Introduction
Oral squamous cell carcinoma (OSCC) is an aggressive
epithelial malignancy with a poor prognosis despite advances in
diagnosis and treatment (1). The
most common risk factor for OSCC is tobacco, alcohol, ultraviolet
light and oral lesions (2,3).
Although surgery is effective, the incidence of this type of cancer
is increasing (4). Accordingly,
the development of optimal treatment or therapeutic strategies for
OSCC, as well as novel therapeutic regimens to prevent and treat
OSCC is mandatory.
Natural products, potential sources of new drugs,
have been applied in the field of medicine, pharmacy and biology
for the past several decades (5).
Histone deacetylase (HDAC) inhibitors, a novel class of
chemotherapeutic drug, have shown potent anticancer activities in
preclinical studies (6). HDAC
inhibitors induce the rapid histone hyperacetylation of nucleosomal
histones and chromatin remodeling, leading to changes in the
expression of genes that control growth, differentiation and
survival (7,8). Specifically, HDAC inhibitors have
been linked to several downstream effects in tumor cell lines,
resulting in cell cycle arrest and the induction of apoptosis
(9–11). A wide range of structurally
diverse HDAC inhibitors has been purified from natural products and
synthetically produced. Therefore, several clinical trials have
been initiated.
Panobinostat (LBH589) is a novel HDAC inhibitor that
blocks multiple cancer-related pathways and reverse epigenetic
events implicated in cancer progression (12). HDACs can be subdivided into two
groups: zinc-dependent HDACs (class I, class II a/b and class IV)
and zinc-independent HDACs (class III) (13). LBH589 is characterized as a
pan-deacetylase (pan-DAC) inhibitor, with activity against class I,
II and IV HDACs (12). LBH589
exerts inhibitory effects at low nanomolar concentrations across a
wide range of hematological malignancies, such as lymphoma, acute
myeloid leukemia and multiple myeloma (14–16). However, its effects on oral cancer
and the mechanisms behind LBH589-induced apoptosis remain poorly
understood.
In the present study, we examined the effects of
LBH589 on two OSCC cell lines, HN22 and HSC4. We demonstrate that
LBH589 inhibits cell growth and induces apoptosis in the HN22 and
HSC4 cells. The results from the present study provide experimental
evidence to support the hypothesis that LBH589 decreases
specificity protein 1 (Sp1) expression and inhibits OSCC cell
viability by inducing cell cycle arrest and activating apoptotic
pathways. Our results also provide evidence for the
chemotherapeutic efficacy of LBH589 in the treatment of OSCC.
Materials and methods
Materials
HN22 and HSC4 cells are human oral squamous cancer
cell lines. HN22 cells were provided by Dankook University
(Cheonan, Korea) and HSC4 cells were provided by Hokkaido
University (Hokkaido, Japan). HN22 and HSC4 cells were cultured in
HyClone Dulbecco’s modified Eagle’s medium (DMEM) (Thermo
Scientific, Logan, UT, USA) containing 10% heat-inactivated fetal
bovine serum and 100 U/ml each of penicillin and streptomycin
(Thermo Scientific) at 37°C with 5% CO2 in a humidified
atmosphere. LBH589 was purchased from SelleckBio (Houston, TX,
USA).
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay
The effect of LBH589 on cell viability was estimated
using the CellTiter 96® AQueous One Solution Cell
Proliferation Assay kit (Promega, Madison, WI, USA) according to
the manufacturer’s instructions for MTS assay. The HN22 and HSC4
cells were seeded in 96-well plates for 24 h and treated with
various concentrations of LBH589 for 24 and 48 h. The absorbance
was measured at 490 nm using a GloMax-Multi Microplate Multimode
Reader (Promega). The data were expressed as the percentage of cell
viability compared with the control.
DAPI staining
The number of cells undergoing apoptosis following
treatment with LBH589 was quantified using DAPI staining. The cells
with nuclear condensation and fragmentation were determined using
nucleic acid stained with 4′-6-diamidino-2-phenylindole (DAPI)
(Sigma-Aldrich). After 48 h of post-treatment with different doses
of LBH589 (5, 10 and 20 nM), the HN22 and HSC4 cells were harvested
and fixed in 100% methanol at room temperature for 20 min. The
cells were seeded on slides, stained with DAPI (2 mg/ml) and then
monitored by a FluoView confocal laser microscope (Fluoview FV10i,
Olympus Corp., Tokyo, Japan).
Western blot analysis
Following the treatment of the cells with LBH589,
the cells were washed twice with ice-cold phosphate buffered saline
(PBS) and harvested in an ice-cold PRO-PREP™ protein extraction
solution (Intron Biotechnology, Seoul, Korea) containing a protease
inhibitor. Protein concentrations were measured using the Bradford
protein assay. Protein samples were separated on SDS-PAGE gels and
transferred onto an Immobilon-P PVDF transfer membrane (Millipore,
Billerica, MA, USA) using a semi-dry blotting apparatus. Western
blot analysis was performed using ECL western blotting detection
reagent according to the manufacturer’s instructions (Thermo
Scientific, Rockford, IL, USA).
Propidium iodide (PI) staining
Following the treatment of the cells with LBH589,
the detached cells were collected separately and the adherent cells
were dissociated by trypsin-EDTA. The cells were washed with cold
PBS and then pooled and centrifuged before being fixed in 70%
ethanol overnight at −20°C. Before flow cytometry analysis, the
cells were centrifuged and incubated for 30 min at 37°C in PBS to
allow for the release of low-molecular weight DNA. Following
centrifugation, the cell pellets were resuspended and treated with
150 mg/ml RNase A and 20 mg/ml PI using a MACSQuant®
analyzer (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany).
Immunocytochemistry
The cells were seeded onto glass cover slips on
6-well tissue culture plates for 24 h and incubated with LBH589 for
48 h. The cells were fixed/permeabilized with cytotoxic solution
for 30 min. For the analysis of the expression of Sp1 and
caspase-3, the cells were blocked with 1% BSA and then incubated
with monoclonal Sp1 and cleaved caspase-3 antibody at 4°C
overnight. After washing with PBS, the cells were incubated with
Alexa Fluor® 546 anti-mouse IgG and Alexa
Fluor® 488 anti-rabbit IgG (1:1,000 dilution; Molecular
Probes) in 1% BSA for 1 h and mounted with VECTSHIELD®
mounting medium for fluorescence with DAPI (Vector Laboratories,
Inc., Burlingame, CA, USA) onto the cells. The cells were
visualized using a laser confocal microscope.
Statistical analysis
The statistical significance of the differences
between groups was assessed using the Student’s t-test. The null
hypothesis was rejected at a p-value <0.05.
Results
LBH589 inhibits cell viability and
induces the apoptosis of human OSCC cells
The aim of this study was to investigate whether
LBH589 exerts growth inhibitory effects on human OSCC cells. The
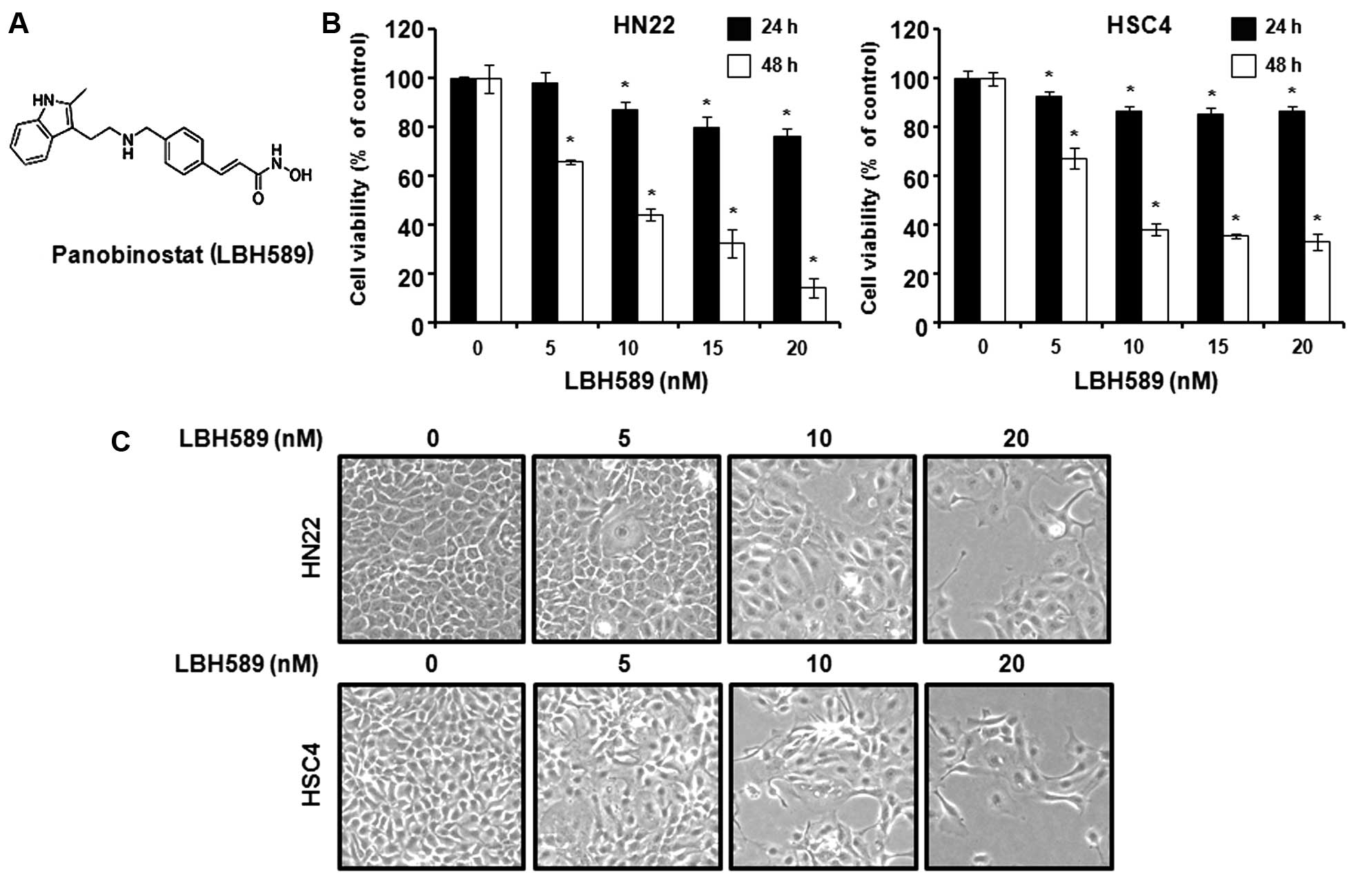
structure of LBH589 is shown in Fig.
1A. To investigate the efficacy of LBH589 as an anticancer
drug, the HN22 and HSC4 cells were treated with LBH589 and cell
viability was determined by MTS assay. As shown in Fig. 1B, MTS assay was carried out
following treatment with LBH589 at various concentrations (5, 10,
15 and 20 nM) for 24 h or 48 h. The cell viability graphs show that
LBH589 reduced HN22 and HSC4 cell viability at 24 h and 48 h, in a
concentration-dependent manner (p<0.05). The maximal decrease
was observed at 48 h relative to 24 h. The morphological changes
were observed under an optical microscope after 48 h; the apoptotic
phenotype showed was a rounded cell, with cytoplasmic blebbing and
irregularities in shape (Fig.
1C). These results indicate that LBH589 inhibits the growth of
human OSCC cells.
LBH589 induces G1 phase cell cycle arrest
and apoptosis in OSCC cells
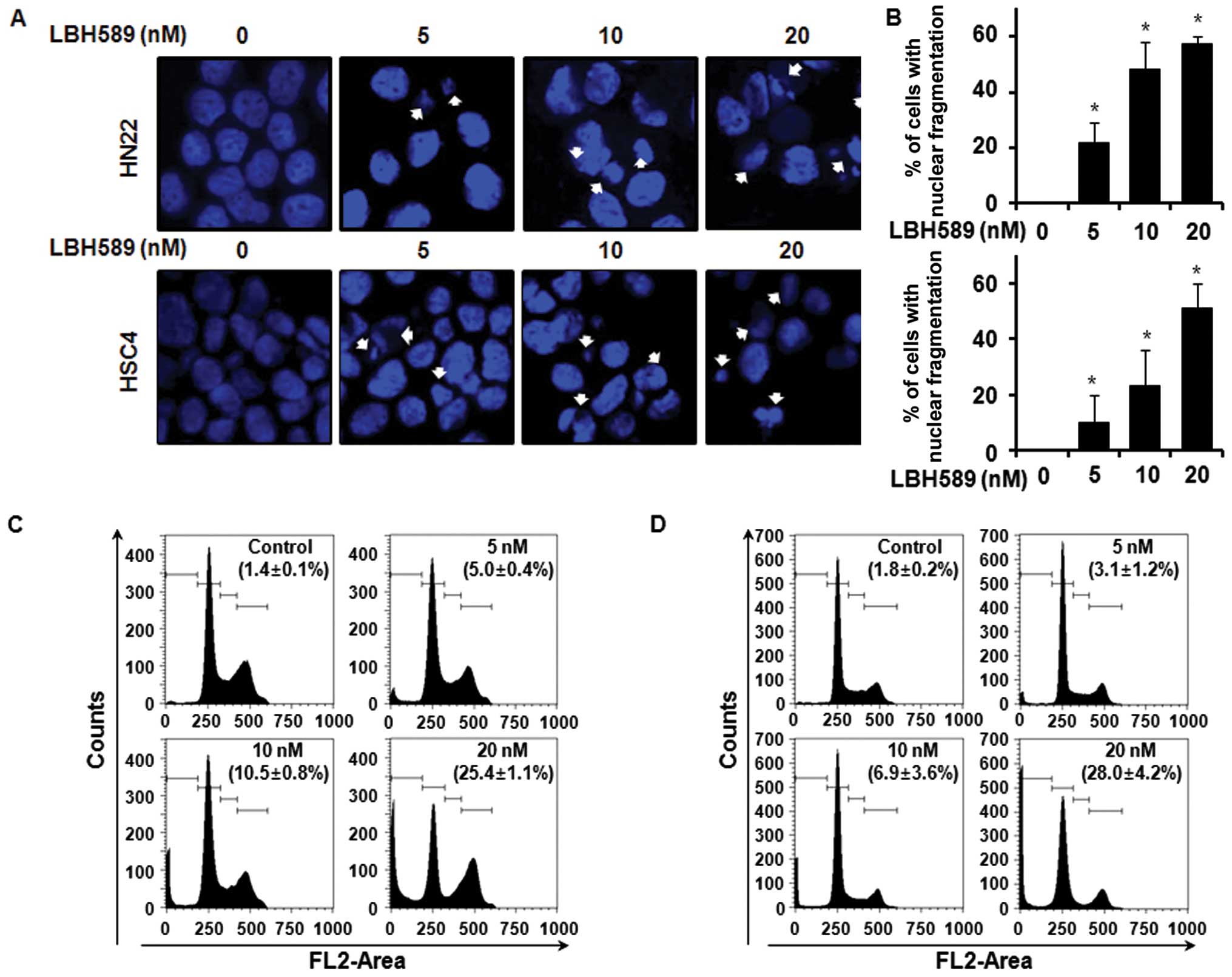
Cancer cell growth can be suppressed by cell cycle
arrest or the induction of apoptosis, or both (17). We carried out a confocal laser
microscopic analysis of the LBH589-treated HN22 or HSC4 cells to
demonstrate the apoptotic morphological changes using DAPI
staining, which specifically stains the nuclei. The results
revealed the presence of nuclear condensation and perinuclear
apoptotic bodies upon LBH589 treatment at a concentration of 5, 10
and 20 nM for 48 h. The percentage of cells with nuclear
fragmentation in the LBH589-treated group compared with the
DMSO-treated group is shown in Fig.
2A and B. To determine whether the LBH589-mediated growth
inhibition of HN22 or HSC4 cells was attributed to cell cycle
arrest, the cell cycle distribution was analyzed by FACS analysis.
As shown in Fig. 2C, in the HN22
cells, there was a significant increase in the number of sub-G1
cells: 5.0±0.4% in the presence of 5 nM of LBH589, 10.5±0.8% with
10 nM and 25.4±1.1% with 20 nM LBH589, compared with the untreated
control cells. In the HSC4 cells, an increase in the number of
sub-G1 cells was also observed: 3.1±1.2% at a concentration of 5 nM
LBH589, 6.9±3.6% at 10 nM and 28.0±4.2% at 20 nM of LBH589 compared
with the untreated control cells. The graph shows the
quantification of the FACS data (Fig.
2D).
LBH589 suppresses Sp1 expression in OSCC
cells
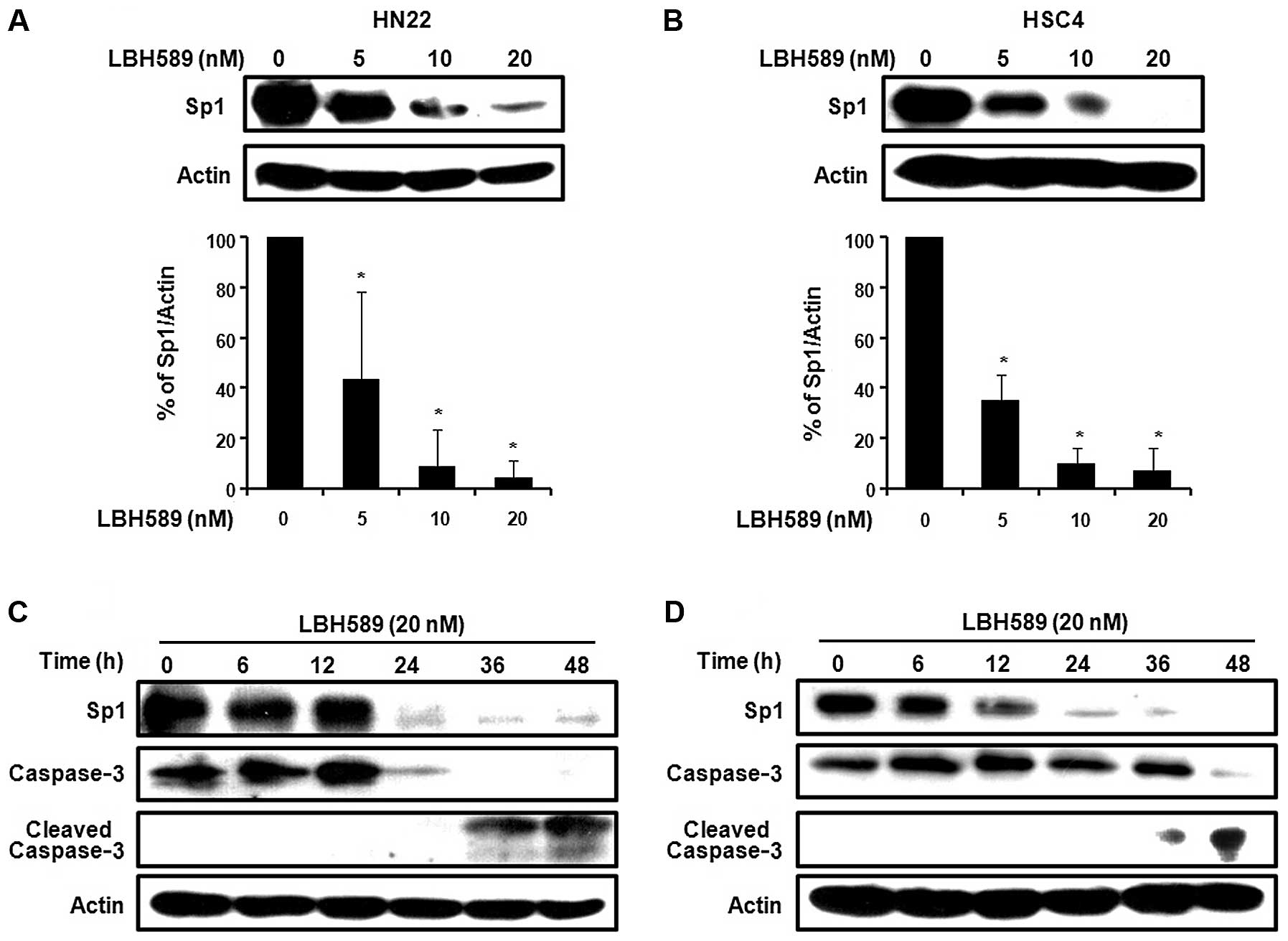
Sp1 plays an important role in oncogenesis.
Therefore, if the expression level of Sp1 protein may be
effectively modulated by a chemotherapeutic agent, then the agent
may be a potent candidate for an anticancer drug by suppressing
tumor progression. To determine whether Sp1 protein expression
levels were reduced by LBH589, the HN22 and HSC4 cell lines were
treated with various concentrations of LBH589 at 0, 5, 10 and 20 nM
for 48 h. As shown in Fig. 3A and
B, treatment with LBH589 induced a significant decrease in the
protein expression levels of Sp1 in the HN22 and HSC4 cells in a
dose-dependent manner. To further investigate the apoptotic effects
of the downregulation of Sp1 by LBH589, the two OSCC cell lines,
HN22 and HSC4, were treated with 20 nM LBH589 for different periods
of time (0, 6, 12, 24, 36 and 48 h). The Sp1 levels significantly
decreased as time progressed. LBH589 also induced the cleavage of
caspase-3, thus inducing apoptosis (Fig. 3C and D). Consistent with these
observations, the immunocytochemistry results also revealed a
decreased level of Sp1 and an increased level of cleaved caspase-3
in a dose-dependent manner in the HN22 and HSC4 cell lines
(Fig. 3E). Collectively, these
results suggest that the downregulation of Sp1 by treatment with
LBH589 leads to apoptotic cell death.
LBH589 modulates the regulator of cell
cycle arrest and apoptosis in OSCC cells
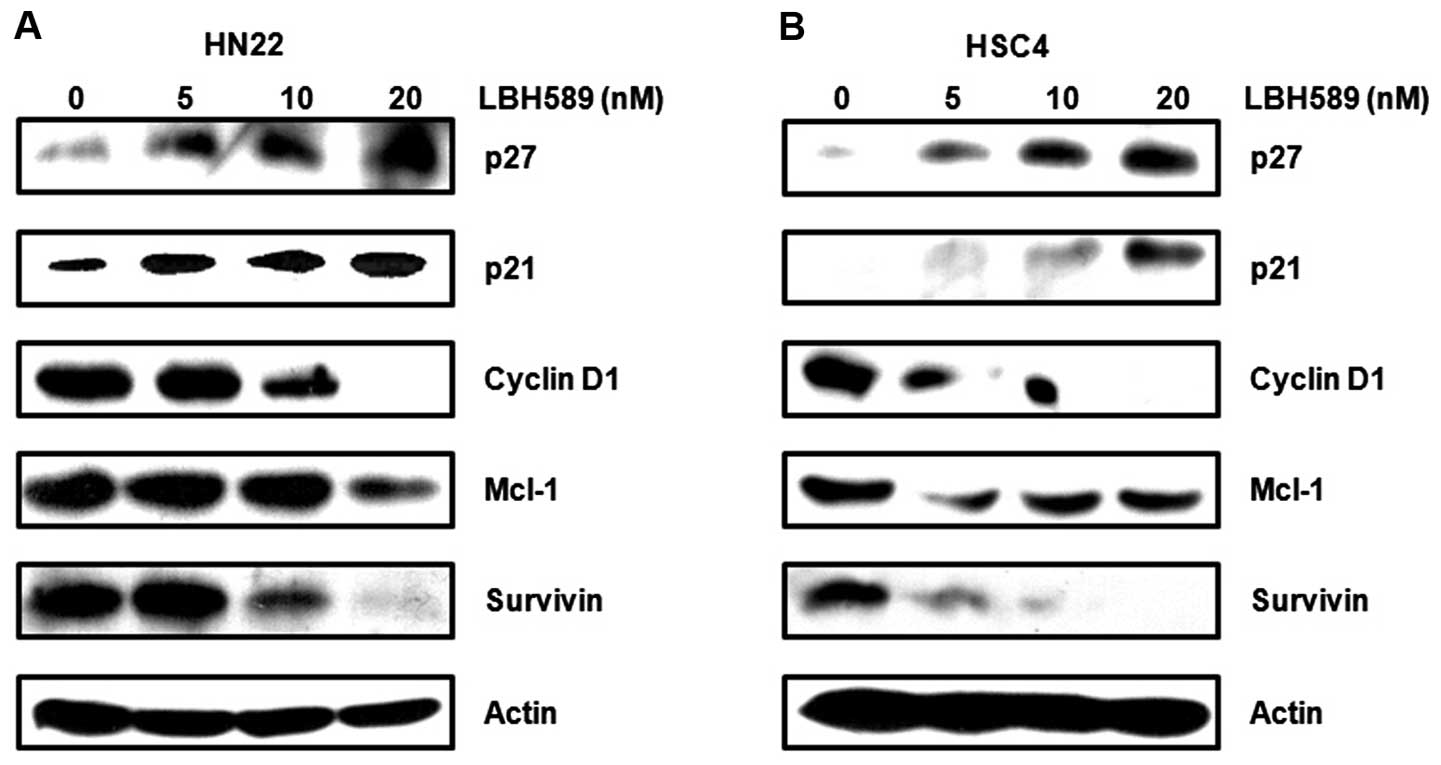
Sp1 has been shown to regulate the expression of
various gene products involved in cell cycle progression, growth
and apoptosis, an important role in oncogenesis (18,19). To further support the association
between LBH589 and Sp1-mediated apoptosis, we investigated Sp1
target proteins and apoptotic proteins. We found that the cell
cycle arrest involved proteins, such as p27 and p21, which were
significantly increased by LBH589, whereas cell proliferation and
survival-related proteins, such as cyclin D1, myeloid cell
leukemia-1 (Mcl-1) and survivin, were remarkably attenuated by
LBH589 treatment in a dose-dependent manner (Fig. 4A and B). Furthermore, we
investigated the expression of proteins involved in apoptosis
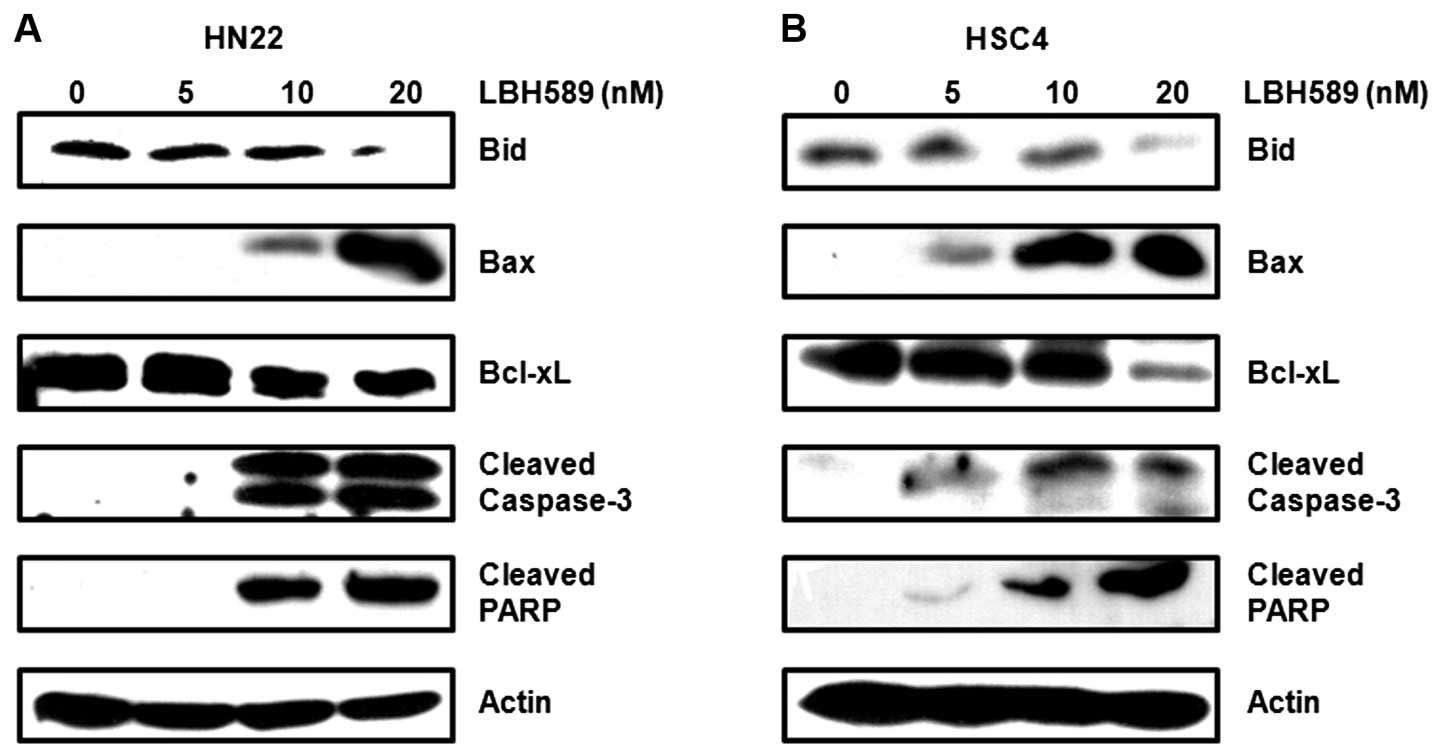
regulation. As shown in Fig. 5A and
B, the downregulation of Bid and Bcl-xL and the upregulation of
Bax appeared to be involved in the apoptotic cell death induced by
LBH589. In addition, the cleavage of caspase-3 and PARP was induced
by LBH589 in a dose-dependent manner. These results indicate that
the treatment of OSCC cells with LBH589 induces the downregulation
of Sp1, resulting in growth arrest and the induction of apoptotic
cell death.
Discussion
While the cellular responses of disparate
chemotherapeutic agents seem to be many and varied, it now seems
certain that a major mechanism of action of such agents is the
induction of endogenous cell death pathways, inducing apoptosis and
thereby eliminating tumor cells (20). The first pathway is the ligation
of death receptors, such as Fas and tumor necrosis factor receptor
(TNFR), inducing a cascade of protein-protein interactions mediated
by caspases (21). The other
pathway, stimulated by stress stimuli, such as growth factors and
chemotherapeutic drugs, uses the mitochondria as a key component
for the induction of cell death, resulting in the release of
mitochondrial proteins and the activation of caspases (22).
The effectiveness of chemotherapeutic agents can be
affected by alterations in apoptotic pathways, and this possibility
is becoming an important factor in determining the most effective
chemotherapeutic drugs for cancer treatment. HDAC inhibitors have
appeared as promising chemotherapeutic agents by their ability to
induce apoptosis and inhibit cell cycle progression (23–25). The antitumorigenic effects of HDAC
inhibitors are notable due to the fact that their cytotoxicity is
specific to cancer cells and not to normal cells or tissues.
Compared with other anticancer drugs, HDAC inhibitors are well
tolerated with a good toxicity profile (26). A number of studies have reported
that LBH589 exerts antitumor effects on various cancer-derived
cells, including epithelial ovarian, prostate and liver cancer
cells, as well as hepatocellular carcinoma cells (27–30). Nevertheless, the anticancer
activities of LBH589 on human OSCC cells are not yet fully
understood. LBH589 triggers ER stress with the activation of
caspase-12, the upregulation of phosphorylated JNK and the
overexpression of CHOP, with the final activation of executioner
caspase-3 (30).
In this study, we determined whether LBH589 is
capable of inhibiting cell growth and decreasing Sp1 expression,
thus inducing apoptosis in OSCC cells. We found that LBH589
inhibited cell growth and induced apoptosis in the HN22 and HSC4
cell lines. Moreover, the LBH589-induced apoptosis was associated
with a decrease in Sp1 expression in the HN22 and HSC4 cells.
The ubiquitous transcription factor, Sp1, is
overexpressed in various human cancer cell lines (31–35) and plays a role in the regulation
of genes which are involved in many cellular processes (36). Sp1 has transcriptional activity on
the promoters of genes involved in cell cycle progression,
differentiation and oncogenesis (37). Several studies have demonstrated
that HDAC inhibitors downregulate different Sp1 target proteins by
inhibiting Sp1 activity (38,39). In this study, Sp1 expression was
significantly decreased in the LBH589-treated cells. LBH589 also
regulated apoptosis-related proteins, such as caspase-3. Treatment
with LBH589 induced the cleavage of caspase-3, thus promoting
apoptosis.
To further characterize the effects of LBH589 on
Sp1, we analyzed the effects of LBH589 on p27, p21, cyclin D1,
Mcl-1 and survivin, a Sp1 target protein, by western blot analysis
(40–42). The results revealed that LBH589,
as a HDAC inhibitor, also inhibited the level of Sp1 and regulated
Sp1 target proteins, such as p27, p21, cyclin D1, Mcl-1 and
survivin in a dose-dependent manner. Consistent with this, LBH589
reduced Bid and Bcl-xL expression and increased Bax expression.
LBH589 also activated caspase-3 and PARP, suggesting that LBH589
regulated Sp1 and ultimately led to apoptotic cell death.
In conclusion, the present study demonstrates that
LBH589 suppresses Sp1 expression, leading to the upregulation of
p27 and p21 and the downregulation of cyclin D1, Mcl-1 and survivin
and a subsequent decrease in cell viability through a
caspase-3-dependent apoptotic signaling pathway in OSCC cells. The
present study delineates, in part, the signaling pathways involved
in the LBH589-induced decrease in the viability of OSCC cells.
Acknowledgements
This study was supported by the Basic Science
Research program through the National Research Foundation Korea
(NRF) funded by the Ministry of Education, Science and Technology
(2011-0008463) and the Next-Generation BioGreen 21 Program
(PJ008116062011), Rural Development Administration, Republic of
Korea.
References
|
1
|
Forastiere A, Koch W, Trotti A and
Sidransky D: Head and neck cancer. N Engl J Med. 345:1890–1900.
2001. View Article : Google Scholar
|
|
2
|
Mashberg A, Boffetta P, Winkelman R and
Garfinkel L: Tobacco smoking, alcohol drinking, and cancer of the
oral cavity and oropharynx among U.S. veterans. Cancer.
72:1369–1375. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Neville BW and Day TA: Oral cancer and
precancerous lesions. CA Cancer J Clin. 52:195–215. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liang XH, Lewis J, Foote R, Smith D and
Kademani D: Prevalence and significance of human papillomavirus in
oral tongue cancer: the Mayo Clinic experience. J Oral Maxillofac
Surg. 66:1875–1880. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gordaliza M: Natural products as leads to
anticancer drugs. Clin Transl Oncol. 9:767–776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rasheed WK, Johnstone RW and Prince HM:
Histone deacetylase inhibitors in cancer therapy. Expert Opin
Investig Drugs. 16:659–678. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vigushin DM and Coombes RC: Histone
deacetylase inhibitors in cancer treatment. Anticancer Drugs.
13:1–13. 2002. View Article : Google Scholar
|
|
8
|
Lin HY, Chen CS, Lin SP, Weng JR and Chen
SC: Targeting histone deacetylase in cancer therapy. Med Res Rev.
26:397–413. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marks P, Rifkind RA, Richon VM, Breslow R,
Miller T and Kelly WK: Histone deacetylases and cancer: causes and
therapies. Nat Rev Cancer. 1:194–202. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Johnstone RW and Licht JD: Histone
deacetylase inhibitors in cancer therapy: is transcription the
primary target? Cancer Cell. 4:13–18. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Atadja P: Development of the pan-DAC
inhibitor panobinostat (LBH589): successes and challenges. Cancer
Lett. 280:233–241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dokmanovic M, Clarke C and Marks PA:
Histone deacetylase inhibitors: overview and perspectives. Mol
Cancer Res. 5:981–989. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shao W, Growney JD, Feng Y, et al:
Activity of deacetylase inhibitor panobinostat (LBH589) in
cutaneous T-cell lymphoma models: Defining molecular mechanisms of
resistance. Int J Cancer. 127:2199–2208. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maiso P, Carvajal-Vergara X, Ocio EM, et
al: The histone deacetylase inhibitor LBH589 is a potent
antimyeloma agent that overcomes drug resistance. Cancer Res.
66:5781–5789. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Giles F, Fischer T, Cortes J, et al: A
phase I study of intravenous LBH589, a novel cinnamic hydroxamic
acid analogue histone deacetylase inhibitor, in patients with
refractory hematologic malignancies. Clin Cancer Res. 12:4628–4635.
2006. View Article : Google Scholar
|
|
17
|
Gupta SC, Kim JH, Prasad S and Aggarwal
BB: Regulation of survival, proliferation, invasion, angiogenesis,
and metastasis of tumor cells through modulation of inflammatory
pathways by nutraceuticals. Cancer Metastasis Rev. 29:405–434.
2010. View Article : Google Scholar
|
|
18
|
Deniaud E, Baguet J, Mathieu AL, Pages G,
Marvel J and Leverrier Y: Overexpression of Sp1 transcription
factor induces apoptosis. Oncogene. 25:7096–7105. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jutooru I, Chadalapaka G, Sreevalsan S, et
al: Arsenic trioxide downregulates specificity protein (Sp)
transcription factors and inhibits bladder cancer cell and tumor
growth. Exp Cell Res. 316:2174–2188. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schmitt CA and Lowe SW: Apoptosis and
therapy. J Pathol. 187:127–137. 1999. View Article : Google Scholar
|
|
21
|
Nagata S: Fas-induced apoptosis. Intern
Med. 37:179–181. 1998. View Article : Google Scholar
|
|
22
|
Green DR: Apoptotic pathways: paper wraps
stone blunts scissors. Cell. 102:1–4. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marks PA, Richon VM and Rifkind RA:
Histone deacetylase inhibitors: inducers of differentiation or
apoptosis of transformed cells. J Natl Cancer Inst. 92:1210–1216.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miller TA, Witter DJ and Belvedere S:
Histone deacetylase inhibitors. J Med Chem. 46:5097–5116. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Drummond DC, Noble CO, Kirpotin DB, Guo Z,
Scott GK and Benz CC: Clinical development of histone deacetylase
inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol.
45:495–528. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chao H, Wang L, Hao J, et al: Low dose
histone deacetylase inhibitor, LBH589, potentiates anticancer
effect of docetaxel in epithelial ovarian cancer via PI3K/Akt
pathway in vitro. Cancer Lett. 329:17–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vallo S, Mani J, Stastny M, et al: The
prostate cancer blocking potential of the histone deacetylase
inhibitor LBH589 is not enhanced by the multi receptor tyrosine
kinase inhibitor TKI258. Invest New Drugs. 31:265–272. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Di Fazio P, Montalbano R, Neureiter D, et
al: Downregulation of HMGA2 by the pan-deacetylase inhibitor
panobinostat is dependent on hsa-let-7b expression in liver cancer
cell lines. Exp Cell Res. 318:1832–1843. 2012.PubMed/NCBI
|
|
30
|
Di Fazio P, Schneider-Stock R, Neureiter
D, et al: The pan-deacetylase inhibitor panobinostat inhibits
growth of hepatocellular carcinoma models by alternative pathways
of apoptosis. Cell Oncol. 32:285–300. 2010.
|
|
31
|
Chiefari E, Brunetti A, Arturi F, et al:
Increased expression of AP2 and Sp1 transcription factors in human
thyroid tumors: a role in NIS expression regulation? BMC Cancer.
2:352002. View Article : Google Scholar
|
|
32
|
Hosoi Y, Watanabe T, Nakagawa K, et al:
Up-regulation of DNA-dependent protein kinase activity and Sp1 in
colorectal cancer. Int J Oncol. 25:461–468. 2004.PubMed/NCBI
|
|
33
|
Wang L, Wei D, Huang S, et al:
Transcription factor Sp1 expression is a significant predictor of
survival in human gastric cancer. Clin Cancer Res. 9:6371–6380.
2003.
|
|
34
|
Yao JC, Wang L, Wei D, et al: Association
between expression of transcription factor Sp1 and increased
vascular endothelial growth factor expression, advanced stage, and
poor survival in patients with resected gastric cancer. Clin Cancer
Res. 10:4109–4117. 2004. View Article : Google Scholar
|
|
35
|
Zannetti A, Del Vecchio S, Carriero MV, et
al: Coordinate up-regulation of Sp1 DNA-binding activity and
urokinase receptor expression in breast carcinoma. Cancer Res.
60:1546–1551. 2000.PubMed/NCBI
|
|
36
|
Davie JR, He S, Li L, et al: Nuclear
organization and chromatin dynamics-Sp1, Sp3 and histone
deacetylases. Adv Enzyme Regul. 48:189–208. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li L and Davie JR: The role of Sp1 and Sp3
in normal and cancer cell biology. Ann Anat. 192:275–283. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Duan H, Heckman CA and Boxer LM: Histone
deacetylase inhibitors down-regulate bcl-2 expression and induce
apoptosis in t(14;18) lymphomas. Mol Cell Biol. 25:1608–1619. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wilson AJ, Chueh AC, Togel L, et al:
Apoptotic sensitivity of colon cancer cells to histone deacetylase
inhibitors is mediated by an Sp1/Sp3-activated transcriptional
program involving immediate-early gene induction. Cancer Res.
70:609–620. 2010. View Article : Google Scholar
|
|
40
|
Blume SW, Snyder RC, Ray R, Thomas S,
Koller CA and Miller DM: Mithramycin inhibits SP1 binding and
selectively inhibits transcriptional activity of the dihydrofolate
reductase gene in vitro and in vivo. J Clin Invest. 88:1613–1621.
1991. View Article : Google Scholar
|
|
41
|
Chintharlapalli S, Papineni S, Lei P,
Pathi S and Safe S: Betulinic acid inhibits colon cancer cell and
tumor growth and induces proteasome-dependent and -independent
downregulation of specificity proteins (Sp) transcription factors.
BMC Cancer. 11:3712011. View Article : Google Scholar
|
|
42
|
Pietrzak M and Puzianowska-Kuznicka M:
p53-dependent repression of the human MCL-1 gene encoding an
anti-apoptotic member of the BCL-2 family: the role of Sp1 and of
basic transcription factor binding sites in the MCL-1 promoter.
Biol Chem. 389:383–393. 2008. View Article : Google Scholar
|