Introduction
Myocardial ischemia/reperfusion (MI/R) injury occurs
inevitably in a wide range of patients, such as survivors of
cardiac arrest, victims of acute myocardial infarction, as well as
in patients undergoing cardiac surgery (1). MI/R leads to oxidative stress, which
subsequently leads to reactive oxygen species (ROS) production, an
increase in malondialdehyde (MDA) levels and subsequent cytotoxic
injury (2,3). Oxidative stress and the accelerated
ROS production induced by MI/R play key roles in the progression of
ischemic heart disease and cardiomyocyte apoptosis (2–4). A
number of studies have suggested that cardiomyocyte apoptosis
induces a spectrum of events, including cardiac remodeling, a
larger infarct size and severe heart failure (5,6).
Although cardiomyocyte and tissue damage induced by oxidative
stress has been extensively investigated in recent years, there is
still a need for effective therapeutic strategies.
Glucagon-like peptide-1 (GLP-1), a gut hormone, has
been confirmed to exert potent insulin-releasing and
glucose-lowering effects (7).
However, its short half-life limits its clinical use. Therefore,
analogues of GLP-1 with much longer half-lives, such as exenatide
have been developed and are currently being used as novel
anti-diabetic drugs (7,8). A large number of studies have
demonstrated that GLP-1 and its analogues have multiple beneficial
effects on the cardiovascular system (9–12).
It has been reported that GLP-1 and its analogues inhibit
cardiomyocyte apoptosis by regulating the c-Jun N-terminal protein
kinase signaling pathway (13),
the phosphoinositide 3-kinase (PI3K) pathway (14), the ERK1/2 pathway (14), as well as others (15). However, to date, to our knowledge,
the cardioprotective effects of exenatide on oxidative
stress-induced injury have not been investigated in depth.
The aim of this study was to determine whether
exenatide is capable of reducing oxidative stress-induced injury.
To establish this, we used a model of oxidative stress induced by
hydrogen peroxide (H2O2) to assess the
effects of exenatide against oxidative stress-induced injury in
H9c2 cells. Furthermore, the rat model of MI/R was used to evaluate
the therapeutic efficacy of exenatide against oxidative damage in
the heart. We also investigated the possible mechanims behind the
anti-apoptotic effects of exenatide by assessing the activation of
the PI3K/Akt signaling pathway.
Materials and methods
Cell culture and
H2O2 treatment
The rat cardiomyoblast cell line, H9c2, was
purchased from the Cell Culture Center of Institute of Basic
Medical Sciences, (Chinese Academy of Medical Sciences). The cells
were cultured in Dulbecco’s modified Eagle’s medium/Ham’s Nutrient
Mixture F12 (DMEM/F12; Thermo Fisher Biochemical Products Co.,
Ltd., Beijing, China) supplemented with 10% fetal bovine serum
(FBS; Invitrogen Life Technologies, Carlsbad, CA, USA), penicillin
(100 U/ml) and streptomycin (10 μg/ml; both from Beyotime Institute
of Biotechnology, Haimen, China) and incubated at 37ºC in a
humidified atmosphere containing 5% CO2.
We first aimed to determine the most effective
concentration of H2O2 to establish the model
of oxidative stress. The H9c2 cells were treated with 4 different
concentrations (50, 100, 200 and 400 μM) of
H2O2 for 6 h. We then evaluated the
protective effects of exenatide (Baxter Pharmaceutical Solutions
LLC, Deerfield, IL, USA). The cells were pre-treated with various
concentrations (0.01, 0.1, 1 and 10 nM) of exenatide for 30 min
prior to exposure to H2O2. When the most
effective concentrations of H2O2 and
exenatide were determined, the cells were randomly assigned to one
of the following 4 groups: i) the control group: cells were
cultured under normal incubation conditions; ii) the
H2O2 group: cells were exposed to
H2O2 without pre-treatment with exenatide;
iii) the exenatide group: cells were pre-treated with exenatide for
30 min prior to exposure to H2O2; iv) the
exenatide + LY294002 (exenatide + L) group: to observe the effects
of exenatide on H9c2 cell apoptosis, the cells were pre-treated
with the PI3K inhibitor, LY294002 (15 μM; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), for 10 min prior to exenatide
treatment.
Viability assay
Cell viability was assessed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell
proliferation and cytotoxicity assay kit (MTT; Beyotime Institute
of Biotechnology, Haimen, China) according to the manufacturer’s
instructions. Briefly, the cells were seeded in a 96-well plate at
a density of 1×104 cells/well and incubated for 24 h.
The cells were then pre-treated with or without exenatide (0.01,
0.1, 1 and 10 nM) for 30 min subsequent to incubation with
H2O2 (50, 100, 200 and 400 μM) for 6 h. The
cells were administered with fresh medium and MTT solution (10 μl)
for 4 h followed by incubation with formazan solution (10 μl) for 4
h at 37ºC. The optical density (OD) values at 570 nm were measured
using a microplate reader (Multiskan MK33; Thermolab Systems,
Helsinki, Finland). Each experiment was repeated 6 times and the
data are expressed as a percentage of the control.
Flow cytometry
Cell apoptosis was detected using flow cytometry as
previously described with some modifications (16). After the indicated treatments, the
cells were collected by centrifugation at 600 × g for 5 min and
resuspended at a density of 1×106 cells/ml. Cells (500
μl) were mixed with fluorescein isothiocyanate (FITC)-Annexin V (5
μl) and propidium iodide (PI; 10 μl, 20 μg/ml) and incubated for 20
min in the dark at room temperature and analyzed immediately on a
flow cytometer. Flow cytometric analysis (excitation 488
nm/emission 530 nm) was performed on a FACSCalibur cell sorter (BD
FACSVantage SE; Beckman Coulter, Brea, CA, USA). Each experiment
was repeated 3 times.
In order to quantitatively analyze the production of
ROS, we measured ROS levels by flow cytometry as previously
described with some modifications (16). After the indicated treatments, the
cells were treated with 2′,7′-dichlorofluorescein diacetate
(DCFH-DA; 1 ml, 60 min, Beyotime Institute of Biotechnology) at
37ºC. The fluorescence intensity was measured by flow cytometry and
analyzed using CellQuestTM software. The experiment was
repeated 3 times and the data are expressed as the mean
fluorescence intensity.
Experimental animals
Male Sprague-Dawley rats (6–8 weeks of age) were
purchased from the Laboratory Animal Center of Chongqing Medical
University, Chongqing, China [certificate: SCXK (YU) 2007-0001].
The rats were housed under optimal conditions for hygiene,
temperature, photoperiods (12L:12D) and standard laboratory chow
and water were provided ad libitum, conforming to the
Guidelines for Care and Use of Laboratory Animals. All procedures
on animals were approved by the Ethics Committee of Chongqing
Medical University.
Experimental model of MI/R-induced
injury
Thirty-two male Sprague-Dawley rats were randomly
divided into 4 groups (n=8): i) sham-operated group; ii) MI/R
group; iii) exenatide group; and iv) exenatide + LY294002
(exenatide + L) group. To observe the anti-apoptotic effects of
exenatide, the animals were pre-treated with the PI3K inhibitor,
LY294002, for 30 min prior to exenatide treatment. Exenatide (10
μg/kg/day) was administered by intraperitoneal injection for 2
weeks. LY294002 (0.3 mg/kg/3 days) was administered by
intraperitoneal injection 30 min before exenatide was injected.
Exenatide and LY294002 were dissolved in dimethyl sulfoxide (DMSO).
The sham-operated group and MI/R group received the same volume of
DMSO for 2 weeks.
After 2 weeks of pre-treatment, all rats were
anesthetized by chloral hydrate (concentration: 3.5%, 10 ml/kg).
Tracheotomy was carried out for ventilation by a respirator
(ALC-V8B, Alcott Biotech Co., Ltd., Shanghai, China) with a stroke
volume of 28 ml/kg, air pressure of 10 mmHg, respiration rate of
1:1 and at a rate of 86 strokes per minute, and a lead II
electrocardiogram (ECG) was performed. Thoracotomy was performed
and the left anterior descending coronary artery was then ligated
using a 6–0 silk suture. Myocardial ischemia was by the presence of
a zone of cyanosis and the elevation of the S-T segment in the ECG.
After completion of the surgical procedure, the animals were
allowed to stabilize for 30 min prior to reperfusion, of the
previously ischemic myocardium, for 2 h. The sham-operated group
rats were subjected to the same surgical procedure, but without
ligation.
Hemodynamic measurements
At the end of the MI/R period, the right common
carotid artery and left femoral artery were isolated. A polystyrene
PE-20 catheter was inserted into the left ventricle via the right
common carotid artery, with one end connected to an MPA-2000
multichannel physiological recorder. The left ventricular
end-systolic pressure (LVESP), the left ventricular end-diastolic
pressure (LVEDP) and the rates of maximum positive and negative
left ventricular pressure development (±LVdp/dtmax) were measured.
LVESP and LVEDP were expressed as mmHg. ±LVdp/dtmax was expressed
as mmHg/sec. All the rats were sacrificed and their hearts were
collected. The blood plasma samples were collected immediately and
stored at −80ºC.
2,3,5-Triphenyl tetrazolium chloride
(TTC) staining
Infarct size was measured using TTC staining as
previously described (17). In
brief, the heart was transected parallel to the atrioventricular
groove at the center of the infarct area and incubated in 1% TTC
solution for 15 min at 37ºC. After staining, the infarct area
appears pallid, whereas the viable myocardium appears red. Infarct
size was expressed as the ratio of the infarct area to the total
volume (volume of infarct area and viable area).
Terminal deoxynucleotidyl
transferase-mediated dUTP-biotin nick end-labeling (TUNEL)
staining
TUNEL staining was performed using a TUNEL staining
assay kit according to the manufacturer’s instructions (Boster
Bio-engineering Co., Ltd., Wuhan, China). Briefly, after
deparaffinization, tissue sections were first treated with
H2O2 (3%) and then digested with proteinase K
(20 μg/ml; pH 7.4) at 25ºC. Following digestion for 10 min, the
tissue sections were incubated with labeling buffer (1:18) at 37ºC.
Following incubation for 120 min, the tissue sections were
incubated with biotinylated anti-digoxin antibody (1:100) for 30
min at 37ºC. Incorporated fluorescein was then detected with
streptavidin-biotin-peroxidase and subsequently the tissue sections
were dyed with 3,3′-diaminobenzidine (DAB). This assay detects
apoptotic cells by labeling the 3′-OH end DNA fragments with
digoxigenin-deoxyuridine triphosphate (Dig-dUTP) using terminal
deoxynucleotidyl transferase. The nuclei of the apoptotic cells
were stained brown and the nuclei of normal cells were stained
blue. The apoptotic index (AI) was determined as the ratio of the
number of brown nuclei to the total number of nuclei. Nuclei in a
total of 10 fields per tissue slice (n=6) were included.
Colorimetry
The activity of lactate dehydrogenase (LDH) in the
culture medium and plasma, the concentrations of MDA and total
superoxide dismutase (T-SOD) in the H9c2 cells and the
concentrations of MDA, T-SOD, catalase and glutathione peroxidase
(GSH-Px) in the heart homogenates were determined by colorimetry.
The experiment was performed using commercially available kits,
according to the manufacturer’s instructions (Jiancheng
Bioengineering Institute, Nanjing, China). Briefly, culture medium
and plasma were collected. The H9c2 cells and heart tissues were
collected and lysed by cell lysis buffer. The cell lysates were
then centrifuged at 1,600 × g for 10 min at 4ºC. The supernatants
of the culture medium, plasma and heart cell lysates were collected
for the detection of LDH, MDA, T-SOD, catalase and GSH-Px.
Following incubation with the reagents included in the kits, the
absorbance values at 340, 532, 550, 450 and 412 nm were measured
using a spectrophotometer (721D; Pudong Shanghai Physical Optics
Instrument Factory, Shanghai, China). The experiment was performed
at least 3 times and the LDH level was expressed as U/l. T-SOD,
catalase and GSH-Px levels were expressed as U/mg protein. The MDA
level was expressed as nmol/mg protein.
ELISA assays
The levels of creatine kinase-MB (CK-MB) in the
culture medium and plasma were measured using a CK-MB ELISA assay
kit (R&D Systems, Minneapolis, MN, USA), according to the
manufacturer’s instructions. After the indicated treatments, the
culture medium and plasma were collected and centrifuged at 1,600 ×
g for 10 min at 4ºC. The supernatants were collected for the
detection of CK-MB. The supernatants were then incubated with the
reagents included in the kits. Finally, the absorbance values were
measured using a microplate reader (Molecular Devices, Downingtown,
PA, USA) at 450 nm. All experiments were performed independently at
least 3 times and the CK-MB level was expressed as U/l.
Western blot analysis
The H9c2 cells and left ventricular myocardium
lysates were homogenized in cell lysis buffer (Beyotime Institute
of Biotechnology). Lysates were kept on ice for 45 min and cleared
by centrifugation at 14,000 × g for 10 min at 4ºC and defined as
total cardiac protein. Proteins were separated by SDS-PAGE and
transferred onto membranes. The membranes were blocked in 5% bovine
serum albumin (BSA) and incubated with primary antibodies against
Akt (1:1,000, Cell Signaling Technology, Inc., Danvers, MA, USA),
phospho-AKTserine473 (1:1,000, Cell Signaling
Technology, Inc.), cleaved caspase-3 (1:1,000, Cell Signaling
Technology, Inc.), phospho-Badserine136 (1:500, Santa
Cruz Biotechnology, Inc.) and anti-GAPDH antibody (1:1,000,
Beyotime Institute of Biotechnology). The membranes were then
incubated with a secondary antibody (Beyotime Institute of
Biotechnology). The signals were detected with the ECL system
(Beyotime Institute of Biotechnology). Blots were scanned using the
Bio-Rad gel imaging system (Bio-Rad, Hercules, CA, USA) and bands
were quantified using QuantityOne software.
Statistical analysis
SPSS 17.0 software was used for statistical
analysis. Data are presented as the means ± standard deviation
(SD). Group data were analyzed using a one-way analysis of variance
(ANOVA) followed by the Student-Newman-Keuls test. When the equal
variance test failed, a Mann-Whitney Rank Sum test was used. Values
of P<0.05 were considered to indicate statistically significant
differences.
Results
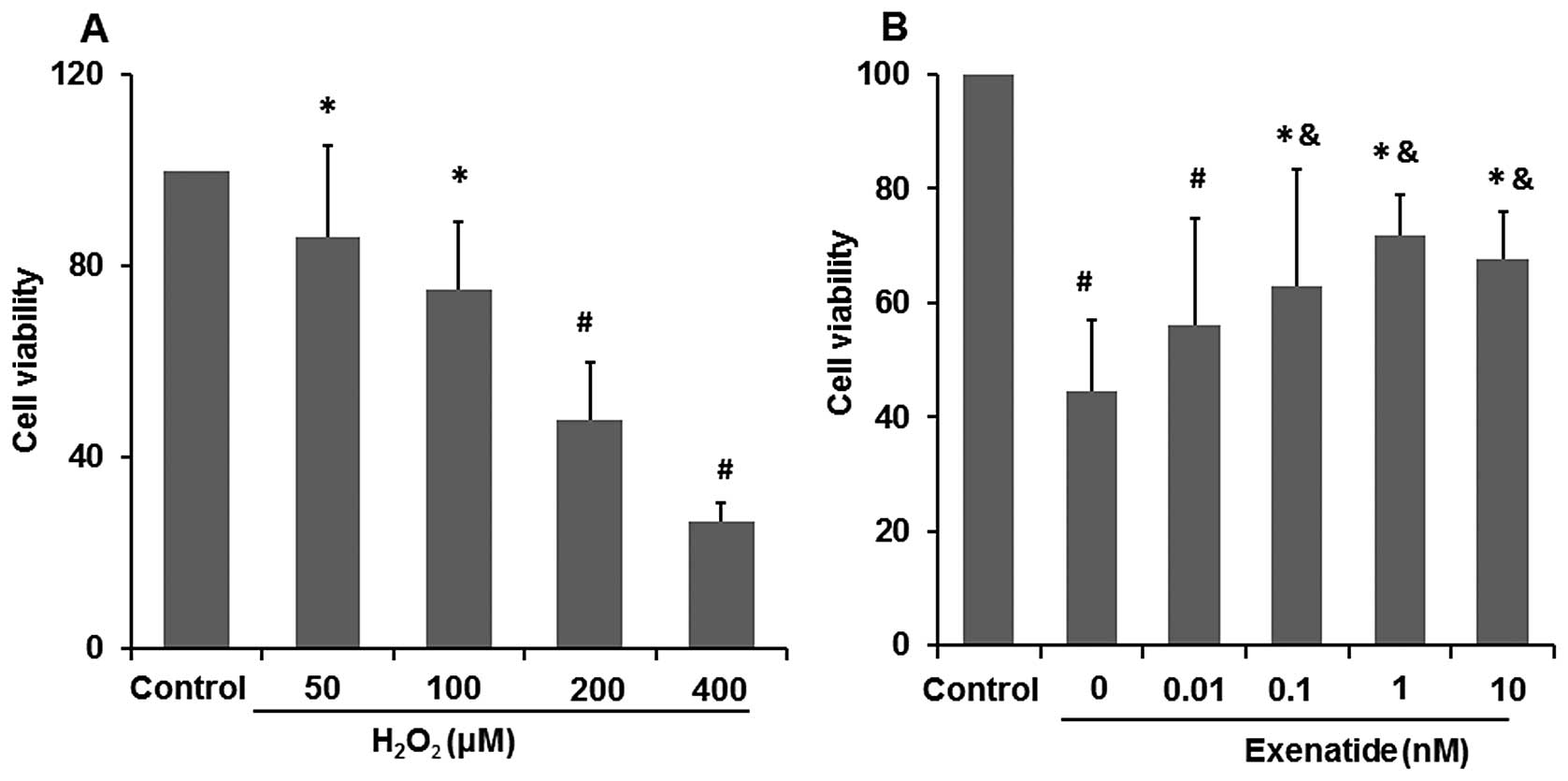
Exenatide increases the viability of
H2O2-treated H9c2 cells
After the H9c2 cells were exposed to various
concentrations of H2O2 (50, 100, 200 and 400
μM) for 6 h, MTT assay was performed to determine the viability of
the H2O2-treated H9c2 cells and to determine
the most effective concentration. The increasing concentration of
H2O2 led to the intensified damage of H9c2
cells (Fig. 1A). When the cells
were exposed to 50 and 100 μM H2O2 their
viability was reduced to 85.08 and 78.08%, respectively compared
with that of the control group (P<0.05), while when exposed to
200 and 400 μM H2O2 their viability was
reduced to 47.6 and 26.35%, respectively compared with that of the
control group (P<0.01). Finally, the concentration of 200 μM
H2O2 was selected for the study of the medium
cellular mortality and of the damage induced by oxidative
stress.
To investigate the possible protective effects of
exenatide on oxidative stress-induced injury, the cells were
pre-treated with exenatide (0, 0.01, 0.1, 1 and 10 nM) for 30 min
prior to exposure to H2O2. We found that
pre-treatment with 0.1, 1 and 10 nM exenatide statistically
increased cell viability which was decreased by oxidative
stress-induced injury (P<0.05) (Fig. 1B). These results strongly suggest
that exenatide exerts cardiomyocyte protective effects against
oxidative stress-induced injury in H9c2 cells. Exenatide, at a
concentration of 1 nM, had the optimal protective effects on cell
viability. Thus, it was selected for the following experiments.
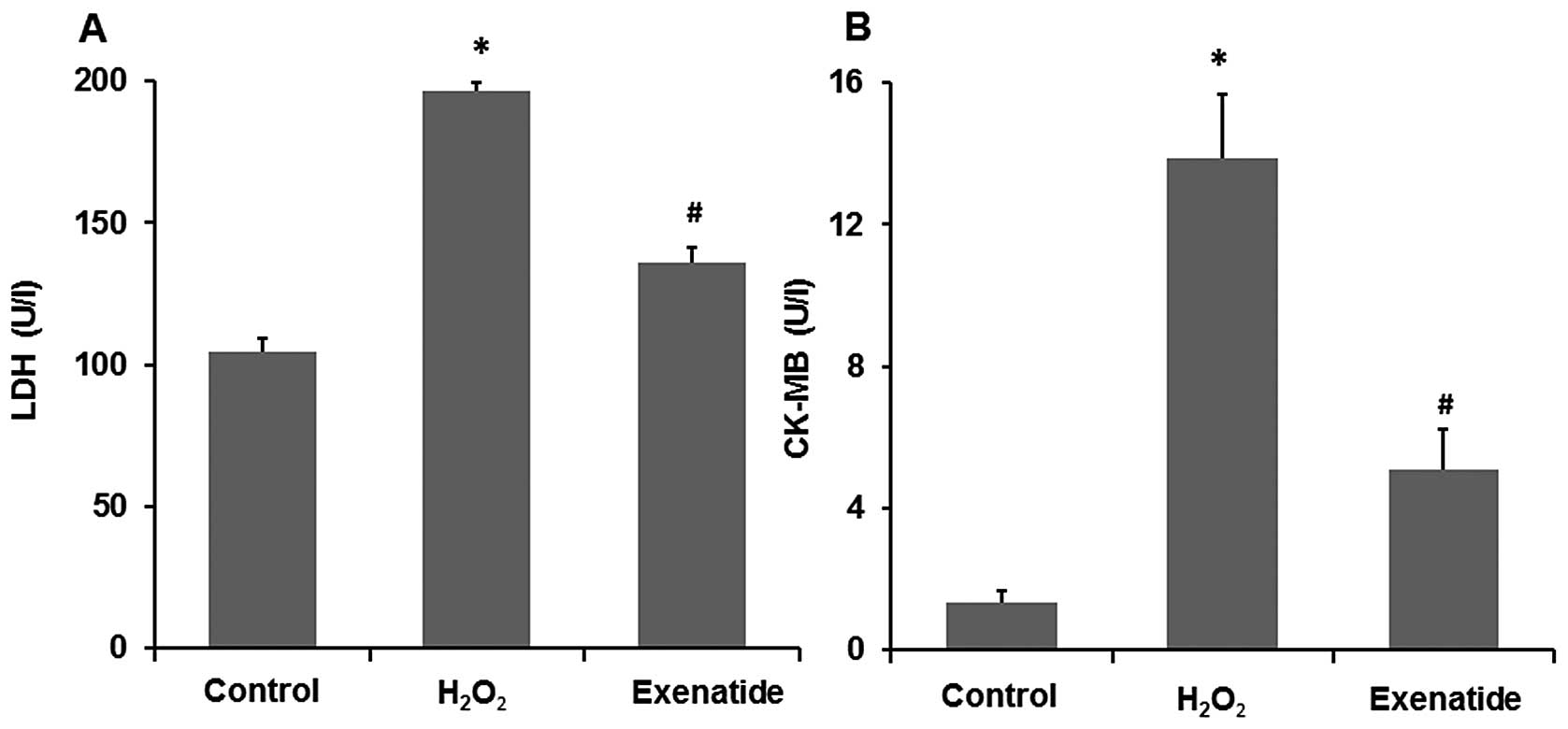
Exenatide protects H9c2 cells against
oxidative stress-induced injury following exposure to
H2O2
As LDH and CK-MB are two acknowledged markers of
cell damage, we further assessed the release of LDH and CK-MB in
the culture medium (Fig. 2A and
B). Compared with the levels in the control group, the LDH and
CK-MB levels were significantly increased in the
H2O2 group (P<0.05), while the H9c2 cells
pre-treated with 1 nM exenatide presented a significant decrease in
the H2O2-induced release of LDH and CK-MB
(P<0.05). These results further indicate that exenatide exerts
cardiomyocyte protective effects against oxidative stress-induced
injury in H9c2 cells.
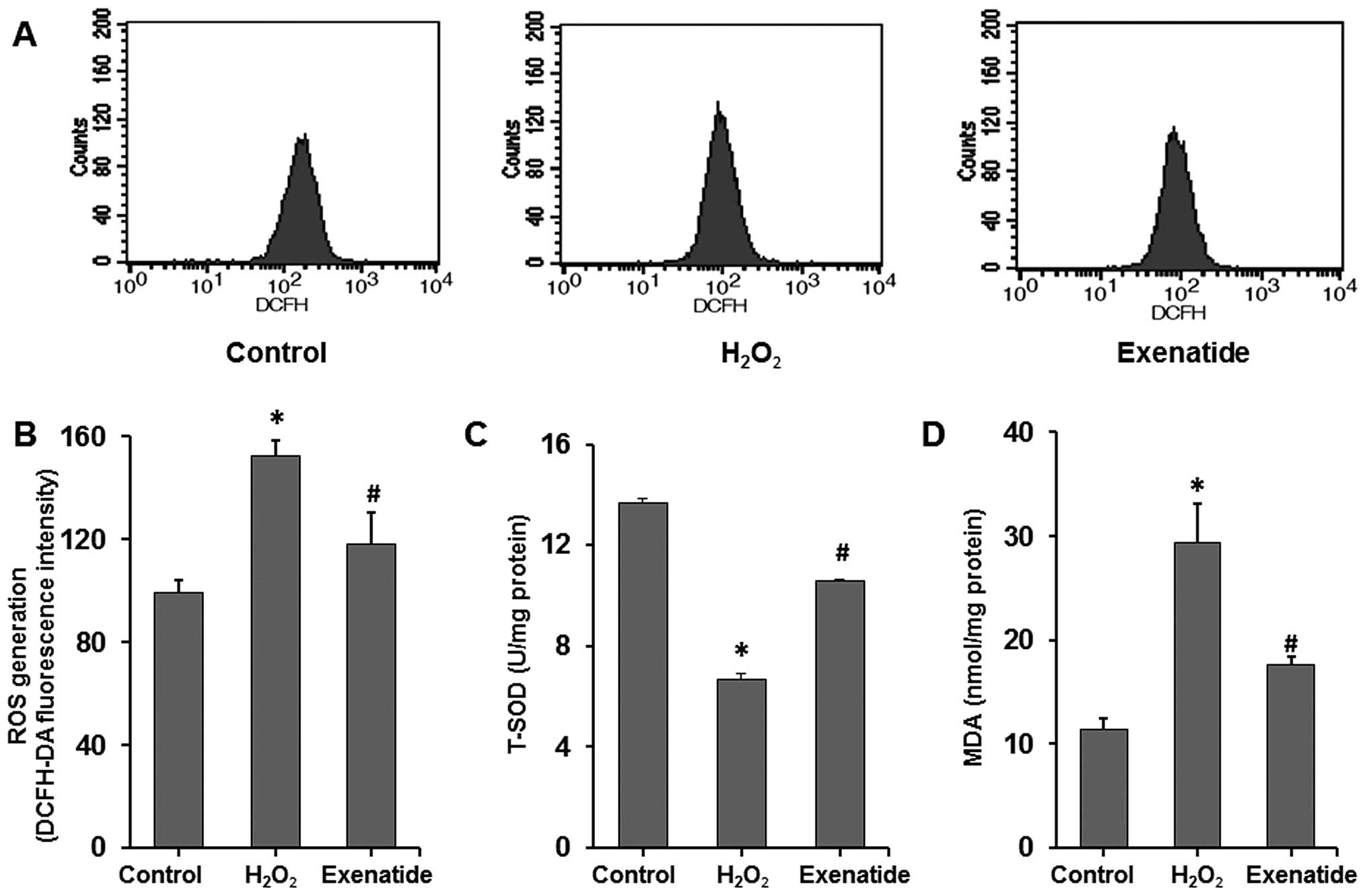
Exenatide reduces
H2O2-mediated oxidative stress in H9c2
cells
To further determine the effects of exenatide on
oxidative stress induced by H2O2, the
intracellular levels of ROS, T-SOD and MDA were measured (Fig. 3A–D). ROS and MDA levels were
significantly increased in the H2O2 group
compared with those in the control group (P<0.05), while the
T-SOD level was significantly decreased (P<0.05). Exenatide at
the concentration of 1 nM, which was administered 30 min prior to
exposure to H2O2, increased the T-SOD level
(P<0.05) and decreased ROS and MDA levels (P<0.05). These
results indicated that exenatide reduced oxidative stress induced
by H2O2 by scavenging oxidative stress
products (ROS and MDA) and increasing the concentration of
antioxidant defense enzymes (SOD) in H9c2 cells.
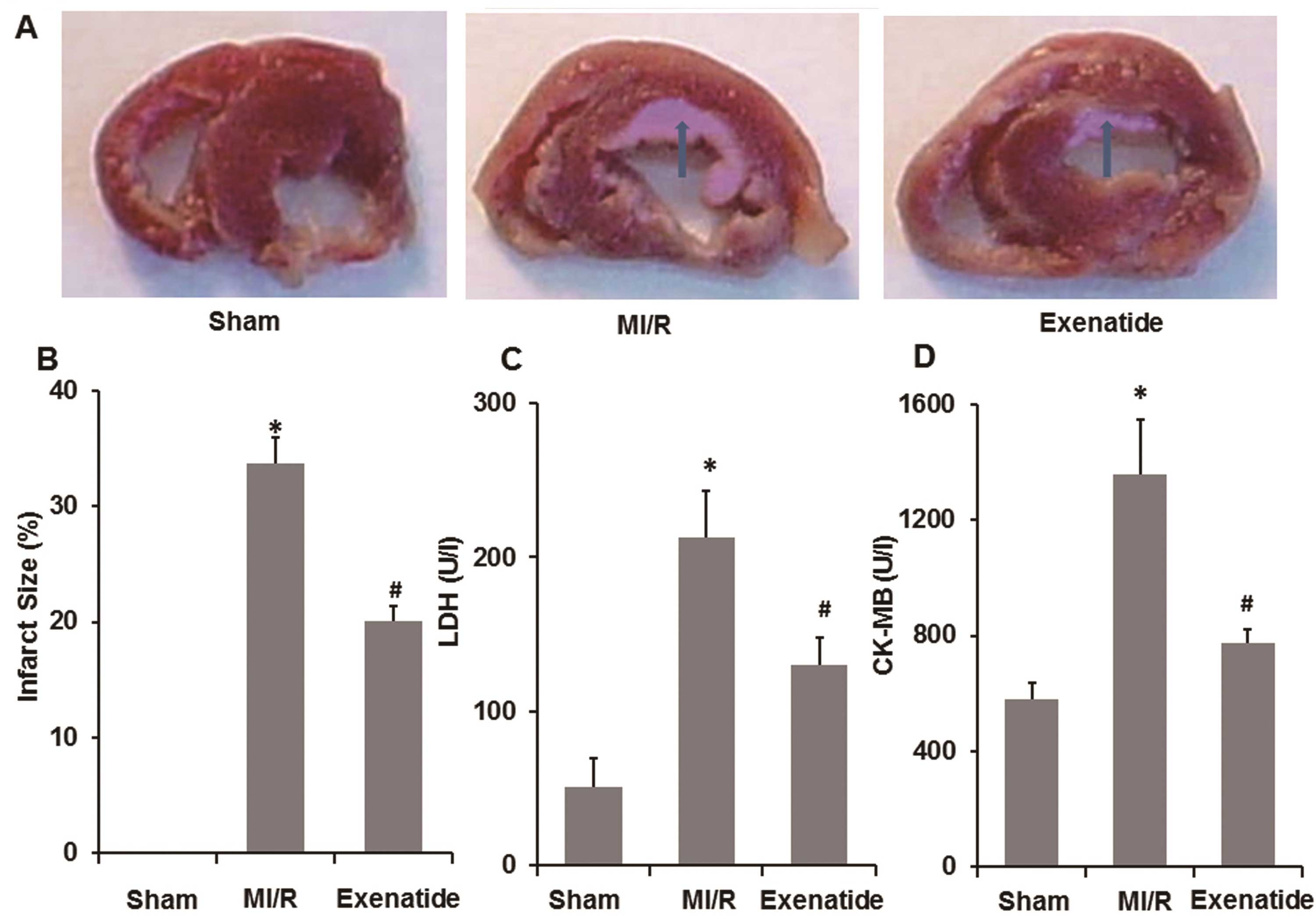
Exenatide decreases myocardial injury in
the rats with MI/R-induced injury
To determine the effects of exenatide on myocardial
injury in the rats with MI/R-induced injury, infarct size and the
release of LDH and CK-MB in the plasma were measured. As shown in
Fig. 4A–D, 30 min of ischemia and
2 h of reperfusion resulted in an increased infarct size, as well
as an increase in LDH and CK-MB levels (P<0.05). Following
pre-treatment with exenatide, infarct size was significantly
decreased compared with the MI/R group (P<0.05). Simultaneously,
LDH and CK-MB levels were significantly reduced in the exenatide
group compared with those in the MI/R group (P<0.05). These
results suggest that exenatide decreases myocardial injury induced
by MI/R in rats.
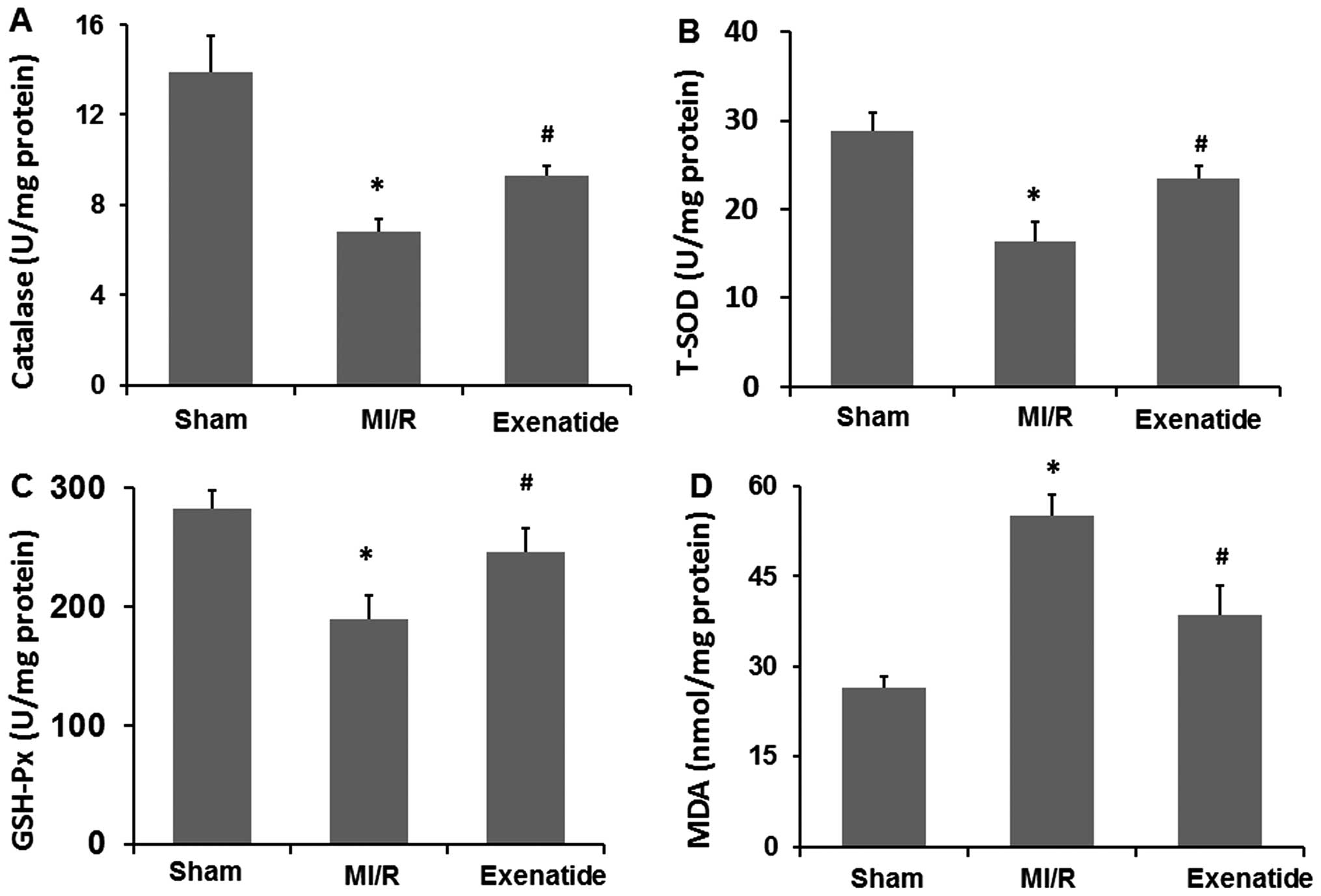
Exenatide reduces MI/R-mediated oxidative
stress in heart homogenates
To further determine the effects of exenatide on
oxidative stress induced by MI/R, the homogenate levels of
catalase, T-SOD, GSH-Px and MDA were measured (Fig. 5A–D). Catalase, T-SOD and GSH-Px
levels were significantly decreased in the MI/R group (P<0.05),
while MDA levels were significantly increased (P<0.05), compared
with those in the sham-operated group. Exenatide at a dose of 10
μg/kg/day increased catalase, T-SOD and GSH-Px levels (P<0.05)
and decreased MDA levels (P<0.05) compared with those in the
MI/R group. These results suggest that exenatide reduces oxidative
stress induced by MI/R by scavenging oxidative stress products
(MDA) and increasing the concentration of antioxidant defense
enzymes (catalase, T-SOD and GSH-Px).
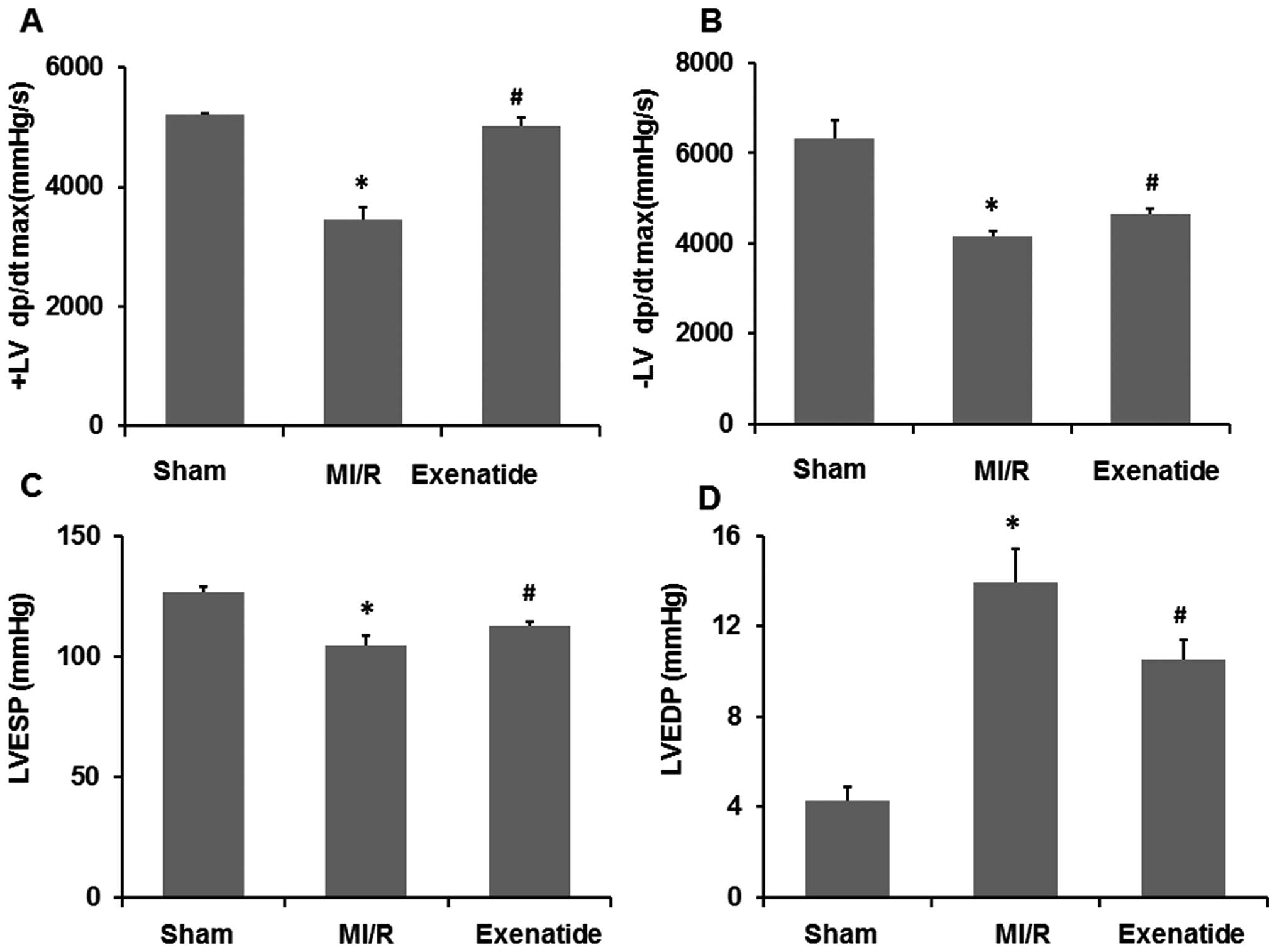
Exenatide enhances left ventricular
function in rats with MI/R-induced injury
To determine the effects of exenatide on cardiac
function in the rats with MI/R-induced injury, hemodynamic
measurements were performed at the end of the MI/R period. As shown
in Fig. 6, compared with the
sham-operated group, MI/R significantly decreased +LVdp/dtmax,
−LVdp/dtmax and LVESP (P<0.05), while it significantly increased
LVEDP (P<0.05). Compared with the MI/R group, exenatide
significantly enhanced +LVdp/dtmax, −LVdp/dtmax, LVESP (P<0.05),
but significantly reduced LVEDP (P<0.05), suggesting that the
left ventricular function was enhanced by exenatide pre-treatment
in the rats with MI/R-induced injury.
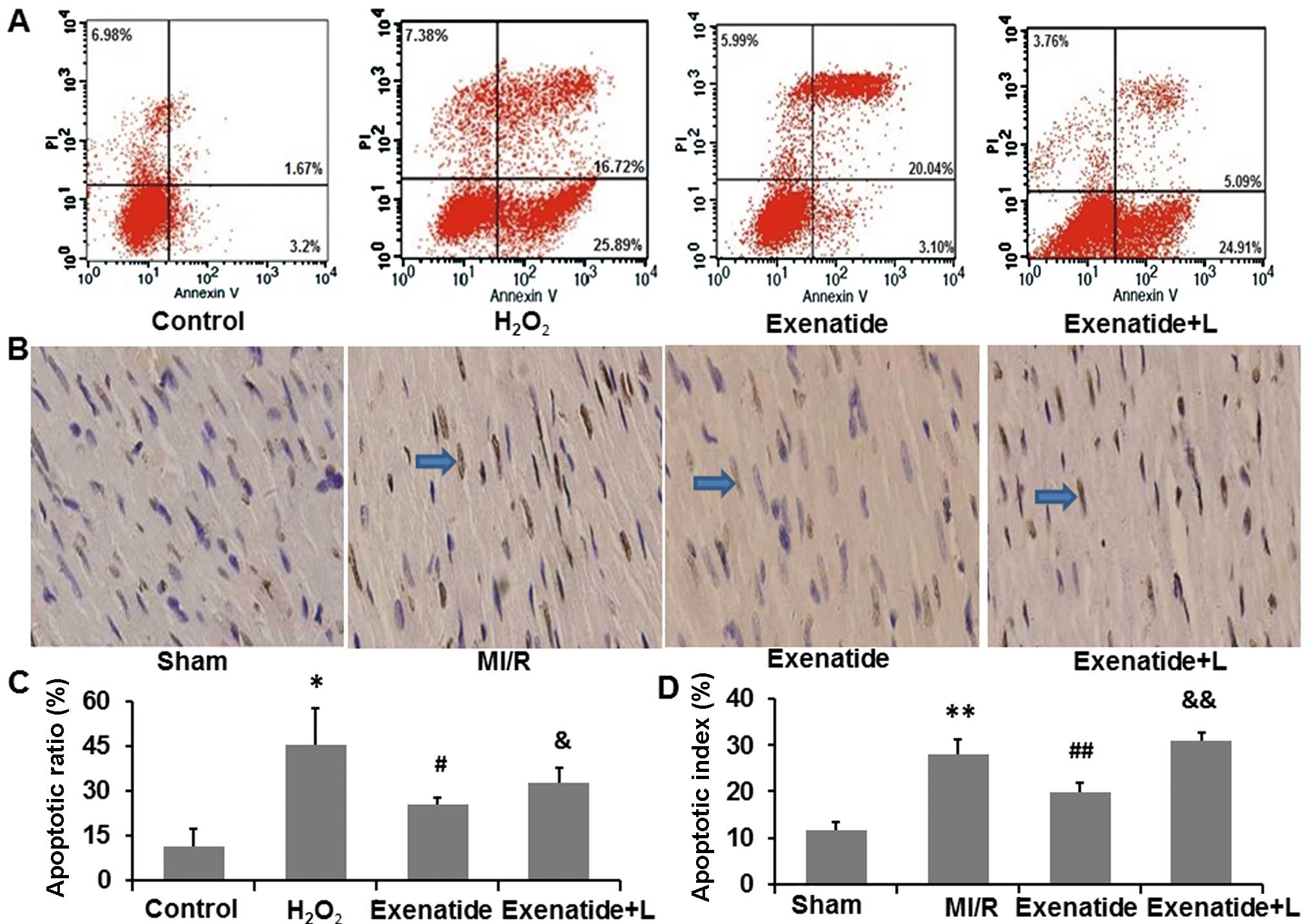
Exenatide reduces H9c2 cell apoptosis
induced by H2O2 and cardiomyocyte apoptosis
induced by MI/R
To investigate the anti-apoptotic effects of
exenatide, we measured the apoptotic ratio of H9c2 cells and
cardiomyocytes in the rats. The PI3K inhibitor, LY294002, was
employed to determine the mechanisms behind the anti-apoptotic
effects of exenatide (Fig.
7).
Firstly, we investigated the anti-apoptotic effects
of exenatide in the H9c2 cells by flow cytometry (Fig. 7A and C). The apoptotic ratio of
H9c2 cells was significantly increased in the
H2O2 group compared with the control group
(P<0.05). Following pre-treatment with exenatide, the apoptotic
ratio was decreased compared with the H2O2
group (P<0.05), indicating that exenatide protected the H9c2
cells from H2O2-induced apoptosis. However,
the anti-apoptotic effects of exenatide were attenuated in the
presence of LY294002, suggesting that LY294002 inhibited the
anti-apoptotic effects of exenatide in H9c2 cells.
We also examined the anti-apoptotic effects of
exenatide on cardiomyocytes in myocardial tissue by TUNEL staining
(Fig. 7B and D). We found that
the apoptotic index was significantly increased in the MI/R group
compared with the sham-operated group (P<0.05). The apoptotic
index was significantly decreased in the exenatide group compared
with that in the MI/R group (P<0.05). Moreover, the apoptotic
index was significantly higher in the exenatide + L group than the
exenatide group (P<0.05), suggesting that LY294002 inhibited the
anti-apoptotic effects of exenatide in the rats with MI/R-induced
injury.
Thus, our flow cytometry and TUNEL staining results
indicated that exenatide suppressed apoptosis in vitro and
in vivo and that these effects were attenuated by the PI3K
inhibitor, LY294002.
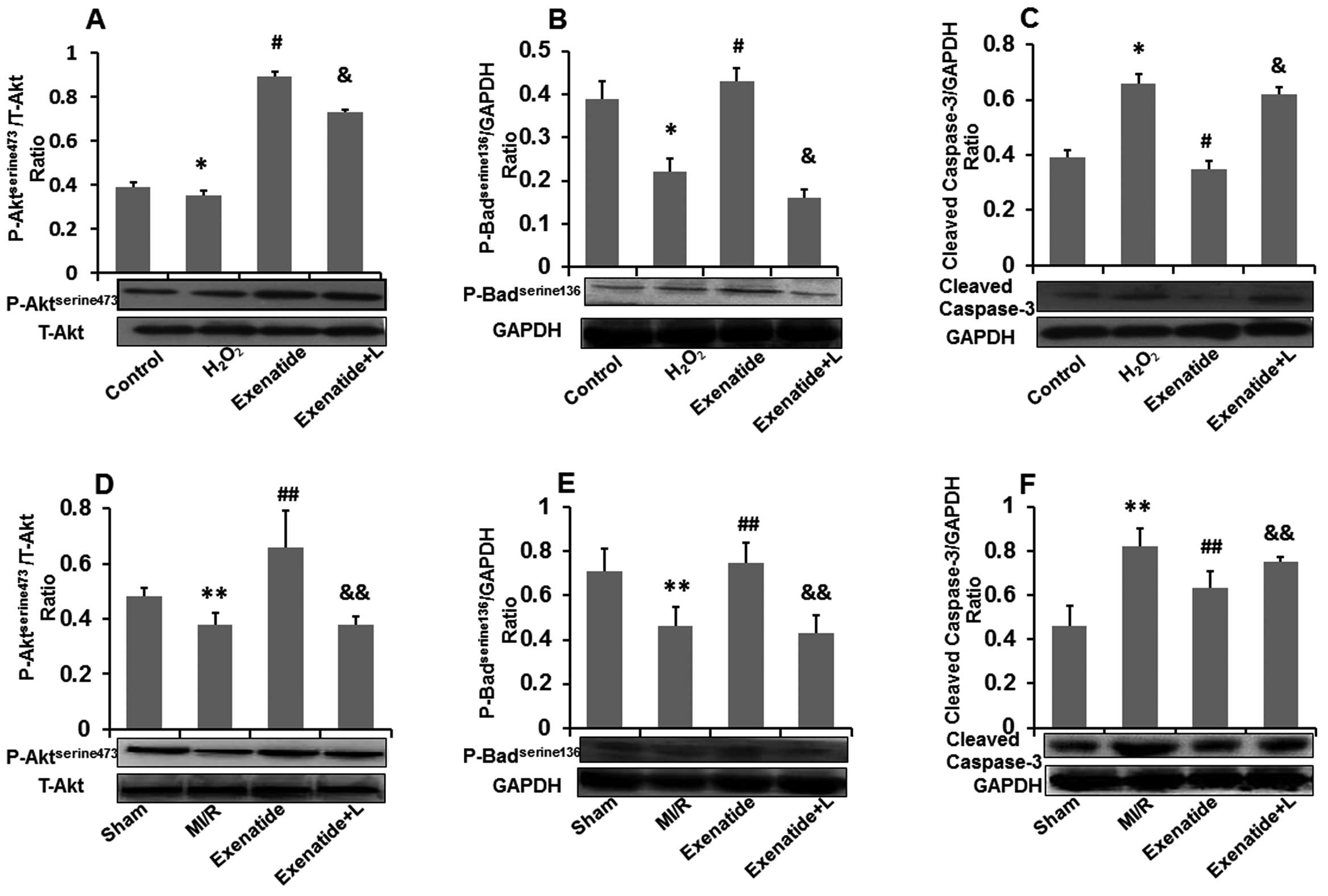
Exenatide increases
Aktserine473 and Badserine136 phosphorylation
and decreases cleaved caspase-3 expression
To investigate the anti-apoptotic effects of
exenatide against oxidative stress-induced injury, we further
assessed the effects of exenatide on
phospho-AktTserine473, phospho-Badserine136
and cleaved caspase-3 levels in H9c2 cells and in myocardial tissue
(Fig. 8). The representative
western blot analysis results and the results from quantitative
analysis are shown in Fig.
8A–F.
As shown in Fig. 8A
and B, H2O2 treatment significantly
reduced the levels of phospho-Aktserine473 (P<0.05)
and phospho-Badserine136 (P<0.05) compared with those
in the control group. Compared with the H2O2
group, pre-treatment with exenatide increased the levels of
phospho-AKTserine473 (P<0.05) and
phospho-Badserine136 (P<0.05). The PI3k inhibitor,
LY29002, attenuated the effects of exenatide on the increased
phospho-AKTserine473 (P<0.05) and
phospho-Badserine136 levels (P<0.05). As shown in
Fig. 8C, exposure to
H2O2 increased cleaved caspase-3 expression
(P<0.05) compared with the control group, whereas pre-treatment
of the H9c2 cells with exenatide reduced cleaved caspase-3
expression (P<0.05). Similarly, the PI3K inhibitor, LY294002,
attenuated the effects of exenatide on the decreased cleaved
caspase-3 expression (P<0.05).
We also examined changes in the levels of
phospho-AKTserine473, phospho-Badserine136
and cleaved caspase-3 in myocardial tissue (Fig. 8D–F). The levels of
phospho-AKTserine473 and phospho-Badserine136
in myocardial tissue were significantly decreased (P<0.05) in
the MI/R group compared with those in the sham-operated group,
contrary to the expression of cleaved caspase-3 which was
significantly increased (P<0.05). The levels of
phospho-AKTserine473 and phospho-Badserine136
in the exenatide group were significantly higher than those in the
MI/R group (P<0.05) and the expression of cleaved caspase-3 in
the exenatide group was significantly lower than that in the MI/R
group (P<0.05). The levels of phospho-AKTserine473
and phospho-Badserine136 were significantly decreased
(P<0.05) in the exenatide + L group, contrary to the expression
of cleaved caspase-3 which was significantly increased
(P<0.05).
Thus, it can be hypothesized that exenatide inhibits
apoptosis in vitro and in vivo, at least in part,
through the PI3K/Akt pathway.
Discussion
In the present study, we evaluated the protective
effects of exenatide on oxidative stress-induced injury. We found
that pre-treatment with exenatide protected cardiomyocytes against
oxidative stress induced by H2O2 and MI/R by
increasing cell viability, decreasing the levels of cardiac injury
makers (LDH and CK-MB), reducing infarct size, enhancing cardiac
function and inhibiting cell apoptosis. The mechanisms behind these
protective effects may be attributed to the scavenging of oxidative
stress products, such as ROS and MDA, the increase in the
concentration of antioxidant defense enzymes, such as catalase, SOD
and GSH-Px and the inhibition of cardiomyocyte apoptosis. Our
results also suggest that exenatide inhibits cardiomyocyte
apoptosis, at least in part, through the PI3K/Akt pathway.
Oxidative stress products, such as ROS, are
considered to be important factors inducing myocardial injury
during MI/R (3,18), whereas treatment with antioxidant
agents or the upregulation of endogenous antioxidant enzymes in
animals have been shown to exert cardioprotective effects against
MI/R-induced injury (18,19). ROS and MDA are the products of
oxidative stress, which reflect the cell damage caused by oxidative
stress. Catalase, SOD and GSH-Px, by inhibiting O2 and
H2O2 interaction, constitute the first line
of cellular defense against oxidative injury (20). In the present study, we
demonstrated that exposure to H2O2 (6 h)
in vitro and MI/R (30 min/2 h) in vivo significantly
increased the levels of oxidative stress products (ROS and MDA),
decreased the concentrations of antioxidant defense enzymes
(catalase, T-SOD and GSH-Px) and aggravated myocardial injury;
these results are in line with previous reports (21,22). Importantly, in this study, the
rats treated with exenatide had enhanced activities of antioxidant
defense enzymes (catalase, T-SOD and GSH-Px), but lower MDA
production in comparison with the rats with MI/R-induced injury.
Similarly, we found that exenatide significantly increased the
levels of T-SOD and decreased the levels of ROS and MDA in the
H2O2-treated H9c2 cells. A recent study
reported that the GLP-1 receptor agonist, exendin-4, increased SOD
levels and decreased MDA levels in neonatal rats with
hyperglycemia-induced cardiomyocytes injury (23). Our results suggest that exenatide
regulates the levels of endogenous antioxidant enzymes and
oxidative stress products in H2O2-treated
H9c2 cells and rats with MI/R-induced injury.
Cytosolic enzymes, such as CK-MB and LDH, which leak
out from damaged tissues to the blood stream when the cell membrane
becomes permeable or ruptures, serve as diagnostic markers of
myocardial cell injury (24). In
the present study, the activities of LDH and CK-MB were
significantly increased in the H2O2-treated
H9c2 cell conditioned medium and in the plasma of rats with
MI/R-induced injury. However, the increased levels of LDH and CK-MB
were significantly suppressed in the exenatide pre-treated H9c2
cells and rats. Our findings in vitro are consistent with
previous reports in which GLP-1 decreased the LDH and CK-MB levels
in neonatal rat cardiomyocytes (23,25). We also found that exenatide
pre-treatment significantly reduced the infarct size in the rats
with MI/R-induced injury; this result is in line with previous
reports (12,26). As previously reported (22,27,28), we also found that MI/R impairs
cardiac function in rats. More importantly, we found that exenatide
pre-treatment significantly improved cardiac function by increasing
±LV dp/dtmax, LVESP and limiting the increase of LVEDP in the rats
with MI/R-induced injury. Similar to our study, Timmers et
al observed that exenatide treatment improved cardiac function
in a porcine model of MI/R-induced injury (12). These results strongly indicate
that exenatide attenuates myocardial injury induced by oxidative
stress.
A number of studies have confirmed that
cardiomyocyte apoptosis is one of the most common
pathophysiological processes in injury induced by oxidative stress
and MI/R (3). Consistent with
these reports, we demonstrated significantly higher cell apoptosis
in H2O2-treated H9c2 cells and in the rats
with MI/R-induced injury. Furthermore, the anti-apoptotic effects
of exenatide were confirmed by the results of Annexin V-FITC and
TUNEL staining. These results strongly indicate that exenatide
inhibits cell apoptosis induced by oxidative stress-induced
injury.
On the basis of the obtained results that exenatide
inhibits cardiomyocyte apoptosis induced by oxidative stress, we
further investigated the possible mechanisms behind the
anti-apoptotic effects of exenatide. Previous studies have
demonstrated that the activation of the reperfusion injury salvage
kinase (RISK) pathway, including PI3K/Akt and ERK1/2 provides an
amenable pharmacological target for cardioprotection (29). Therefore, we speculate that the
anti-apoptotic effects of exenatide may be responsible for the
activation of the PI3K/Akt pathway. Accumulating evidence has shown
that the PI3K/Akt signaling pathway can be activated by the
phosphorylation of Akt to protect the myocardium from apoptosis
following MI/R (30–32). The mechanisms behind the
anti-apoptotic effects of PI3K/Akt signaling pathways are varied,
such as inhibiting caspase activation, affecting glucose
metabolism, regulating Bcl-2 family activity and inhibiting death
gene expression. In the present study, we found that LY294002 (a
specific inhibitor of PI3K) inhibits the functions of downstream
target kinases of PI3K) and reverses the anti-apoptotic effects of
exenatide, suggesting that the anti-apoptotic effects of exenatide
are dependent on the PI3K/Akt pathway. More importantly, our data
demonstrated that exenatide upregulated Aktserine473 and
Badserine136 phosphorylation levels in
H2O2-treated H9c2 cells and in the rats with
MI/R-induced injury, which in turn led to the decreased expression
of cleaved caspase-3. However, these effects of exenatide were
attenuated in the presence of LY294002. These results indicate that
the anti-apoptotic effects of exenatide may be, at least in part,
associated with the activation of the PI3K/Akt signaling
pathway.
In conclusion, the prominent finding of this study
was that exenatide exerts significant cardioprotective effects
against oxidative stress induced by H2O2 and
MI/R. The mechanisms involved may be attributed to the scavenging
of oxidative stress products, increasing the concentration of
antioxidant defense enzymes and inhibiting cardiomyocyte apoptosis.
Moreover, the anti-apoptotic effects of exenatide are, at least in
part, associated with the activation of the PI3K/Akt signaling
pathway. The data from our study may provide a new and deeper
insight into the therapeutic targets for ischemic heart
disease.
Acknowledgements
This study was supported by the National Natural
Science Funds for Youths (grant no. 81100196). We are grateful to
Jianyong Wu and Dezhang Zhao (Institute of Life Sciences, Chongqing
Medical University) for providing excellent technical support for
the flow cytometry analysis.
References
|
1
|
Acar E, Ural D, Bildirici U, Sahin T and
Yılmaz I: Diabetic cardiomyopathy. Anadolu Kardiyol Derg.
11:732–737. 2011.
|
|
2
|
Zweier JL and Talukder MA: The role of
oxidants and free radicals in reperfusion injury. Cardiovasc Res.
70:181–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhao ZQ: Oxidative stress-elicited
myocardial apoptosis during reperfusion. Curr Opin Pharmacol.
4:159–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gottlieb RA: Cell death pathways in acute
ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther.
16:233–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kajstura J, Cheng W, Reiss K, Clark WA,
Sonnenblick EH, Krajewski S, Reed JC, Olivetti G and Anversa P:
Apoptotic and necrotic myocyte cell deaths are independent
contributing variables of infarct size in rats. Lab Invest.
74:86–107. 1996.PubMed/NCBI
|
|
6
|
Palojoki E, Saraste A, Eriksson A, Pulkki
K, Kallajoki M, Voipio-Pulkki LM and Tikkanen I: Cardiomyocyte
apoptosis and ventricular remodeling after myocardial infarction in
rats. Am J Physiol Heart Circ Physiol. 280:H2726–H2731.
2001.PubMed/NCBI
|
|
7
|
Garber AJ: Novel GLP-1 receptor agonists
for diabetes. Expert Opin Investig Drugs. 21:45–57. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davidson JA: Advances in therapy for type
2 diabetes: GLP-1 receptor agonists and DPP-4 inhibitors. Cleve
Clin J Med. 76(Suppl 5): S28–S38. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lorber D: GLP-1 receptor agonists: effects
on cardiovascular risk reduction. Cardiovasc Ther. Jul
30–2012.(Epub ahead of print).
|
|
10
|
Mundil D, Cameron-Vendrig A and Husain M:
GLP-1 receptor agonists: a clinical perspective on cardiovascular
effects. Diab Vasc Dis Res. 9:95–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chiquette E, Toth PP, Ramirez G, Cobble M
and Chilton R: Treatment with exenatide once weekly or twice daily
for 30 weeks is associated with changes in several cardiovascular
risk markers. Vasc Health Risk Manag. 8:621–629. 2012.PubMed/NCBI
|
|
12
|
Timmers L, Henriques JP, de Kleijn DP,
Devries JH, Kemperman H, Steendijk P, Verlaan CW, Kerver M, Piek
JJ, Doevendans PA, Pasterkamp G and Hoefer IE: Exenatide reduces
infarct size and improves cardiac function in a porcine model of
ischemia and reperfusion injury. J Am Coll Cardiol. 53:501–510.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Laviola L, Leonardini A, Melchiorre M,
Orlando MR, Peschechera A, Bortone A, Paparella D, Natalicchio A,
Perrini S and Giorgino F: Glucagon-like peptide-1 counteracts
oxidative stress-dependent apoptosis of human cardiac progenitor
cells by inhibiting the activation of the c-Jun N-terminal protein
kinase signaling pathway. Endocrinology. 153:5770–5781. 2012.
View Article : Google Scholar
|
|
14
|
Ravassa S, Zudaire A, Carr RD and Díez J:
Antiapoptotic effects of GLP-1 in murine HL-1 cardiomyocytes. Am J
Physiol Heart Circ Physiol. 300:H1361–H372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Younce CW, Burmeister MA and Ayala JE:
Exendin-4 attenuates high glucose-induced cardiomyocyte apoptosis
via inhibition of endoplasmic reticulum stress and activation of
SERCA2a. Am J Physiol Cell Physiol. 304:C508–C518. 2013. View Article : Google Scholar
|
|
16
|
Kumar S, Kain V and Sitasawad SL: High
glucose-induced Ca2+ overload and oxidative stress
contribute to apoptosis of cardiac cells through mitochondrial
dependent and independent pathways. Biochim Biophys Acta.
1820:907–920. 2012.
|
|
17
|
Fishbein MC, Meerbaum S, Rit J, Lando U,
Kanmatsuse K, Mercier JC, Corday E and Ganz W: Early phase acute
myocardial infarct size quantification: validation of the triphenyl
tetrazolium chloride tissue enzyme staining technique. Am Heart J.
101:593–600. 1981. View Article : Google Scholar
|
|
18
|
Loesser KE, Kukreja RC, Kazziha SY, Jesse
RL and Hess ML: Oxidative damage to the myocardium: a fundamental
mechanism of myocardial injury. Cardioscience. 2:199–216.
1991.PubMed/NCBI
|
|
19
|
Suzuki K, Murtuza B, Sammut IA, Latif N,
et al: Heat shock protein 72 enhances manganese superoxide
dismutase activity during myocardial ischemia-reperfusion injury,
associated with mitochondrial protection and apoptosis reduction.
Circulation. 106:I270–I276. 2002.
|
|
20
|
Peng X and Li Y: Induction of cellular
glutathione-linked enzymes and catalase by the unique
chemoprotective agent, 3H-1,2-dithiole-3-thione in rat
cardiomyocytes affords protection against oxidative cell injury.
Pharmacol Res. 45:491–497. 2002. View Article : Google Scholar
|
|
21
|
Li C, Liu Z, Tian J, Li G, Jiang W, Zhang
G, Chen F, Lin P and Ye Z: Protective roles of Asperosaponin VI, a
triterpenesaponin isolated from Dipsacusasper Wall on acute
myocardial infarction in rats. Eur J Pharmacol. 627:235–241. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li J, Shao ZH, Xie JT, Wang CZ,
Ramachandran S, Yin JJ, Aung H, Li CQ, Qin G, Vanden Hoek T and
Yuan CS: The effects of ginsenoside Rb1 on JNK in oxidative injury
in cardiomyocytes. Arch Pharm Res. 35:1259–1267. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jin HB, Yang YB, Song YL, Zhang YC and Li
YR: Protective roles of quercetin in acute myocardial ischemia and
reperfusion injury in rats. Mol Biol Rep. 39:11005–11009. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cai Y, Hu X, Yi B, Zhang T and Wen Z:
Glucagon-like peptide-1 receptor agonist protects against
hyperglycemia-induced cardiocytes injury by inhibiting high
mobility group box 1 expression. Mol Biol Rep. 39:10705–10711.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fontes JP, Gonçalves M and Ribeiro VG:
Serum markers for ischemic myocardial damage. Rev Port Cardiol.
18:1129–1136. 1999.(Portuguese).
|
|
26
|
Xie Y, Wang SX, Sha WW, Zhou X, Wang WL,
Han LP, Li DQ and Yu DM: Effects and mechanism of glucagon-like
peptide-1 on injury of rats cardiomyocytes induced by
hypoxia-reoxygenation. Clin Med J. 121:2134–2138. 2008.PubMed/NCBI
|
|
27
|
Chinda K, Chattipakorn S and Chattipakorn
N: Cardioprotective effects of incretin during
ischaemia-reperfusion. Diab Vasc Dis Res. 9:256–269. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Khalil PN, Neuhof C, Huss R, Pollhammer M,
Khalil MN, Neuhof H, Fritz H and Siebeck M: Calpain inhibition
reduces infarct size and improves global hemodynamics and left
ventricular contractility in a porcine myocardial
ischemia/reperfusion model. Eur J Pharmacol. 528:124–131. 2005.
View Article : Google Scholar
|
|
29
|
Hausenloy DJ and Yellon DM: New directions
for protecting the heart against ischaemia-reperfusion injury:
targeting the reperfusion injury s alvage kinase (RISK)-pathway.
Cardiovasc Res. 61:448–460. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mullonkal CJ and Toledo-Pereyra LH: Akt in
ischemia and reperfusion. J Invest Surg. 20:195–203. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fujio Y, Nguyen T, Wencker D, Kitsis RN
and Walsh K: Akt promotes survival of cardiomyocytes in
vitro and protects against ischemia-reperfusion injury in mouse
heart. Circulation. 101:660–667. 2000.PubMed/NCBI
|
|
32
|
Matsui T, Tao J, del Monte F, Lee KH, Li
L, Picard M, Force TL, Franke TF, Hajjar RJ and Rosenzweig A: Akt
activation preserves cardiac function and prevents injury after
transient cardiac ischemia in vivo. Circulation.
104:330–335. 2001. View Article : Google Scholar : PubMed/NCBI
|