Introduction
Muscular dystrophy (MD) refers to a genetically
heterogeneous group of degenerative muscle disorders that are
characterized by the progressive loss of muscle strength and
integrity (1), and the most
common and severe type of MD is Duchenne muscular dystrophy (DMD;
MIM#310200), which accounts for more than half of all MD cases
(2–4) and affects approximately 1 in 3,500
live male newborns (5,6). This disease is associated with
continuous cycles of muscle cell regeneration and degeneration,
ultimately resulting in the failure of muscle regeneration; the
muscle is substituted by fat and connective tissue. Clinical
symptoms include constant falling, waddling and out-turned knees,
which appear as early as the age of 2. Pathological studies have
shown that DMD is triggered primarily by the decreased function of
a vital muscle protein known as dystrophin, which is a long,
rod-shaped molecule composed of 3,685 amino acid residues. It
contains 4 distinct domains: an N-terminal actin binding domain, a
central rod domain containing a second actin binding domain, a
cysteine-rich (CR) domain and a C-terminal (CT) domain (7).
In 1987, the dystrophin gene was cloned by Koenig
et al (8,9) approximately 150 years after the
discovery of DMD, which is the largest human gene, which spans
>3,000 kb on the X-chromosome and encodes a 14-kb transcript
that consists of 79 exons, 78 introns and 8 promoters (10–13). Genotype analysis has revealed that
DMD is a common X-linked recessive neuromuscular disorder caused by
mutations of this gene inherited through the mothers who are
carriers or that arise from germ line mosaicism. To date, there are
2 types of identified mutations of the dystrophin gene: deletions,
which constitute approximately 60% of the mutations and generally
involve the region of a major 3′-hotspot and a minor 5′-hotspot,
exons 1–11 and 41–54 regions and non-deletions, which constitute
40% of the mutations, including point mutations, small deletions
and insertions of the dystrophin gene (14,15).
In this study, we analyzed the coding sequences of
the dystrophin gene in a Chinese family and identified a novel
duplication mutation in exon 56 of the dystrophin gene that causes
DMD, expanding the spectrum of mutations causing DMD. To our
knowledge, we are the first to demonstrate that a novel duplication
of a single base can cause severe DMD.
Materials and methods
Patients
The study participants were identified and enrolled
at Huazhong University of Science and Technology Union Hospital,
Wuhan, China. Informed consent was obtained from the participants
in accordance with the study protocols approved by the Ethics
Committee of Huazhong University of Science and Technology.
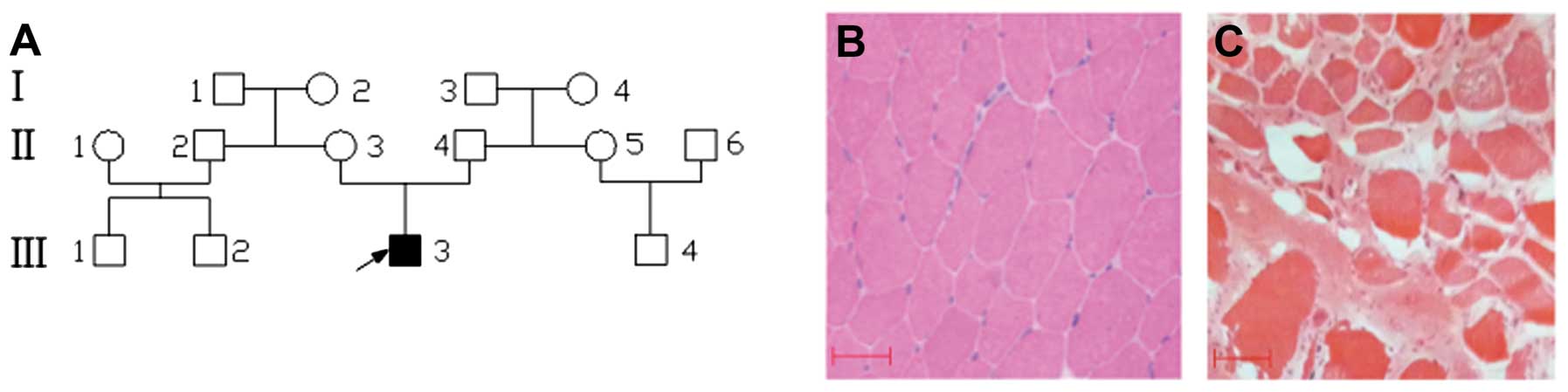
Fourteen family members, including 9 males and 5 females
participated in this study (Fig.
1A). Detailed records of their medical history, physical
examinations and histopathological analysis of muscle tissue were
obtained. The diagnosis of DMD was made on the basis of symptoms,
physical signs and blood creatine kinase (CK) levels. Among all the
family members, only the proband presented the clinical criteria of
the DMD phenotype.
Direct DNA sequencing analyses
As previously described (16), venous blood (5 ml) was collected
from the participants, and total human genomic DNA was isolated
using the DNA Isolation kit for Mammalian Blood (Roche Diagnostics,
Indianapolis, IN, USA). Considering that the dystrophin gene
mutation is the genetic factor in DMD, we carried out mutation
screening in the dystrophin gene directly without performing
linkage analysis. The entire 79 exons and the exon-intron
boundaries of the dystrophin gene of the proband were amplified by
polymerase chain reaction (PCR). Primers were designed to amplify
DNA fragments to span all 79 exons (range, of 200–800 bp; primers
not shown). The nucleotide sequence of the DMD gene was obtained
from GenBank (sequences of the primers are available upon request).
Each amplicon was designed for an optimal size, with the exon
centered within the amplicon. As a result, a total of 79 amplicons
were used to sequence the coding region of the gene. Briefly, for
PCR amplification, amplification was performed in a PTC-200 thermal
cycler (MJ Research Inc., Waterdown, MA, USA) in a 25-μl reaction
mixture containing 1.5 mM MgCl2, 0.2 mM of dNTP (Qiagen,
Hilden, Germany), 0.5 μM primers, 1 U of TaqDNA polymerase (Qiagen)
and 50 ng of genomic DNA. PCR was performed as follows: an initial
denaturation step was carried out for 5 min at 94°C, 9 cycles of 45
sec at 94°C, 45 sec at 61.5°C and 45 sec at 72°C, followed by the
same 29 cycles with a separate annealing temperature at 55°C.
Direct bidirectional resequencing of all PCR-amplified products was
performed using the BigDye Terminator Cycle Sequencing v3.1 kit
(Applied Biosystems, Foster City, CA, USA) and electrophoresed on
an ABI PRISM 3730 Genetic Analyzer (Applied Biosystems). Sequencing
results from the subjects and dystrophin gene consensus sequences
from GenBank (GenBank accession no. M18533) were compared using
BLAST analysis. Mutation description followed the nomenclature
recommended by the Human Genomic Variation Society. Resequencing of
the mutated exon 56 of the dystrophin gene was performed on the 13
family members and the 100 unrelated controls and the primers used
were as follows: forward, GGCACTGGGGTACACTTTATCATAGAA; and reverse,
GCTGCACTCCTCATTTAAATTCACTCT.
Single-strand conformation polymorphism
(SSCP) analysis
To confirm the mutation and determine whether the
mutation co-segregates with the disease in the family, the novel
variation detected in exon 56 of the dystrophin gene was further
evaluated in the 14 available family members, as well as the normal
control subjects using SSCP analysis, as previously described
(17). Briefly, as mentioned
above, PCR amplification was performed on exon 56 of the dystrophin
gene and the primers used were as follows: forward,
GAAAAGGGATTTGAGATGTA; and reverse, GTGCTAAGACAATGAGGAAA, and an
initial denaturation annealing temperature at 60°C and a separate
annealing temperature at 53°C. Subsequently, 2 μl of undigested PCR
products were mixed with 4 μl of the degenerating loading buffer,
denatured at 95°C for 10 min and immediately placed on ice; they
were then loaded on 6% polyacrylamide gels and the DNA samples were
separated by electrophoresis overnight at 150 V. The DNA bands were
visualized by silver staining.
Results
Pedigree and clinical features of the
family
The patient was a 4-year-old boy of Chinese origin,
born after a normal term pregnancy. He developed normally until the
age of 2. Family history was negative and his parents were not
consanguineous. He began to walk when at 18 months of age. However,
at age 3 he presented his first symptom, a tendency to fall, and
had difficulty in rising from the floor and in walking on his toes.
At age 4 he had a waddling gait and could no longer climb stairs. A
physical examination revealed proximal muscle weakness, calf
hypertrophy, a mild weakness of the limb-girdle muscles, deep
tendon hyporflexia, hyperlordosis and a positive Gower’s sign.
Serum muscle enzyme concentrations were markedly increased to
33,400 U/l (normal: 37–174 U/l). His muscle biopsy specimen
revealed dystrophic features with a wide variation in fiber size,
including fiber hypertrophy, degeneration, atrophy and an increase
in endomysial connective tissue (Fig.
1C). However, all the clinical features of the proband were not
discovered in the other members of his family, particularly his
mother.
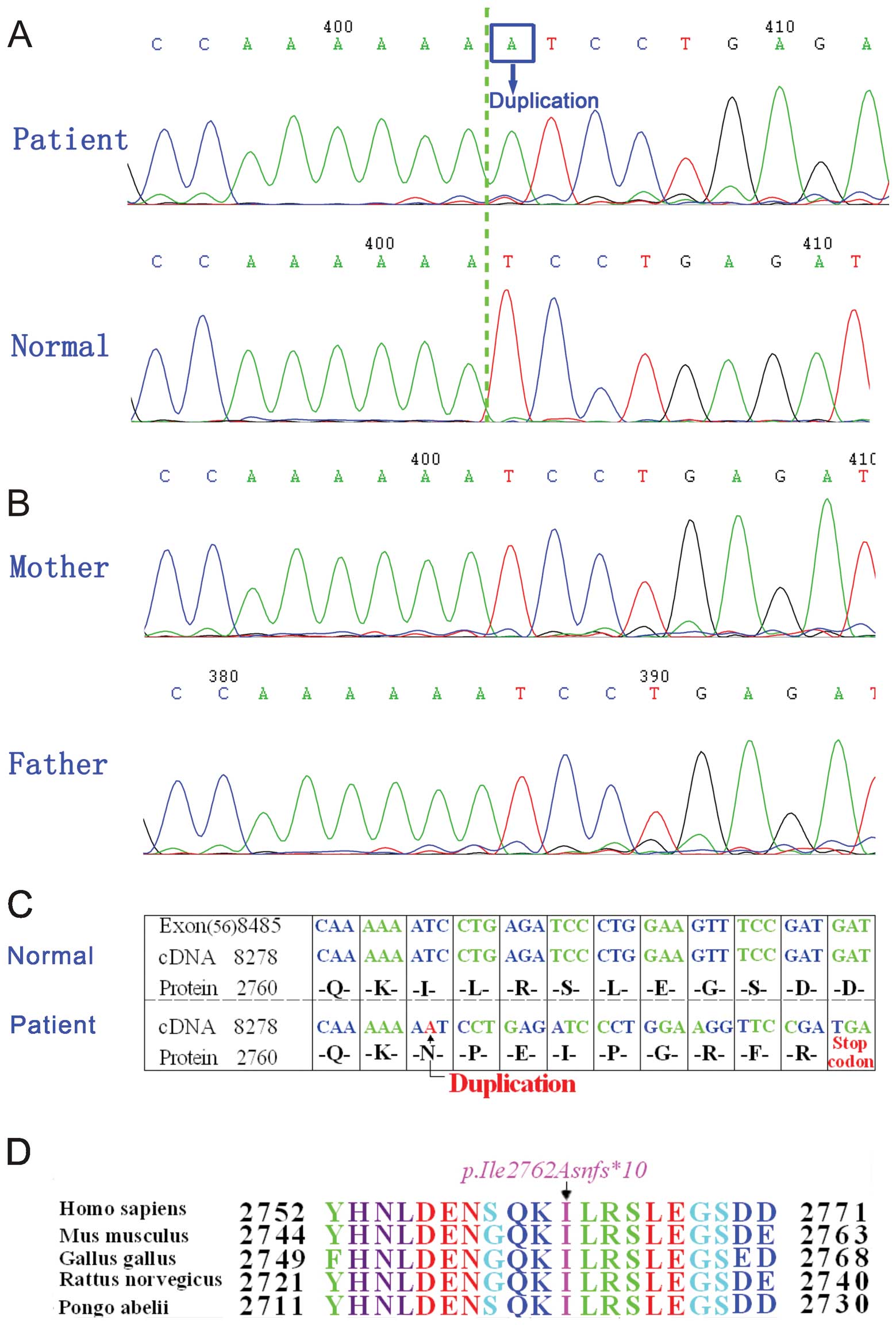
Mutation analysis
To identify the molecular basis of DMD in the
proband, exons 1–79 of the dystrophin gene were amplified by PCR.
By direct bidirectional sequencing of the PCR products in the
proband, a duplication at position 8284 (c.8284dupA) in exon 56 of
the dystrophin gene (Fig. 2A) was
revealed; this was confirmed by repeating the experiment. We did
not find any other mutation or polymorphism in the exons of the
dystrophin gene. Since DMD is a common X-linked recessive
neuromuscular disorder, direct sequence analysis of DNA from his
parents and brother was also performed on exon 56 of the dystrophin
gene. However, the sequence was the same as that of the normal one
(Fig. 2B).
Analysis of the changes in dystrophin
protein sequences after mutation
A duplication (c.8284dupA) in the dystrophin gene
causes a termination codon TGA that results in a truncated product
of 2,770 amino acid residues (p.Ile2762Asnfs*10)
(Fig. 2C) instead of the
wild-type length of 3,685 amino acid residues. Ile2762 is highly
conserved among humans (Homo sapiens), mice (Mus
musculus), chickens (Gallus gallus), rats (Rattus
norvegicus) and orangutans (Pongo abelii) (Fig. 2D).
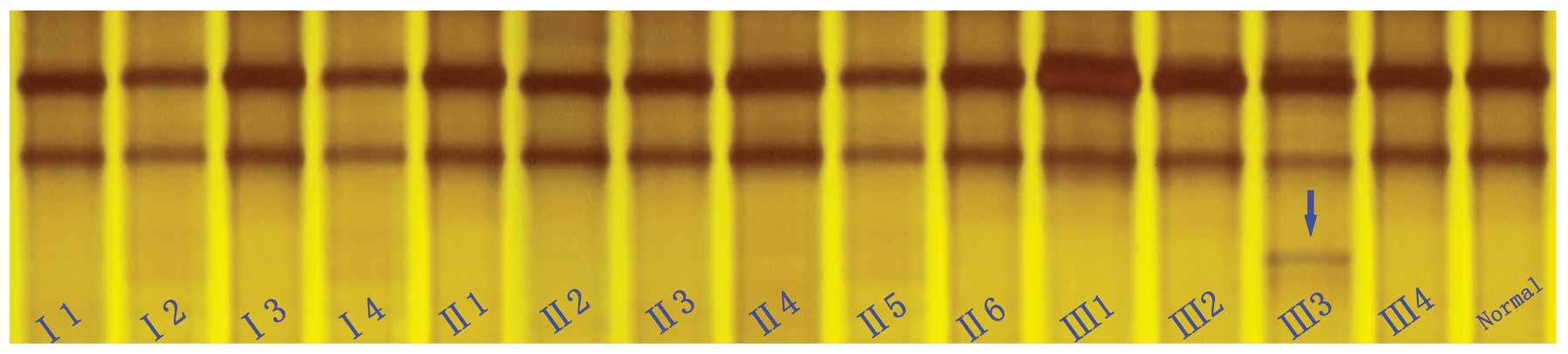
SSCP analysis
To confirm the mutation and test whether the
mutation co-segregates with the disease in the family, the novel
variation detected in exon 56 of the dystrophin gene was further
evaluated in the 14 available family members, as well as in the
normal control subjects using SSCP analysis (Fig. 3). The SSCP results revealed that
an extra band was found in exon 56 of the dystrophin gene. However,
the DNA samples from the 100 normal males and all the family
members were also analyzed by SSCP, and the results revealed that
the unaffected members of the family and the 100 normal Chinese Han
controls did not carry this mutation. These results further suggest
that this novel mutation (c.8284dupA, p.Ile2762Asnfs*10)
of the dystrophin gene is not a rare polymorphism, but a causative
mutation for DMD in the proband.
Discussion
The present study demonstrates that the newly
identified mutation in exon 56 of the dystrophin gene (c.8284dupA)
is associated with DMD in a child in a Chinese family, and that
this mutation is a de novo mutation, but not an X-linked
recessive mode of inheritance from the mother. First, the fact that
the clinical manifestations of proximal muscle weakness, calf
hypertrophy, mild weakness of the limb-girdle muscles, deep tendon
hyporflexia, hyperlordosis and a positive Gower’s sign occurred
only in the proband. The other members did not present these
clinical features. Second, the genotype revealed a duplication at
position 8284 (c.8284dupA) in exon 56 of the dystrophin gene, but
the mother and brother had a normal genotype at this position.
Furthermore, the fact that the mutation found in the proband was
not detected in the 100 unrelated control subjects without DMD by
SSCP excludes the possibility that this mutation is a
polymorphism.
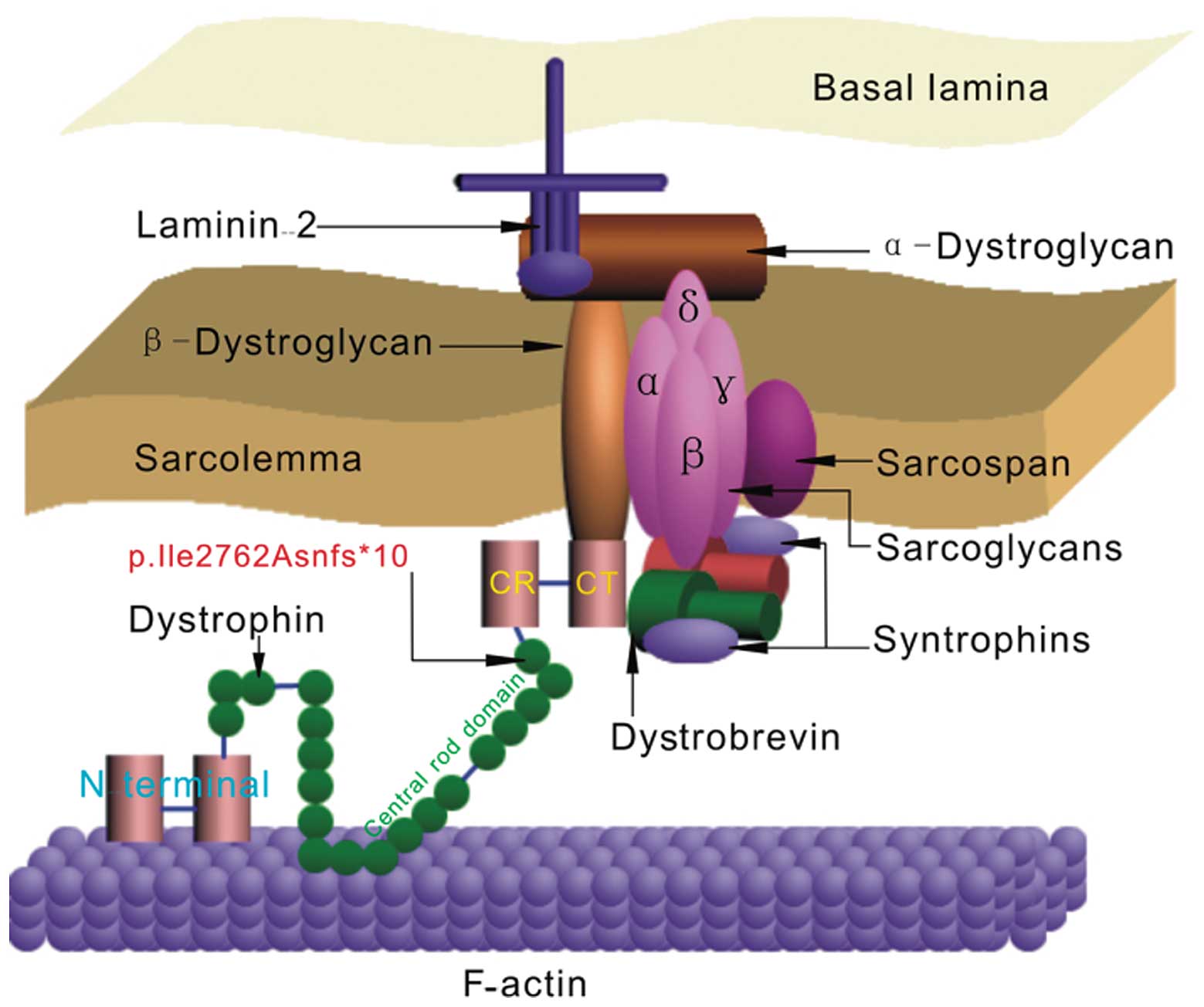
Full-length dystrophin is a large rod-shaped protein
with a molecular weight of 427 kDa that contains 3,685 amino acid
(AA) residues and comprises 4 domains (Fig. 4), which include the actin-binding
(N-terminal) domain (AA #14–240, exons 2–8), the central rod domain
(AA #253–3040, exons 8–61) consisting of 24 spectrin repeats and 4
hinge regions, the CR domain (AA #3080–3360, exons 62–69) and the
CT domain (AA #3361–3685, exons 69–79) (18–21). In this study, we find the newly
identified mutation in exon 56 of the dystrophin gene (c.8284dupA),
which results in a frameshift mutation and a premature termination
10 codons downstream (p.Ile2762Asnfs*10) by stop codon
TGA, which is thus far the most frequent point mutation found in
the DMD gene (10/47) (22). Due
to this mutation, the entire CR and CT domain was lacking. In the
context of a full-length dystrophin protein, the essential CR and
CT domain was not retained; the protein is completely
non-functional (23), which seems
to cause severe symptoms (24).
These results are consistent with the conclusion of Koenig et
al (8,9), who pointed out that the nucleotide
sequence corresponding to the CR and CT domain was lacking in most
DMD cases based on their dystrophin gene mutation analysis.
Dystrophin is associated with the plasma membrane of cardiac and
skeletal muscle (sarcolemma) and its main role in the sarcolemma is
to interact with integral membrane proteins (sarcoglycan,
dystroglycans, syntrophin and dystrobrevin complexes) that are
assembled in the dystrophin-glycoprotein complex (DGC) through its
CR and CT domains (Fig. 4)
(25), including the last 54
amino acid residues of the rod to the CR domain (amino acid
residues #3026–3345) (26).
Therefore, the lack of these domains in the dystrophin gene leads
to destabilization and loss of the DGC (27). Thus, as long as the CR and CT are
deleted, the binding between syntrophin and dystrobrevin is
eliminated. Consequently, the function of the muscle is
diminished.
In conclusion, although several point mutations in
the dystrophin gene associated with DMD have been identified
worldwide (28,29), in this study, we identified a
novel duplication in exon 56 of the dystrophin gene in a Chinese
child with DMD. To our knowledge, this is the first report of
Chinese DMD patients with a duplication in the region of exon 56,
which results in truncated dystrophin lacking the CR and CT. This
finding expands the mutation spectrum of the dystrophin gene and
may prove useful and valuable for genetic counseling and prenatal
diagnosis in families with DMD.
Abbreviations:
|
CR
|
cysteine-rich domain
|
|
CT
|
C-terminal domain
|
|
DGC
|
dystrophin-glycoprotein complex
|
|
DMD
|
Duchenne muscular dystrophy
|
|
MD
|
muscular dystrophy
|
|
SSCP
|
single-strand conformation
polymorphism analysis
|
References
|
1
|
Emery AE: The muscular dystrophies.
Lancet. 359:687–695. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hoffman EP, Brown RH and Kunkel LM:
Dystrophin: The protein product of the Duchenne Muscular Dystrophy
locus. Cell. 51:919–928. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Emery AE: Population frequencies of
inherited neuromuscular disorders - a world survey. Neuromuscul
Disord. 1:19–29. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Blake DJ, Weir A, Newey SE and Davies KE:
Function and genetics of dystrophin and dystrophin-related proteins
in muscle. Physiol Rev. 82:291–329. 2002.PubMed/NCBI
|
|
5
|
van Essen AJ, Kneppers AL, van der Hout
AH, et al: The clinical and molecular genetic approach to Duchenne
and Becker muscular dystrophy: an updated protocol. J Med Genet.
34:805–812. 1997.PubMed/NCBI
|
|
6
|
Sura T, Eu-ahsunthornwattana J,
Pingsuthiwong S and Busabaratana M: Sensitivity and frequencies of
dystrophin gene mutations in Thai DMD/BMD patients as detected by
multiplex PCR. Dis Markers. 25:115–121. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Batchelor CL and Winder SJ: Sparks,
signals and shock absorbers: How dystrophin loss causes muscular
dystrophy. Trends Cell Biol. 16:198–205. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koenig M, Hoffman EP, Bertelson CJ, et al:
Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and
preliminary genomic organization of the DMD gene in normal and
affected individuals. Cell. 50:509–517. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Koenig M, Monaco AP and Kunkel LM: The
complete sequence of dystrophin predicts a rod-shaped cytoskeleton
protein. Cell. 53:219–228. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ahn AH and Kunkel LM: The structural and
functional diversity of dystrophin. Nat Genet. 3:283–291. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nishio H, Takeshima Y, Narita N, et al:
Identification of a novel first exon in the human dystrophin gene
and of a new promoter located more than 500 kb upstream of the
nearest known promoter. J Clin Invest. 94:1037–1042. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Magri F, Del Bo R, D’Angelo MG, et al:
Clinical and molecular characterization of a cohort of patients
with novel nucleotide alterations of the dystrophin gene detected
by direct sequencing. BMC Med Genet. 12:372011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dent KM, Dunn DM, von Niederhausern AC, et
al: Improved molecular diagnosis of dystrophinopathies in an
unselected clinical cohort. Am J Med Genet A. 134:295–298. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tuffery-Giraud S, Chambert S, Demaille J
and Claustres M: Point mutations in the dystrophin gene: evidence
for frequent use of cryptic splice sites as a result of splicing
defects. Hum Mutat. 14:359–368. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Q, Li-Ling J, Lin C, et al:
Characteristics of dystrophin gene mutations among Chinese patients
as revealed by multiplex ligation-dependent probe amplification.
Genet Test Mol Biomarkers. 13:23–30. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cai F, Zhu J, Chen W, et al: A novel PAX6
mutation in a large Chinese family with aniridia and congenital
cataract. Mol Vis. 16:1141–1145. 2010.PubMed/NCBI
|
|
17
|
Wang Q, Shen J, Splawski I, et al: SCN5A
mutations associated with an inherited cardiac arrhythmia, long QT
syndrome. Cell. 80:805–811. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sadoulet-Puccio HM and Kunkel LM:
Dystrophin and its isoforms. Brain Pathol. 6:25–35. 1996.
View Article : Google Scholar
|
|
19
|
Roberts RG: Dystrophins and dystrobrevins.
Genome Biol. 2:REVIEWS30062001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Davies KE, Tinsley JM and Blake DJ:
Molecular analysis of Duchenne muscular dystrophy: past, present,
and future. Ann NY Acad Sci. 758:287–296. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ozawa E: Our trails and trials in the
subsarcolemmal cytoskeleton network and muscular dystrophy
researches in the dystrophin era. Proc Jpn Acad Ser B Phys Biol
Sci. 86:798–821. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang J, Wang W, Li R, et al: The diploid
genome sequence of an Asian individual. Nature. 456:60–65. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nishida A, Kataoka N, Takeshima Y, et al:
Chemical treatment enhances skipping of a mutated exon in the
dystrophin gene. Nat Commun. 2:3082011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hall N: Advanced sequencing technologies
and their wider impact in microbiology. J Exp Biol. 210:1518–1525.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Singh SM, Kongari N, Cabello-Villegas J
and Mallela KM: Missense mutations in dystrophin that trigger
muscular dystrophy decrease protein stability and lead to
cross-beta aggregates. Proc Natl Acad Sci USA. 107:15069–15074.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ishikawa-Sakurai M, Yoshida M, Imamura M,
et al: ZZ domain is essentially required for the physiological
binding of dystrophin and utrophin to beta-dystroglycan. Hum Mol
Genet. 13:693–702. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kemaladewi DU, Hoogaars WM, van Heiningen
SH, et al: Dual exons kipping in myostatin and dystrophin for
Duchenne muscular dystrophy. BMC Med Genomics. 4:362011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hwa HL, Chang YY, Huang CH, et al: Small
mutations of the DMD gene in Taiwanese families. J Formos Med
Assoc. 107:463–469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sitnik R, Campiotto S, Vainzof M, et al:
Novel point mutations in the dystrophin gene. Hum Mutat.
10:217–222. 1997. View Article : Google Scholar : PubMed/NCBI
|