, SNHL,
brothers, medium + high frequencies |
| T3504C | ND1 | wt/wt | wt/wt | wt/wt | 5 | 80 | 81.25 | No, sporadic | A1555G; C7471T | No | Mit76 | 47 | F | Italy, SNHL |
A3511G
Thr→Ala | ND1 | wt/wt | wt/wt | wt/wt | 0 | 54 | 79 | Adopted | G709A;
T1193C
T3394C; G3591A | No | Mit87 | 8 | F | India |
| C3546A | ND1 | wt/wt | wt/wt | nd | 0 | 35 | 40 | Uncle? | C712A; A1811G | Yes | Mit184 | 13 | M | Sardinia, SNHL
perinatal asphyxia. Normoacusic brother |
T3644C
Val→Ala | ND1 | wt/wt | wt/wt | nd | 1.2 | 122.5 | 122.5 | No, sporadic | T3336C; T3396C | Yes | Mit61 | 10 | F | Ecuador, SNHL not
progressive |
| T3645C | ND1 | nd | nd | nd | nd | Moderate | Moderate | Familial | G1719A; T3645C;
G7521A | No | Mit151 | 56 | F | Italy, SNHL |
| A3672G | ND1 | wt/wt | wt/wt | wt/wt | 0 | nd | nd | No, sporadic | A1811G; T7705C | Yes | Mit167 | 0 | F | Italy, SNHL |
| G3705A | ND1 | wt/wt | wt/wt | wt/wt | 2.5 | 115 | 16 | No, sporadic | G709A; G1888A;
G3705A | No | Mit127 | nd | M | Albania, SNHL |
| A3720G | ND1 | wt/wt | wt/wt | nd | 4 | 71 | 61 | No | A1811G; A3720G | Yes | Mit129 | 44 | F | Italy, SNHL
progressive, otitis |
| C3741T | ND1 | wt/wt | wt/wt | wt/wt | 6 months | nd | nd | No, sporadic | T980C; A1811G | No | Mit180 | 3 | M | Italy,
SNHL
CT/MRI ok
Normoacusic brother |
| A3808G | ND1 | wt/wt | wt/wt | nd | nd | 80 | 82.5 | nd | G1719A | Yes | Mit24–25 | 41–46 | F | Italy, SNHL,
sisters |
| T3847C | ND1 | nd | nd | nd | 30 | 38.75 | 38.75 | No, sporadic | No | Yes | Mit72 | 37 | F | Italy, SNHL |
| C3936T | ND1 | wt/wt | wt/wt | wt/wt | 2.5 | 34 | 75 | No, sporadic | No | Yes | Mit153 | 4 | M | Italy, SNHL |
| A7385G | CO1 | wt/wt | wt/wt | nd | nd | 70 | 35 | No, sporadic | A7768G | Yes | Mit57 | 51 | M | Italy, SNHL |
T7440G
Ser→Ala | CO1 | wt/wt | wt/wt | nd | 7 | nd | nd | No, sporadic | G709A; G1888A;
T7440G | No | Mit143 | 8 | M | Italy SNHL IQ ok.
premature |
| C7471T |
S(UCN) | wt/wt | wt/wt | wt/wt | nd | 80 | 81.2 | No, sporadic | A1555G; T3504C | No | Mit76 | 47 | F | Italy SNHL, close
to 7,472 known to cause HL |
| G7642A | CO2 | wt/wt | wt/wt | 6, 7, 8, 10, 19
wt | Congenital | 95 | 95 | nd | A3213G; A3348G;
G3591A; A3714G; G7805A | No | Mit70 | 4 | F | Morocco, SNHL
negative anamnesis |
| A7720G | CO2 | wt/wt | wt/wt | wt/wt | nd | 19 | 26 | No, sporadic | No | Yes | Mit89 | 3 | M | Not Italy,
SNHL |
| C7792T | CO2 | wt/wt | wt/wt | nd | 22 | 69 | 68 | nd | G8020A | Yes | Mit142 | 42 | M | Italy SNHL
progressive |
G7805A
Val→Ile | CO2 | wt/wt | wt/wt | 6, 7, 8, 10, 19
wt | Congenital | 95 | 95 | nd | A3213G; A3348G;
G3591A; A3714G; G7642A | No | Mit70 | 4 | F | Morocco, SNHL
negative anamnesis |
G7830A
Arg→His | CO2 | wt/wt | wt/wt | wt/wt | nd | 60 | 60 | nd | G709A; G1888A;
G7984A | Yes | Mit94 | 46 | F | Italy, SNHL |
| G7853A | CO2 | nd | nd | nd | nd | 19 | 26.25 | nd | G709A; G1888A;
G7853A | No | Mit187 | 5 | M | Italy, SNHL |
| C7873T | CO2 | wt/wt | wt/wt | nd | nd | 40.00 | 43.75 | No | A644G; G709A;
G1888A; C7873T | No | Mit22 | 13 | F | Italy, SNHL |
| G7984A | CO2 | wt/wt | wt/wt | wt/wt | nd | 60 | 60 | nd | G709A; G1888A;
G7830A | No | Mit94 | 46 | F | Italy, SNHL |
Connexin 26 and mtDNA mutations
We searched for a correlation between mutations in
connexin 26 and mtDNA mutations. Eighteen patients only harboured
mutations in the GJB2 gene and 25 of them harboured both the
GJB2 and mtDNA variants (10 of whom were homozygotic for
35delG).
In our subjects, we noticed a higher presence of the
G3915A polymorphism, as 5 out of the total 7 probands with this
polymorphism in NDI were associated with GJB2
mutations. In two siblings with SNHL and 35delG/35delG in
GJB2, we identified the missense mutation, T3308C
(Met>Thr), at the highly conserved amino acid position 1 in
MT-ND1. Among the patients with homozygotic 35delG in
GJB2 we found some rare mutations that may worsen their
condition of hearing impairment (Table V). The mutations found were
A3447G, C3903T, A7717G and G8027A, all in conserved positions in
the genes MT-ND1 and MT-CO2. In the literature these
were found to be more involved in Leber’s hereditary optic
neuropathy (LHON) than in HL. Another patient homozygotic for
35delG showed two additional variants: the missense mutation,
A3505G, causing the Thr>Ala substitution in MT-ND1 and
the conserved T1243C mutation in the MT-RNR1
gene.
| Table VmtDNA and GJB2 mutations. |
Table V
mtDNA and GJB2 mutations.
| Sample | mtDNA mutation | Gene | GJB2 | Notes |
|---|
| Mit1–2 | C1405T | RNR1 | 35delG/35delG | Severe SNHL,
homozygous twins |
| Mit46–47 | T3308C | ND1 | 35delG/35delG |
Mild-moderate
SNHL, brothers |
| Mit73 | A3447G; G8027A | ND1; CO2 | 35delG/35delG | Profound SNHL,
familial |
| Mit74 | G3915A | ND1 | 35delG/35delG | SNHL |
| Mit135 | T7645C | CO2 | 35delG/35delG | Profound SNHL |
| Mit154 | T1243C; A3505G;
C3792T | RNR1; ND1 | 35delG/35delG | Profound SNHL |
| Mit185 | A942G; T3394C | RNR1; ND1 | 35delG/35delG | Moderate SNHL,
progressive |
| Mitpds7 | C3903T; A7717G | ND1; CO2 | 35delG/35delG | SNHL, congenital,
familial |
| Mit4 | G3915A | ND1 | L90P/M34T | SNHL,
congenital |
| Mit5 | G3915A | ND1 | 35delG/L90P | SNHL,
congenital |
| Mit6 | G3915A | ND1 | 35delG/L90P | SNHL,
congenital |
| Mit32 | G1719A | RNR2 | 35delG/wt | SNHL, progressive,
familial |
| Mit49 | T1189C; A1811G;
A3480G | RNR1; RNR2;
ND1 | R127H/wt | Profound SNHL,
familial, onset at age 4 |
| Mit83–115 | G7521A | TD | L90P/wt | EVA,
transmissive
HL, onset at age 3, brothers |
| Mit123 | G3915A | ND1 | M34T/wt | Moderate SNHL |
| Mit133 | A1811G; A3480G | RNR2; ND1 | 35delG/R184P | Profound SNHL,
congenital |
| Mit139 | C959T; G1719A | RNR1; RNR2 | wt/35delG | Normoacusic |
| Mit145 | G709A; C7534T;
A8014T | RNR1; TD; CO2 | wt/del120E | Normoacusic |
| Mit155 | A7768G | CO2 | wt/M34T | SNHL, sisters |
| Mit156 | A7768G | CO2 | 35delG/M34T | SNHL, sisters |
| Mit162 | T721C | RNR1 | 35delG/wt | Normoacusic |
Discussion
In the present study, we analysed four fragments of
mtDNA in 169 subjects with non-syndromic SNHL, both familial and
sporadic without a clear aetiology. We compared our data with the
DNA of some of their relatives who were normoacusic in order to
define whether the mutations were sporadic or genetically
transmitted. We also considered the mutations in the GJB2,
GJB6 and SLC26A4 genes which are recognised to be
among the most frequent causes of hearing impairment. In total, 43
patients harboured GJB2 mutations and 18 were affected by
GJB2 mutations only (no mtDNA mutations). We thus decided to
exclude this group from our analysis.
The hearing-impaired patients showed a wide range of
penetrance, severity and age-at-onset of HL. We searched for
mutations in the regions corresponding to the hotspots for
deafness: the MT-RNR1 and the MT-TS1 genes, as
the presence of mutations in these two genes in particular, is
known to cause both syndromic and non-syndromic forms of hearing
impairment; we also focused on the region of MT-TL1
as previous studies report its possible role in non-syndromic
disease (29). In order to
establish the potential pathogenicity of the mutations encountered,
we analysed the evolutionary conservation comparing our sequences
to those of other organisms. Furthermore, considering that the
biological functions of 16S rRNA and tRNAs and other structural
RNAs are dictated by their three dimensional structures, we
analysed the possible RNA secondary structure of the mutated
samples and predicted the folding using the Vienna RNA package. Our
aim was to detect and correlate the frequency of mtDNA alterations
in the cases of deafness showing the typical audiological
manifestations of mitochondrial SNHL.
In our cohort of patients, we identified three
subjects harbouring the A1555G mutation. This mutation in the
MT-RNR1 gene is one of the most common mtDNA variants
associated with both non-syndromic progressive SNHL and
aminoglycoside-induced SNHL. Sequence analysis of the
MT-RNR1 gene in our subjects identified three
genetically unrelated individuals harbouring the A1555G mutation
who showed the typical mitochondrial HL audiometric features. The
incidence of the mutation in hypoacusic subjects was 2%, a little
lower than the one recognised by Berrettini et al(29) in 2008, but similar to the data
presented in the studies by Jacobs et al(2) and Lingala et al(30). We could not state if the use of
aminoglycosides had any effect on these subjects as they could not
recall any exposure to antibiotics in the past; however, one of
these patients with severe and progressive HL harboured a novel
mutation in position 7471 in tRNASer(UCN),
close to position 7472, which has shown to cause both syndromic and
non-syndromic deafness (31),
suggesting that this variant somehow functions as a modifier, in
synergy with the primary mutation, thus modulating its phenotypic
manifestations as observed for other tRNA mutations (32).
We identified another mutation in the
MT-RNR1 gene: seven patients harboured the T961G
mutation with a frequency corresponding to data reported in the
literature. Its pathogenicity is quite controversial: the mutations
at position 961 have been detected in subjects affected by
aminoglycoside-induced NSHL. The delT961Cn mutation is more
frequent in Caucasian and Asian subjects (16,26,27,33), as well as the 961C insertion
(17,19,27,28), T961C mutation in Chinese subjects
(4) and T961G mutation in the
Caucasian population (17). In a
previous study, Li et al(28) found the T961G substitution in
5/164 hearing-impaired paediatric patients of Caucasian descent
without a history of exposure to aminoglycoside, while the 226
Caucasian and 324 Chinese control subjects did not harbour this
mutation; thus, it was hypothesised that this variant may be
associated with SNHL. In contrast to these results indicating a
possible pathogenic nature of the mutations around position 961 in
NSHL and aminoglycoside-induced HL, Herrnstadt et al stated
that it could be a typical polymorphism of the H2 haplogroup
(34). The localization of
position 961 is at the C-cluster of the region between loop 21 and
22 of MT-RNR1(35);
compared with A1555G this region is not evolutionarily conserved
and is in fact highly polymorphic in mammalian interspecies
comparisons. Its function is not well defined; in particular, its
pathogenic mechanisms of action in the predisposition of carriers
to aminoglycoside toxicity remain unclear (17,36). Elstner et al performed a
single nucleotide polymorphism (SNP) analysis of the nucleotide 961
in a control group of 320 German samples, finding six T>C and
five T>G nucleotide changes (37). Thus, the effects of this mutation
have yet not been defined; we confirmed this mutation in our
screening; six out of seven patients with T961G showed variable
degrees of hearing impairment, suggesting at least a minor role in
the HL onset; however, at the same time the mother of two
hearing-impaired children harbouring the same mutation did not
present with HL. Thus, it can be hypothesised that T961G is either
a polymorphism, or a pathogenic mutation with an extremely low
penetrance.
One of the subjects in our cohort of patients
harboured a novel mutation in position 786 in
MT-RNR1. She did not harbour any other mutations in
the genes usually associated with HL or any malformations. This
alteration, in our opinion, could be the reason of her HL since its
conserved site in the hotspot gene for HL and also as the RNA
structure shows a clearly different folding compared with the
wild-type one, suggesting a possible malfunctioning of the
ribosome. From a clinical point of view, the patient presented with
sporadic progressive SNHL with post-lingual onset; her audiometry
was compatible with mitochondrial-associated HL and the fact that
she was treated with aminoglycosides in the past confirms our
hypothesis.
In our patient cohort, some other novel mtDNA
variants in genes that are not usually involved in HL or have an
association with other pathological conditions were recorded,
though their exact role is unclear; thus, they should be
investigated, further studied and compared with new cases.
We suggest that some of the rare mutations harboured
by patients with audiometric data compatible with a mitochondrial
HL are possible candidates for genetic risk factors of NSHL. Among
these, we considered T1119C in MT-RNR1. We suggest
that this variant detected in 36-year-old female may be responsible
for her mild progressive bilateral SNHL, which began three years
earlier. T1119C, already found in four subjects suffering from
hearing impairment by Li et al(4), located in a conserved site and
presenting with a different RNA structure, may be the cause of HL
at high frequencies, confirmed by an audiogram. It should be noted
that the late onset and gradual worsening of the impairment may
reflect the tendency of the mitochondrion to accumulate mutations
with aging due to its genomic instability.
Of note, we observed the non-pathogenicity of the
T721C mutation in MT-RNR1 that we thought could be
responsible for the progressive HL of a 36-year-old female which
began at age 22. In fact, we eventually detected the same mutation
in a 34-year-old male heterozygotic for connexin 26 35delG who was
normoacusic.
In patients harbouring mutations in the most common
HL-associated genes (connexin 26), we focused on the mtDNA
mutations, in particular T3308C, which results in a change in the
initiation codon of NADH dehydrogenase. In a study on mutant cells,
Li et al(38).
demonstrated that T3308C induces a significant decrease in the
levels of MT-ND1, resulting in a decreased complex I
activity; furthermore, the T3308C mutation may also alter the
hydrophobicity and antigenicity of the N-terminal peptide of
MT-ND1(39). These facts
suggest that a combination of a mtDNA mutation with other genomic
DNA mutations may increase the penetrance of deafness.
In conclusion, our data confirm a frequency of 2%
for the A1555G mutation and its role in NSHL; however, the
pathogenicity of all the other mtDNA variants encountered should be
established: the variability of the frequency in different
haplogroups, the occurrence in normal hearing individuals and the
correlation with other conditions and mutations should be taken
into account; thus, further genetic and functional studies are
required in order to define their possible additional correlation
with NSHL and/or aminoglycoside-induced HL.
Acknowledgements
The present study was supported by ‘Ospedale
Infantile e Pie Fondazioni Burlo Garofolo e dott. Alessandro ed
Aglaia de Manussi’ Trieste.
References
|
1
|
Morton CC: Genetics, genomics and gene
discovery in the auditory system. Hum Mol Genet. 11:1229–1240.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jacobs HT, Hutchin TP, Käppi T, Gillies G,
Minkkinen K, Walker J, Thompson K, Rovio AT, Carella M, Melchionda
S, Zelante L, Gasparini P, Pyykkö I, Shah ZH, Zeviani M and Mueller
RF: Mitochondrial DNA mutations in patients with postlingual,
nonsyndromic hearing impairment. Eur J Hum Genet. 13:26–33. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martini A: Genetica della Funzione Uditiva
Normale e Patologica. Edizioni Omega; Torino: 2006
|
|
4
|
Li Z, Li R, Chen J, Liao Z, Liao Z, Zhu Y,
Qian Y, Xiong S, Heman-Ackah S, Wu J, Choo DI and Guan MX:
Mutational analysis of the mitochondrial 12S rRNA gene in Chinese
pediatric subjects with aminoglycoside-induced and non-syndromic
hearing loss. Hum Genet. 117:9–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sinnathuray AR, Raut V, Awa A, Magee A and
Toner JG: A review of cochlear implantation in mitochondrial
sensorineural hearing loss. Otol Neurotol. 24:418–426. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ballana E, Morales E, Rabionet R,
Montserrat B, Ventayol M, Bravo O, Gasparini P and Estivill X:
Mitochondrial 12S rRNA gene mutations affect RNA secondary
structure and lead to variable penetrance in hearing impairment.
Biochem Biophys Res Commun. 341:950–957. 2006. View Article : Google Scholar
|
|
7
|
Gürtler N, Schmuziger N, Kim Y, Mhatre AN,
Jungi M and Lalwani AK: Audiologic testing and molecular analysis
of 12S rRNA in patients receiving aminoglycosides. Laryngoscope.
115:640–644. 2005.PubMed/NCBI
|
|
8
|
Scaglia F, Hsu CH, Kwon H, Bai RK, Perng
CL, Chang HM, Dai P, Smith EO, Whiteman DA, Feigenbaum A, Gropman A
and Wong LJ: Molecular bases of hearing loss in multi-systemic
mitochondrial cytopathy. Genet Med. 8:641–652. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen JN, Ho KY and Juan KH: Sensorineural
hearing loss in MELAS syndrome - case report. Kaohsiung J Med Sci.
14:519–523. 1998.PubMed/NCBI
|
|
10
|
Matsunaga T, Kumanomido H, Shiroma M,
Ohtsuka A, Asamura K and Usami S: Deafness due to A1555G
mitochondrial mutation without use of aminoglycoside. Laryngoscope.
114:1085–1091. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Simdon J, Watters D, Bartlett S and
Connick E: Ototoxicity associated with use of nucleoside analog
reverse transcriptase inhibitors: a report of 3 possible cases and
review of the literature. Clin Infect Dis. 32:1623–1627. 2001.
View Article : Google Scholar
|
|
12
|
Bravo O, Ballana E and Estivill X:
Cochlear alterations in deaf and unaffected subjects carrying the
deafness-associated A1555G mutation in the mitochondrial 12S rRNA
gene. Biochem Biophys Res Commun. 344:511–516. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rodriguez-Ballesteros M, Olarte M, Aguirre
LA, Galan F, Galan R, Vallejo LA, Navas C, Villamar M,
Moreno-Pelayo MA, Moreno F and del Castillo I: Molecular and
clinical characterisation of three Spanish families with maternally
inherited non-syndromic hearing loss caused by the 1494C->T
mutation in the mitochondrial 12S rRNA gene. J Med Genet.
43:e542006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Young WY, Zhao L, Qian Y, Li R, Chen J,
Yuan H, Dai P, Zhai S, Han D and Guan MX: Variants in mitochondrial
tRNAGlu, tRNAArg, and tRNAThr may influence the phenotypic
manifestation of deafness-associated 12S rRNA A1555G mutation in
three Han Chinese families with hearing loss. Am J Med Genet A.
140:2188–2197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Prezant TR, Agapian JV, Bohlman MC, Bu X,
Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI,
Shohat M and Fischel-Ghodsian N: Mitochondrial ribosomal RNA
mutation associated with both antibiotic-induced and non-syndromic
deafness. Nat Genet. 4:289–294. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bacino C, Prezant TR, Bu X, Fournier P and
Fischel-Ghodsian N: Susceptibility mutations in the mitochondrial
small ribosomal RNA gene in aminoglycoside induced deafness.
Pharmacogenetics. 5:165–172. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li R, Xing G, Yan M, Cao X, Liu XZ, Bu X
and Guan MX: Cosegregation of C-insertion at position 961 with
A1555G mutation of mitochondrial 12S rRNA gene in a large Chinese
family with maternally inherited hearing loss. Am J Med Genet A.
124A:113–117. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yoshida M, Shintani T, Hirao M, Himi T,
Yamaguchi A and Kikuchi K: Aminoglycoside-induced hearing loss in a
patient with the 961 mutation in mitochondrial DNA. ORL J
Otorhinolaryngol Relat Spec. 64:219–222. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao H, Li R, Wang Q, Yan Q, Deng JH, Han
D, Bai Y, Young WY and Guan MX: Maternally inherited
aminoglycoside-induced and nonsyndromic deafness is associated with
the novel C1494T mutation in the mitochondrial 12S rRNA gene in a
large Chinese family. Am J Hum Genet. 74:139–152. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reid FM, Vernham GA and Jacobs HT: A novel
mitochondrial point mutation in a maternal pedigree with
sensorineural deafness. Hum Mutat. 3:243–247. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fischel-Ghodsian N, Prezant TR, Fournier
P, Stewart IA and Maw M: Mitochondrial mutation associated with
nonsyndromic deafness. Am J Otolaryngol. 16:403–408. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tiranti V, Chariot P, Carella F, Toscano
A, Soliveri P, Girlanda P, Carrara F, Fratta GM, Reid FM, Mariotti
C and Zeviani M: Maternally inherited hearing loss, ataxia and
myoclonus associated with a novel point mutation in mitochondrial
tRNASer(UCN)gene. Hum Mol Genet. 4:1421–1427. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Verhoeven K, Ensink RJ, Tiranti V, Huygen
PL, Johnson DF, Schatteman I, Van Laer L, Verstreken M, Van de
Heyning P, Fischel-Ghodsian N, Zeviani M, Cremers CW, Willems PJ
and Van Camp G: Hearing impairment and neurological dysfunction
associated with a mutation in the mitochondrial
tRNASer(UCN)gene. Eur J Hum Genet. 7:45–51. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Majamaa K, Moilanen JS, Uimonen S, Remes
AM, Salmela PI, Kärppä M, Majamaa-Voltti KA, Rusanen H, Sorri M,
Peuhkurinen KJ and Hassinen IE: Epidemiology of A3243G, the
mutation for mitochondrial encephalomyopathy, lactic acidosis, and
strokelike episodes: prevalence of the mutation in an adult
population. Am J Hum Genet. 63:447–454. 1998. View Article : Google Scholar
|
|
25
|
Van den Ouweland JM, Lemkes HH, Ruitenbeek
W, et al: Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large
pedigree with maternally transmitted type II diabetes mellitus and
deafness. Nat Genet. 1:368–371. 1992.PubMed/NCBI
|
|
26
|
Casano RA, Johnson DF, Bykhovskaya Y,
Torricelli F, Bigozzi M and Fischel-Ghodsian N: Inherited
susceptibility to aminoglycoside ototoxicity: genetic heterogeneity
and clinical implications. Am J Otolaryngol. 20:151–156. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang HY, Hutcheson E, Neill S,
Drummond-Borg M, Speer M and Alford RL: Genetic susceptibility to
aminoglycoside ototoxicity: how many are at risk? Genet Med.
4:336–345. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li R, Greinwald JH Jr, Yang L, Choo DI,
Wenstrup RJ and Guan MX: Molecular analysis of mitochondrial 12S
rRNA and tRNASer(UCN)genes in paediatric subjects with
non-syndromic hearing loss. J Med Genet. 41:615–620. 2004.
View Article : Google Scholar
|
|
29
|
Berrettini S, Forli F, Passetti S, Rocchi
A, Pollina L, Cecchetti D, Mancuso M and Siciliano G: Mitochondrial
non-syndromic sensorineural hearing loss: a clinical, audiological
and pathological study from Italy, and revision of the literature.
Biosci Rep. 28:49–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lingala HB and Sankarathi Penagaluru PR:
Role of connexin 26 (GJB2) & mitochondrial small ribosomal RNA
(mt 12S rRNA) genes in sporadic & aminoglycoside-induced non
syndromic hearing impairment. Indian J Med Res. 130:369–378.
2009.
|
|
31
|
Hutchin TP and Cortopassi GA:
Mitochondrial defects and hearing loss. Cell Mol Life Sci.
57:1927–1937. 2000. View Article : Google Scholar
|
|
32
|
Zheng J, Ji Y and Guan MX: Mitochondrial
tRNA mutations associated with deafness. Mitochondrion. 12:406–413.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Konings A, Van Camp G, Goethals A, Van
Eyken E, Vandevelde A, Ben Azza J, Peeters N, Wuyts W, Smeets H and
Van Laer L: Mutation analysis of mitochondrial DNA 12SrRNA and
tRNASer(UCN) genes in non-syndromic hearing loss patients.
Mitochondrion. 8:377–382. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Herrnstadt C, Elson JL, Fahy E, Preston G,
Turnbull DM, Anderson C, Ghosh SS, Olefsky JM, Beal FM, Davis RE
and Howell N: Reduced-median-network analysis of complete
mitochondrial DNA coding-region sequences for the major African,
Asian, and European haplogroups. Am J Hum Genet. 70:1152–1171.
2002. View
Article : Google Scholar
|
|
35
|
Neefs JM, Van de Peer Y, De Rijik P, Goris
A and De Wachter R: Compilation of small ribosomal subunit RNA
sequences. Nucleic Acids Res. 19 Suppl:1987–2015. 1991. View Article : Google Scholar
|
|
36
|
Del Castillo FJ, Rodriguez-Ballesteros M,
Martin Y, Arellano B, Gallo-Terán J, Morales-Angulo C,
Ramirez-Camacho R, Cruz Tapia M, Solanellas J, Martinez-Conde A,
Villamar M, Moreno-Pelayo MA, Moreno F and del Castillo I:
Heteroplasmy for the 1555A>G mutation in the mitochondrial 12S
rRNA gene in six Spanish families with non-syndromic hearing loss.
J Med Genet. 40:632–636. 2003.
|
|
37
|
Elstner M, Schmidt C, Zingler VC, Prokisch
H, Bettecken T, Elson JL, Rudolph G, Bender A, Halmagyi GM, Brandt
T, Strupp M and Klopstock T: Mitochondrial 12S rRNA susceptibility
mutations in aminoglycoside-associated and idiopathic bilateral
vestibulopathy. Biochem Biophys Res Commun. 377:379–383. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li X, Fischel-Ghodsian N, Schwartz F, Yan
Q, Friedman RA and Guan MX: Biochemical characterization of the
mitochondrial tRNASer(UCN)T7511C mutation associated
with nonsyndromic deafness. Nucleic Acids Res. 32:867–877. 2004.
View Article : Google Scholar
|
|
39
|
Campos Y, Martín MA, Rubio JC, Gutiérrez
del Olmo MC, Cabello A and Arenas J: Bilateral striatal necrosis
and MELAS associated with a new T3308C mutation in the
mitochondrial ND1 gene. Biochem Biophys Res Commun. 238:323–325.
1997. View Article : Google Scholar : PubMed/NCBI
|