Introduction
Early reperfusion has been widely utilized for
impending acute myocardial infarction (AMI) by thrombolysis or
primary percutaneous coronary angioplasty (1,2).
Although ventricular remodeling and dysfunction subsequent to AMI
are considerably improved by restoration of the blood flow,
reperfusion itself results in additional myocardial
ischemia-reperfusion (I/R) injury (3–5),
which counteracts the protective effect of reperfusion therapy.
Mounting evidence has demonstrated that apoptosis contributes
significantly to myocardial I/R injury (4,5).
Angiotensin converting enzyme (ACE) is an important
member of renin-angiotensin system (RAS). ACE cleaves angiotensin I
(Ang I) to produce the biologically active angiotensin II (Ang II),
a vital peptide molecule in RAS, which may stimulate apoptosis
following I/R (6). In general,
the RAS exists as classical circulating RAS and as a local tissue
RAS (7,8). Classically, Ang II can be generated
in the circulation by ACE and delivered to target tissues or cells.
Furthermore, ACE2 is capable of hydrolyzing Ang II to Ang-(1–7)
and Ang I to Ang-(1–9) (9–11),
and regulating the balance of RAS activation. Findings of a
previous study demonstrated that ACE inhibitors prevented
cardiomyocytes from apoptosis induced by I/R via the inhibition of
ACE with a decrease of Ang II levels (12). However, the effect of ACE cannot
be completely blocked as ACE inhibitors may upregulate ACE levels
in myocardium and plasma through a feedback mechanism (13). In addition, the local tissue RAS
includes two forms: intracellular and extracellular (7,8).
Evidence revealed that the morbidity and mortality were
significantly reduced by using ACE inhibitors in patients with MI
(14–16). Notably, it was found that the
concentrations of Ang II in local tissue may exceed those in
plasma. However, it is difficult to separate the effects of
inhibiting intracellular ACE from systemic ACE inhibition in
vivo, whereas it is easy to evaluate the effects of
intracellular RAS inhibition in vitro.
Gene therapy has been increasingly applied to
inherited or acquired diseases at their genetic levels (17,18). As a new knocking gene-technique,
RNA interference (RNAi) has been demonstrated to be useful for the
treatment of diseases by reducing the production of target RNA and
proteins that are a hindrance to identification of disease
(19). To avoid the feedback
elevation of ACE when using ACE inhibitors and more completely
block the effects of ACE, silencing of the ACE gene by RNAi may be
an ideal option.
Results of previous studies demonstrated that the
injury of cardiomyocytes induced by anoxia/reoxygenation (A/R) is a
useful in vitro model for examining myocardial I/R injury
(20,21). The aim of this study was to
investigate whether the apoptosis of H9c2 cells subjected to A/R
would be improved through the silencing of intracellular ACE by
RNAi in order to regulate the intracellular RAS, thereby regulating
the intrinsic pathway of apoptosis.
Materials and methods
ACE-shRNA plasmid construction
A series of 21-nucleotide siRNA duplexes (AAC CTA
ACA TGT CAG CCT CTG) against the rat ACE consensus coding sequence
(GenBank accession no. NM012544) was designed. Sequences were
determined to be unique to the rat gene by basic local alignment
search tool (BLAST) searches of the GenBank database. The target
sequence was designed with a randomly selected nonsense sequence to
serve as the negative control. The series was designed into a shRNA
oligonucleotide template consisting of sense, hairpin loop,
antisense and terminator sequences, all of which were flanked by
restriction enzyme sites to facilitate directional subcloning. The
DNA sequence coding for the rat ACE-shRNA was synthesized and
cloned into a pGenesil-1 plasmid encoding for enhanced green
fluorescent protein (EGFP). The shRNA was confirmed by
sequencing.
Cell culture and transfection
H9c2 cardiomyocytes (Chinese Academy of Sciences
Cell Bank, Shanghai, China), a sub-clone of the original cell line
derived from embryonic BD1X rat heart tissue, were cultured in
Dulbecco’s modified Eagle’s medium (DMEM, Hyclone, Logan, UT, USA)
containing 10% (v/v) fetal bovine serum (FBS, Invitrogen Corp.,
Carlsbad, CA, USA), penicillin (100 U/ml) and streptomycin (100
μg/ml). The cells were maintained at 37°C in a humidified
atmosphere with 5% CO2. The medium was replaced every
2–3 days, and cells were subcultured or subjected to experimental
procedures.
H9c2 cardiomyocytes were seeded (1.0×105
cells/well) in 6-well plates 1 day prior to transfection.
Transfection was performed strictly according to the manufacturer’s
instructions regarding the Lipofectamine® LTX and Plus™
transfection reagent (Life Technologies, Grand Island, NY, USA).
Six hours after transfection, the medium was replaced with fresh
DMEM (10% FBS, v/v). The cells were incubated for 48 h and was
observed under a fluorescence microscope (Olympus IX51; Olympus,
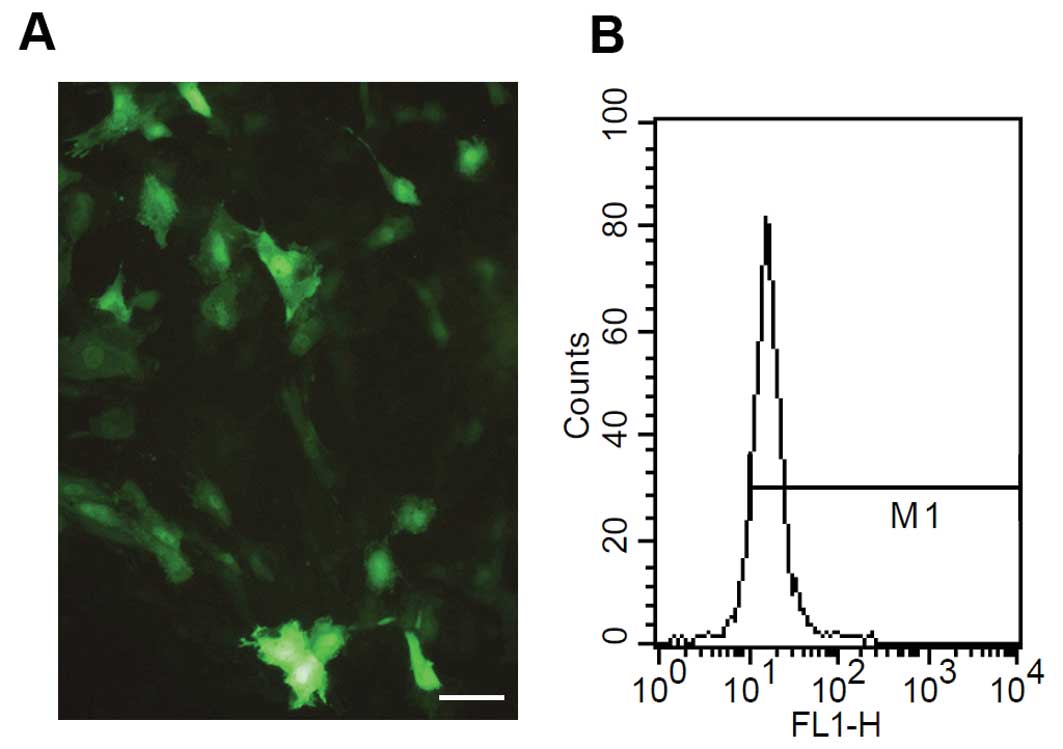
Tokyo, Japan). The percentages of EGFP-expressing cells following
the plasmid transfection were quantified using a flow cytometer
(FACSort; Becton-Dickinson, San Jose, CA, USA). H9c2 cardiomyocytes
were collected for EGFP assay at 48 h post-transfection. EGFP
fluorescence was calculated as the percentage of EGFP cells present
in a total of 104 cells.
A/R injury model
The A/R procedures used in this study were similar
to those described in a previous study (22). H9c2 cardiomyocytes were washed
with PBS and incubated in serum-free DMEM. The cells in DMEM were
placed in a gas transfusive apparatus (Changjing Biotech Co.,
Beijing, China), and anoxic gas (95% N2/5%
CO2) was flushed into the gas transfusive apparatus to
reduce the pO2 to 0 mmHg. Subsequent to 3 h of anoxia at
37°C, reoxygenation was achieved by changing the medium into DMEM
with 10% (v/v) FBS followed by exposure of cells to room air
(CO2 incubator).
Experimental groups and protocols
The cultured H9c2 cardiomyocytes were randomly
divided into different groups. In the control (Con) group, the H9c2
cardiomyocytes were cultured under normal conditions for 12 h. The
A/R group was managed as described in the preceding section. In the
negative control (NC) group and the ACE-shRNA plasmid-treated group
(shRNA), the H9c2 cardiomyocytes were subjected to A/R 48 h
following transfection with the negative control ACE-shRNA plasmid
or ACE-shRNA plasmid.
Cell viability assay
Cell viability was determined by the cell counting
kit (CCK)-8 assay (Dojindo, Tokyo, Japan). The experimental
procedure was conducted according to the manufacturer’s
instructions. H9c2 cardiomyocytes were subcultured at
1×104 cells/well in 96-well plates and incubated for 1
day prior to being divided into the Con, A/R, NC and shRNA groups.
Each group included 9 assays and each assay was averaged from the
absorbance of 4 wells. Prior to the anoxia or following the A/R, 10
μl of WST-8 solution
(2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium,
mono- sodium salt) was added to each well, and the H9c2
cardiomyocytes were incubated for an additional 2 h at 37°C. The
absorbance of each well at 450 nm was measured with a reference at
630 nm using a microplate reader (Bio-Rad Laboratories, Hercules,
CA, USA). The percentage of cell viability was calculated using the
formula: cell viability (%) = (At/Ac) × 100, where At is the mean
absorbance in test wells and Ac is the mean absorbance in control
well.
Apoptosis
Apoptosis was determined by Annexin V and propidium
iodide (PI) double staining. H9c2 cardiomyocytes were centrifuged
to remove the medium, washed with PBS, and stained with Annexin V
and PI in binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM
CaCl2), according to the manufacturer’s instructions
(BioVision, Inc., Palo Alto, CA, USA). Ten thousand events were
collected for each sample. Stained cells in the FL1-H and FL2-H
channels were analyzed using a flow cytometer (FACSort,
Becton-Dickinson).
Measurement of caspase-3 activity
Caspase-3 activity was evaluated by using caspase-3
colorimetric assay kit (BioVision, Inc.). A total of 106
cells were collected by centrifugation, and the pellet was
resuspended in lysis buffer. Protein levels were determined with
bicinchoninic acid assay (Beyotime Biotechnology, Co., Ltd.,
Shanghai, China). Caspase-3 activity was detected in equal amounts
of cell lysates with synthetic peptide substrate Ac-DEVD-pNA, as
described in the manufacturer’s instructions. Caspase-3 activity
was expressed as the optical density, with absorbance at 405 nm of
the released pNA being monitored using a spectrophotometer (UV762;
Shanghai Precision and Scientific Instrument Co., Shanghai,
China).
Quantitative reverse transcription PCR
analysis (qRT-PCR)
Total-RNA was prepared from cells with TRIzol
reagent (Invitrogen Corp.). For quantitative PCR analysis, reverse
transcription was performed to produce cDNA from total RNA with
oligo(dT), and the fragments were amplified with SYBR-Green-based
assays kit (Invitrogen Life Technologies) according to the
manufacturer’s instructions. The RT-PCR conditions were 42°C/15
min, 95°C/2 min for reverse transcription; 95°C/30 sec, 58.9°C
(ACE) or 60°C (GAPDH)/30 sec, and 72°C/60 sec, over 40 cycles for
PCR. ACE mRNA levels were calculated based on the method of
2−ΔΔCT between the intervention and control groups.
GAPDH was used for normalization, and the comparative threshold
method was used to assess the relative abundance of ACE mRNA. The
specific primer sequence and amplicon size of the selected genes
used were: ACE, sense: 5′-GCCTCCCAACGAGTTAGAAGAG-3′, antisense:
5′-CGGGACGTGGCCATTATATT-3′; GAPDH, sense:
5′-GACAACTTTGGCTCGTGGA-3′, antisense: 5′-ATGC AGGGGTTCTGG-3′.
Primers were synthesized by Shanghai Sangon Biological Engineering
Technology Company Limited. Correctness of the gene order was
proven in GenBank.
Western blot analysis
Membranous protein was prepared using membranous
extraction reagents (Pierce Biotechnology, Inc., Rockford, IL, USA)
and mitochondrial protein was extracted using a
mitochondria/cytosol fractionation kit (BioVision, San Francisco,
CA, USA) according to the manufacturer’s instructions. Protein
concentration was determined by the bicinchoninic acid protein
assay (Beyotime Biotechnology, Co., Ltd.,). Equal amounts (50 μg)
of denatured proteins were separated on 10% SDS-polyacrylamide gels
and transferred to nitrocellulose membrane. The membranes were
blocked with 5% non-fat dry milk in TBST (containing 0.05%
Tween-20), and incubated overnight at 4°C with the primary antibody
(ACE, 1:500, Cell Signaling Technology, Inc., Beverly, MA, USA;
ACE2, 1:200, Cell Signaling Technology, Inc.; Bax, 1:500, Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA; Bcl-2, 1:500, Santa
Cruz Biotechnology, Inc.). The blots were then washed and incubated
with horseradish peroxidase-conjugated second antibody (goat
anti-rabbit IgG, 1:2,000, Beyotime Biotechnology) for 1 h under
room temperature. Immunoreactivity was enhanced by
chemiluminescence kit (Beyotime Biotechnology, Co., Ltd.) and
exposed to film. β-actin was used as an internal control to correct
the variations of different samples. The density of bands on
western blots was quantified by using a Bio-Rad image system.
Ang II measurement
The levels of Ang II in the culture medium were
measured by enzyme-linked immunosorbent assay (ELISA), using
commercially available kits (Zhong Shan-Golden Bridge Biological
Technology Co., Beijing, China) according to the manufacturer’s
instructions.
Statistical analysis
Data are presented as the means ± SD. Statistical
analyses of data were performed by one-way ANOVA followed by the
Student-Newman-Keuls test. P<0.05 was considered statistically
significant.
Results
Efficiency of H9c2 cells
transfection
Forty-eight hours after ACE-shRNA plasmid
transfection, (76.8±5.1)% of H9c2 cardiomyocytes were positive for
green fluorescence (Fig. 1).
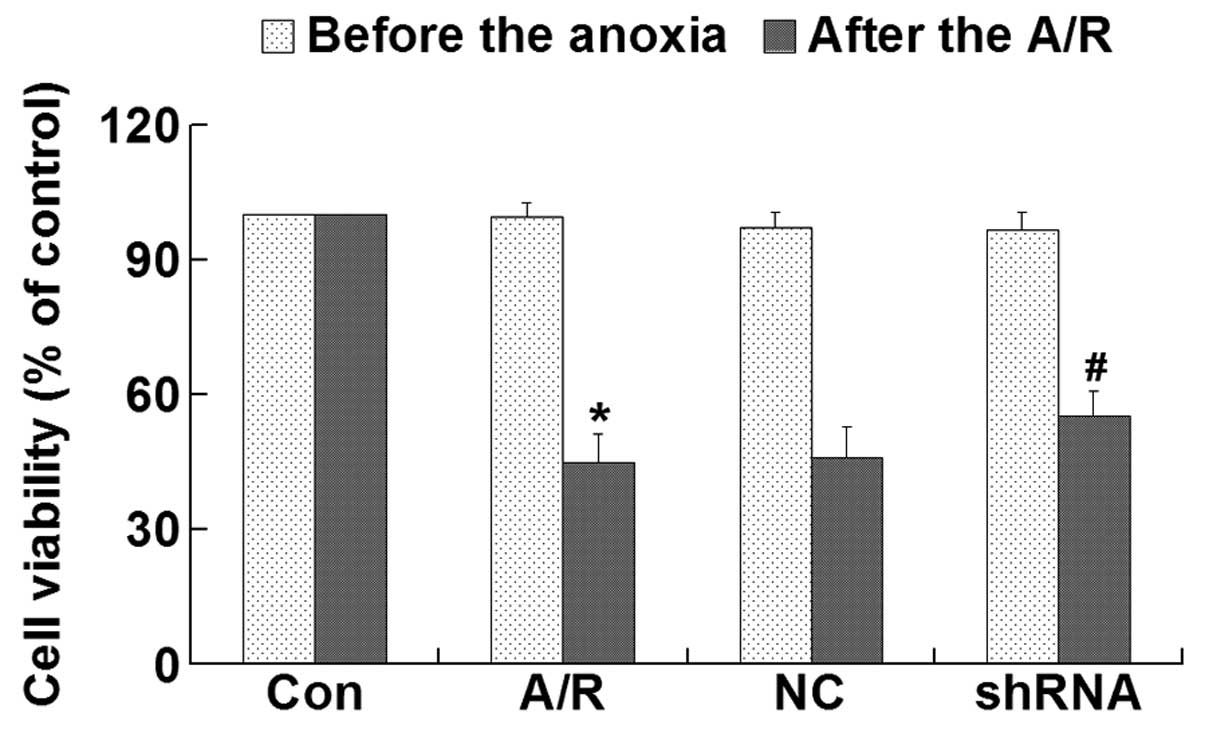
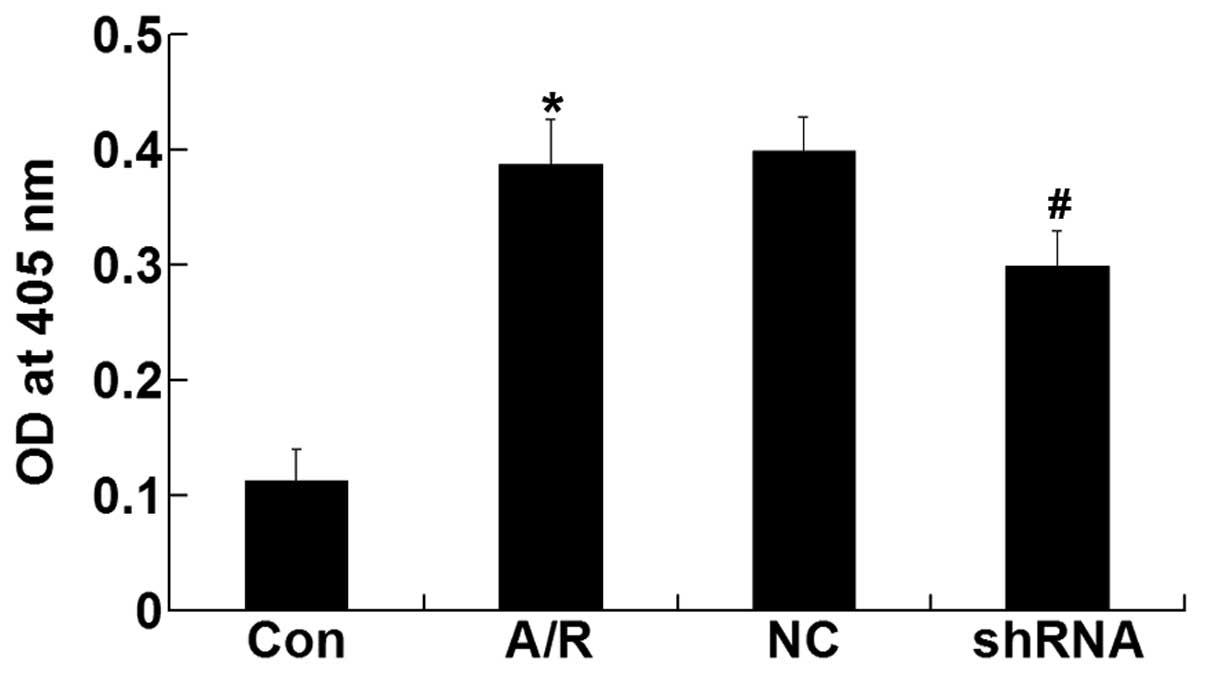
Cell viability
CCK-8 assay was performed to investigate the
cytotoxicity of plasmid-LTX-Plus in H9c2 cardiomyocytes. The
results demonstrated that there were no significant differences in
viability between experimental groups prior to A/R (Fig. 3). However, transfection of the
ACE-shRNA plasmids significantly prevented the loss of H9c2
cardiomyocyte viability induced by A/R (P<0.05) (Fig. 2).
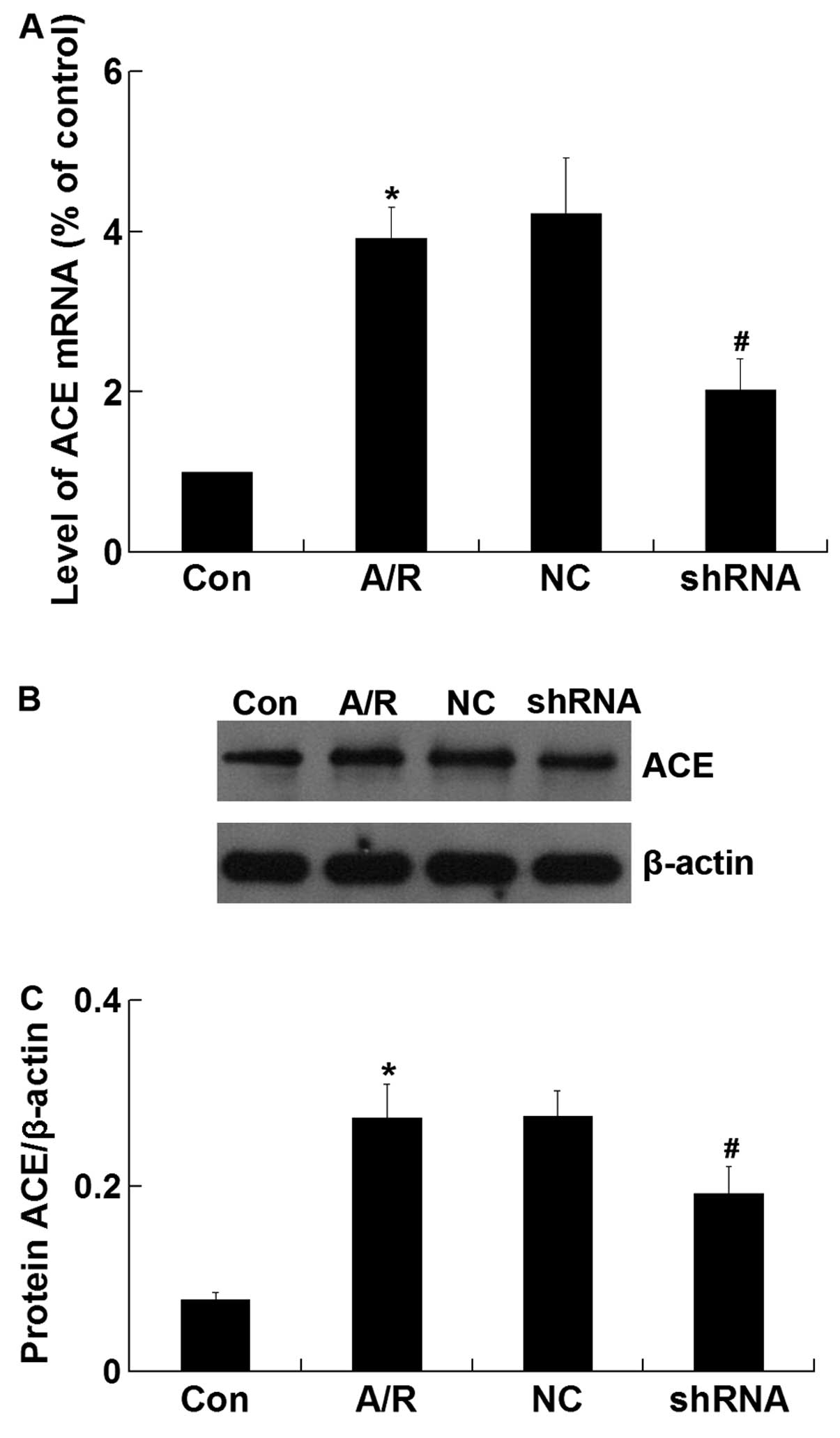
ACE mRNA and protein expression
The expression of ACE mRNA was significantly
increased in H9c2 cardiomyocytes undergoing A/R (P<0.05).
However, ACE-shRNA plasmid transfection significantly decreased the
upregulation of ACE mRNA expression induced by A/R in H9c2
cardiomyocytes (P<0.05) (Fig.
3A). Similar to the qRT-PCR result, western blot analysis
revealed that the ACE protein level was significantly lower in the
shRNA group compared with that of the A/R group (P<0.05)
(Fig. 3B and C). The negative
control ACE-shRNA plasmid transfection did not affect A/R-induced
ACE mRNA and protein expression.
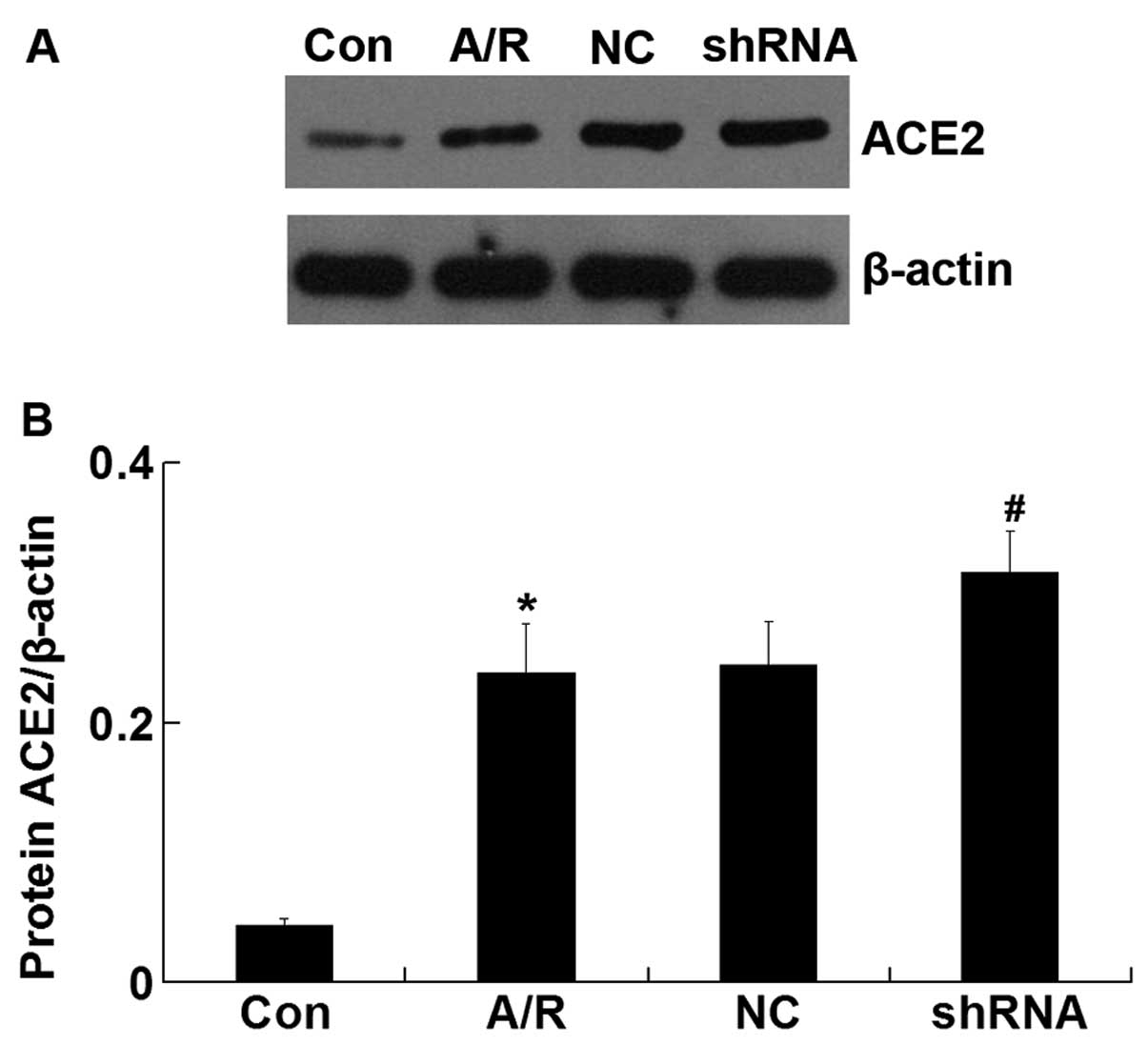
ACE2 protein expression
The ACE2 protein level was upregulated in H9c2
cardiomyocytes following A/R (P<0.05). However, transfection of
ACE-shRNA plasmids significantly promoted elevation of the ACE2
level induced by A/R (P<0.05) but remained unchanged by
transfection of the negative control ACE-shRNA plasmids (Fig 4).
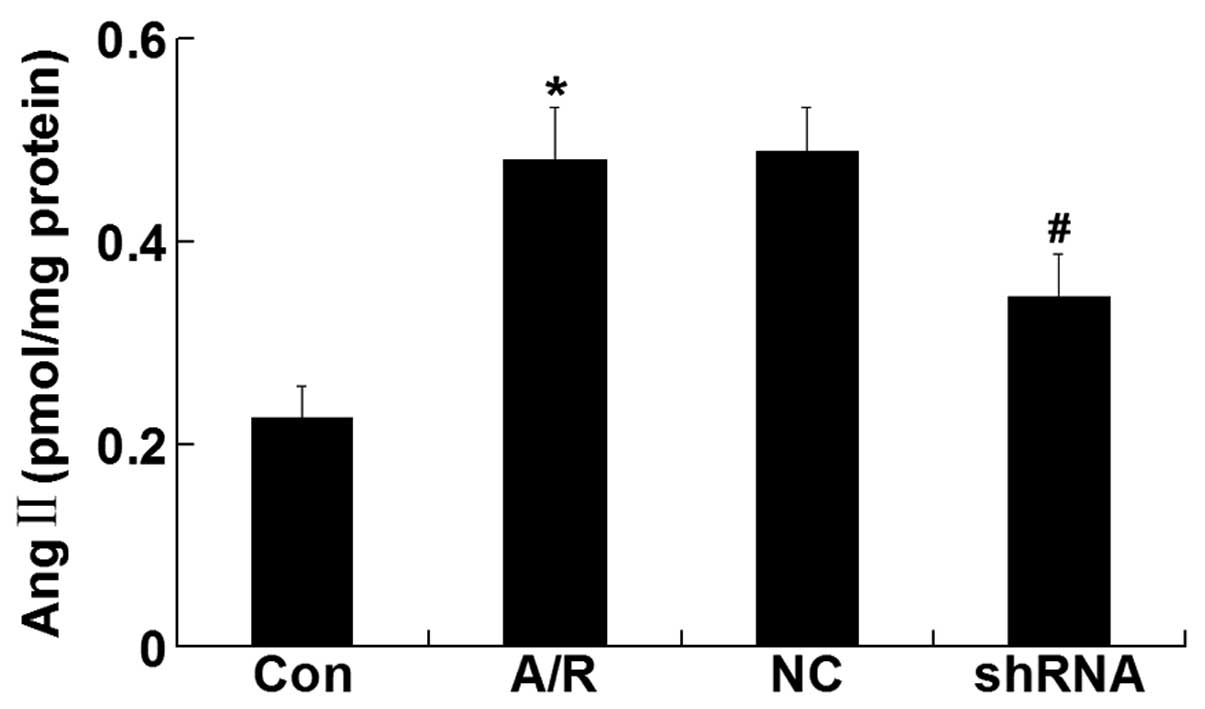
Ang II level
The Ang II level was elevated in H9c2 cardiomyocytes
undergoing A/R (P<0.05). However, ACE-shRNA plasmid transfection
significantly inhibited the elevation of Ang II level induced by
A/R (P<0.05) although it remained unchanged by transfection of
the negative control ACE-shRNA plasmids (Fig. 5).
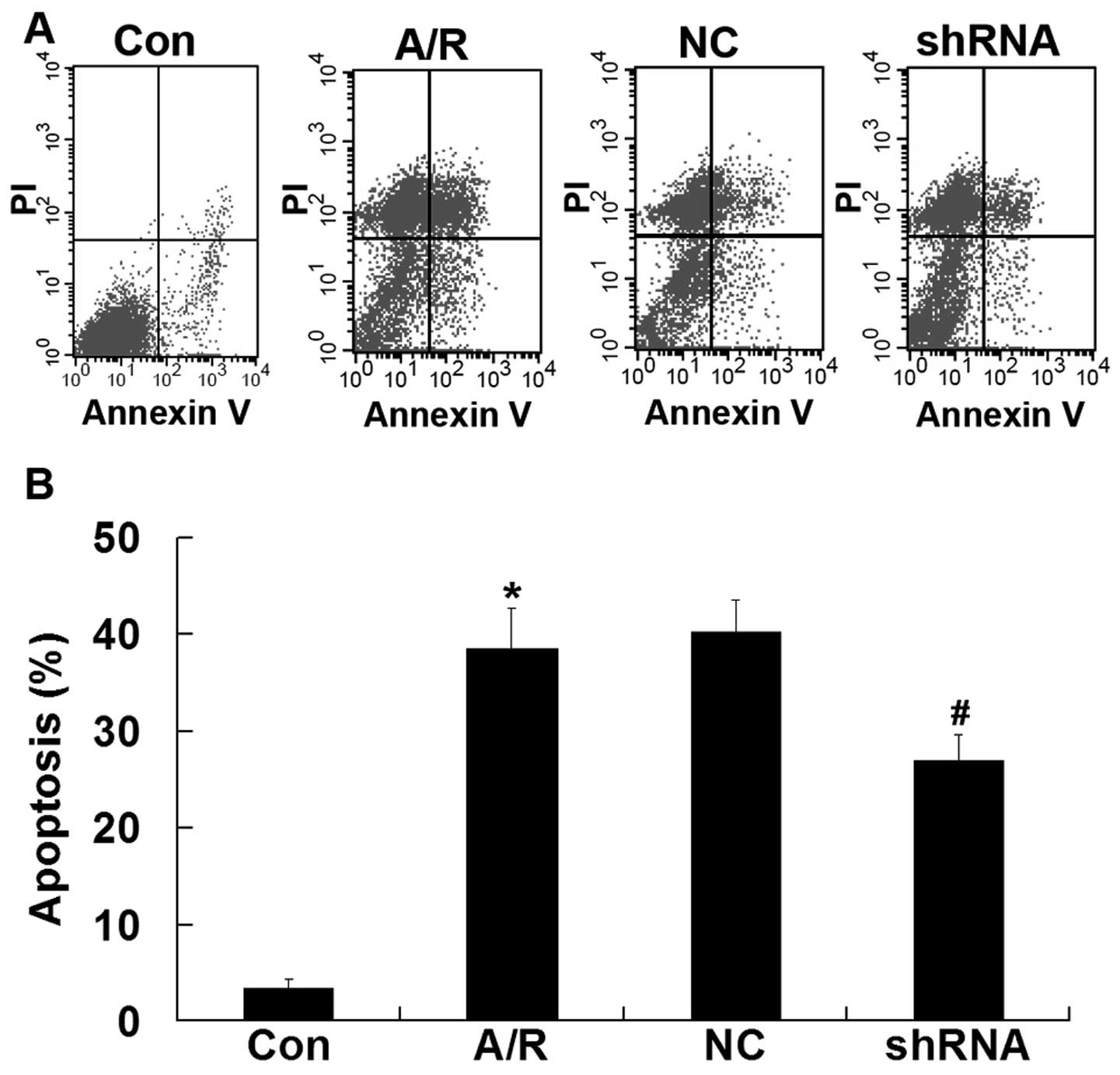
Apoptosis
Flow cytometry was used to quantify the rate of cell
apoptosis. Apoptosis was promoted in H9c2 cardiomyocytes undergoing
A/R (P<0.05). Transfection ACE-shRNA plasmids showed a
significant resistance in apoptosis in H9c2 cardiomyocytes
undergoing A/R (P<0.05). The negative control ACE-shRNA plasmid
treatment was without effect on A/R-induced apoptosis (Fig. 6).
Activity of caspase-3
The activity of caspase-3 was enhanced in H9c2
cardiomyocytes after A/R (P<0.05). However, transfection of the
ACE-shRNA plasmids significantly inhibited the activation of
caspase-3 induced by A/R (P<0.05) but remained unchanged by
treatment with the negative control ACE-shRNA plasmids (Fig. 7).
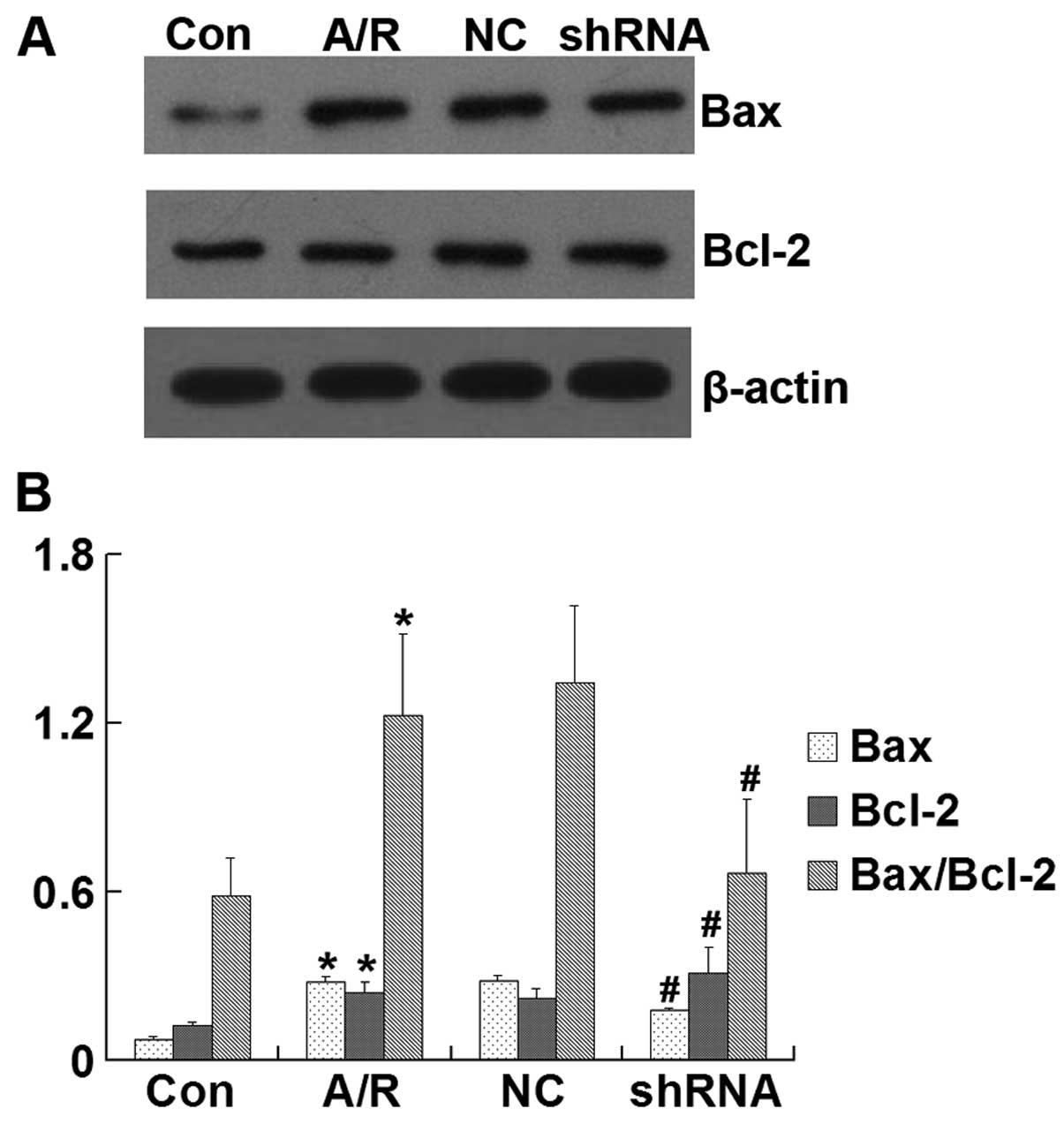
Bcl-2 and Bax protein expression
Bax, Bcl-2 and the Bax/Bcl-2 ratio were
significantly increased in H9c2 cardiomyocytes subjected to A/R
(P<0.05). However, transfection of the ACE-shRNA plasmids
significantly inhibited the elevation of BAX and Bax/Bcl-2 ratio
and promoted the upregulation of Bcl-2 induced by A/R (P<0.05)
but remained unchanged by transfection of the negative control
ACE-shRNA plasmids (Fig. 8).
Discussion
To the best of our knowledge, the present study has
demonstrated for the first time that pretreatment with ACE-shRNA
plasmids markedly suppressed the increase of intracellular ACE
expression and Ang-II level induced by A/R in H9c2 cardiomyocytes.
Gene silencing of intracellular ACE significantly inhibited the
decrease of cell viability and increase of apoptotic H9c2
cardiomyocytes undergoing A/R. At the same time, we demonstrated
that the gene silencing of intracellular ACE significantly promoted
the expression of ACE2, decreased caspase-3 activity and Bax
levels, and enhanced the expression of Bcl-2 in H9c2 cardiomyocytes
subjected to A/R. The results suggest that the gene silencing of
intracellular ACE has great potential in the treatment of
cardiomyocyte apoptosis after I/R injury by regulating the
intracellular RAS, and regulating the mitochondrial pathway of
apoptosis.
The measure of cell viability is usually used as
indicator of cell damage. In this study, we demonstrated that the
cell viability was decreased in H9c2 cardiomyocytes subjected to
A/R. However, inhibition of the intracellular ACE gene expression
significantly attenuated cell injury induced by A/R in H9c2
cardiomyocytes, suggesting that the gene silencing of intracellular
ACE is capable of protecting H9c2 cardiomyocytes against A/R
injury.
Following MI, the apoptotic process of
cardiomyocytes is initiated and becomes markedly accelerated during
early reperfusion (23,24). Extracellular stimuli trigger
apoptosis by the death receptor- and mitochondrial-mediated
pathways (25). Caspase-3
activity, a key terminal effector of apoptosis, was found to
increase subsequent to I/R injury (26). Consistent with previous results
(4,23,24), the results of the present study
demonstrated that apoptosis and caspase-3 activity were
significantly increased in H9c2 cardiomyocytes subjected to A/R.
Transfection of the ACE-shRNA plasmids for 48 h prior to the onset
of A/R significantly inhibited the apoptosis and caspase-3 activity
by decreasing the levels of ACE and Ang II, suggesting that
intracellular RAS is important in cardiomyocyte apoptosis induced
by A/R. Gene silencing of intracellular ACE protects cardiomyocytes
against A/R injury by anti-apoptosis. ACE gene silencing and ACE
inhibitors are capable of reducing Ang II levels, whereas ACE
inhibitors increase ACE expression in cardiomyocytes by a feedback
mechanism (13). Additionally,
ACE gene silencing decreased ACE expression during A/R, which
constitute the essential differences in the underlying mechanism of
anti-apoptosis between ACE gene silencing and ACE inhibitors during
I/R. In addition to systemic ACE inhibition, the gene silencing of
intracellular ACE may be another important way to protect
cardiomyocytes against I/R injury.
The Bcl-2 family proteins are significant regulators
in the intrinsic pathway of apoptosis (27,28). Bcl-2, one of the anti-apoptotic
members of this family, is able to decrease apoptosis by preventing
the release of cytochrome c (an apoptosis-inducing factor)
(29,30). Conversely, Bax, one of the
pro-apoptotic members of this family, is able to induce apoptosis
by promoting cytochrome c release (31). The ratio of BCL-2/Bax plays a
pivotal role in the mitochondrial pathway of apoptosis (32,33). Findings of previous studies showed
that the infarct size and apoptosis were reduced by the
overexpression of Bcl-2 or knockout of Bax in mice after I/R injury
(34,35). Hypoxia is capable of inducing the
p53 expression followed by the upregulation of the Bax level in
cardiomyocytes (36,37). Similar to those results, our
results demonstrated that, with the increase of Bax/Bcl-2 ratio in
H9c2 cardiomyocytes subjected to A/R, apoptosis was significantly
increased. However, the gene silencing of intracellular ACE
significantly inhibited the increase of Bax/Bcl-2 ratio induced by
A/R in H9c2 cardiomyocytes, thereby inhibiting caspase-3 activation
and resulting in the lower apoptosis. This finding suggests that
the gene silencing of intracellular ACE, with the inhibition of
apoptosis induced by A/R, may be related to the regulation of the
mitochondrial pathway of apoptosis in cardiomyocytes.
In addition to the classical circulating RAS,
previous studies have demonstrated the existence of an
intracellular RAS in several cell types, such as cardiomyocytes,
fibroblasts and vascular smooth muscle cells (7,38).
Moreover, the production of intracellular Ang II generally includes
ACE-dependent and/or ACE-independent (cathepsin D and chymase)
mechanisms in various pathological conditions (39,40). Singh et al demonstrated
that the chymase is responsible for Ang II synthesis in
cardiomyocytes in high-glucose conditions (41). However, both ACE and chymase are
responsible for Ang II synthesis in cardiomyocytes subjected to I/R
(39). Our data revealed that the
synthesis of Ang II was significantly reduced in H9c2
cardiomyocytes after A/R by ACE gene silencing. This suggested
that, at least in part, the ACE is responsible for Ang II synthesis
in H9c2 cells under A/R conditions.
ACE2, an enzyme sharing some homology with ACE, was
identified in 2000 (9,10). However, unlike ACE, the major
substrate of ACE2 appears to be Ang II, rather than Ang I (11). In addition, ACE2 activity cannot
be inhibited by ACE inhibitors (9,10).
ACE2 is able to catalyze the conversion of Ang II to Ang1–7, which
opposes the actions of Ang II (42,43). It has been suggested that ACE2 may
be crucial in regulating the local levels of Ang II and Ang1–7
(44). Previous studies showed
that ACE2 gene silencing increased the level of Ang II in the heart
of mice (45,46). In addition, the expression of ACE2
was increased in patients with heart failure induced by ischemia
(47). In their study, Ferrario
et al found that treatment with ACE inhibitors increased
ACE2 activity (48). Consistent
with that result, our study has shown that ACE gene silencing
significantly upregulated the expression of ACE2 in H9c2
cardiomyocytes undergoing A/R. These results suggest that gene
silencing of intracellular ACE, with upregulation of Ang II level
induced by A/R inhibited, may be associated with the increase of
ACE2. Previously, it was demonstrated that aldosterone inhibited
ACE2 gene expression by a minarelocorticoid receptor and Ang II did
not reduce ACE2 expression in cultured neonatal rat cardiomyocytes
(49,50). However, Ang II might indirectly
decrease the ACE2 gene expression by increasing the aldosterone
level in vivo (50). The
underlying mechanism of the elevation of ACE2 expression in H9c2
cardiomyocytes after A/R by ACE gene silencing, however, remains to
be elucidated.
Taken together, the present study has shown that the
inhibition of apoptosis induced by A/R in H9c2 cardiomyocytes by
ACE gene silencing is associated with the downregulation of ACE and
upregulation of ACE2, resulting in the decrease of Ang II level,
and consequently the regulation of the intrinsic pathway of
apoptosis, suggesting that the gene silencing of intracellular ACE
has great potential in the treatment of cardiomyocyte apoptosis
after I/R injury by regulation of intracellular RAS. In the future,
in vivo studies should be carried out to determine the
potentially protective role of intracellular ACE gene silencing for
the treatment of myocardial I/R injury.
Acknowledgements
This study was supported by the National Nature
Science Foundation of China (81170307) and National Key Basic
Research Program of China (2005CB623903).
References
|
1
|
Zijlstra F, Hoorntje JC, de Boer MJ,
Reiffers S, Miedema K, Ottervanger JP, van’t Hof AW and
Suryapranata H: Long-term benefit of primary angioplasty as
compared with thrombolytic therapy for acute myocardial infarction.
N Engl J Med. 341:1413–1419. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boersma E: Does time matter? A pooled
analysis of randomized clinical trials comparing primary
percutaneous coronary intervention and in-hospital fibrinolysis in
acute myocardial infarction patients. Eur Heart J. 27:779–788.
2006. View Article : Google Scholar
|
|
3
|
Braunwald E and Kloner RA: Myocardial
reperfusion: a double-edged sword? J Clin Invest. 76:1713–1719.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peuhkurinen K: Myocardial reperfusion-a
double-edged sword? Duodecim. 105:822–830. 1989.(In Finnish).
|
|
6
|
Burniston JG, Saini A, Tan LB and
Goldspink DF: Angiotensin II induces apoptosis in vivo in skeletal,
as well as cardiac, muscle of the rat. Exp Physiol. 90:755–761.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Re R: Intracellular renin-angiotensin
system: the tip of the intracrine physiology iceberg. Am J Physiol
Heart Circ Physiol. 293:H905–H906. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zablocki D and Sadoshima J: Knocking out
angiotensin II in the heart. Curr Hypertens Rep. 13:129–135. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Donoghue M, Hsieh F, Baronas E, Godbout K,
Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan
R, et al: A novel angiotensin-converting enzyme-related
carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9.
Circ Res. 87:E1–E9. 2000.
|
|
10
|
Tipnis SR, Hooper NM, Hyde R, Karran E,
Christie G and Turner AJ: A human homolog of angiotensin-converting
enzyme. Cloning and functional expression as a
captopril-insensitive carboxypeptidase. J Biol Chem.
275:33238–33243. 2000. View Article : Google Scholar
|
|
11
|
Vickers C, Hales P, Kaushik V, Dick L,
Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, et al:
Hydrolysis of biological peptides by human angiotensin-converting
enzyme-related carboxypeptidase. J Biol Chem. 277:14838–14843.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang XP, Liu YH, Shesely EG, Bulagannawar
M, Liu F and Carretero OA: Endothelial nitric oxide gene knockout
mice: cardiac phenotypes and the effect of angiotensin-converting
enzyme inhibitor on myocardial ischemia/reperfusion injury.
Hypertension. 34:24–30. 1999. View Article : Google Scholar
|
|
13
|
Costerousse O, Allegrini J, Clozel JP,
Menard J and Alhenc-Gelas F: Angiotensin I-converting enzyme
inhibition but not angiotensin II suppression alters angiotensin
I-converting enzyme gene expression in vessels and epithelia. J
Pharmacol Exp Ther. 284:1180–1187. 1998.PubMed/NCBI
|
|
14
|
Houston Miller N: Cardiovascular risk
reduction with renin-angiotensin aldosterone system blockade. Nurs
Res Pract. 2010:1017492010.PubMed/NCBI
|
|
15
|
Sim DS, Jeong MH, Kim YH, Choi S, Lim KS,
Kim JH, Cho KH, Kim MC, Kim HK, Kim SS, et al: Effects of
sildenafil in combination with angiotensin-converting enzyme
inhibitor on limiting infarct expansion in a porcine model of acute
myocardial infarction. Int J Cardiol. 146:459–460. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Serneri GG, Boddi M, Cecioni I, Vanni S,
Coppo M, Papa ML, Bandinelli B, Bertolozzi I, Polidori G, Toscano
T, et al: Cardiac angiotensin II formation in the clinical course
of heart failure and its relationship with left ventricular
function. Circ Res. 88:961–968. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Both GW: Recent progress in gene-directed
enzyme prodrug therapy: an emerging cancer treatment. Curr Opin Mol
Ther. 11:421–432. 2009.PubMed/NCBI
|
|
18
|
Suckau L, Fechner H, Chemaly E, Krohn S,
Hadri L, Kockskämper J, Westermann D, Bisping E, Ly H, Wang X, et
al: Long-term cardiac-targeted RNA interference for the treatment
of heart failure restores cardiac function and reduces pathological
hypertrophy. Circulation. 119:1241–1252. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brosnahan MM, Damiani A, van de Walle G,
Erb H, Perkins GA and Osterrieder N: The effect of siRNA treatment
on experimental equine herpesvirus type 1 (EHV-1) infection in
horses. Virus Res. 147:176–181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rui T, Feng Q, Lei M, Peng T, Zhang J, Xu
M, Abel ED, Xenocostas A and Kvietys PR: Erythropoietin prevents
the acute myocardial inflammatory response induced by
ischemia/reperfusion via induction of AP-1. Cardiovasc Res.
65:719–727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen HP, He M, Huang QR, Liu D and Huang
M: Sasanquasaponin protects rat cardiomyocytes against oxidative
stress induced by anoxia-reoxygenation injury. Eur J Pharmacol.
575:21–27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koyama T, Temma K and Akera T:
Reperfusion-induced contracture develops with a decreasing [Ca2+]i
in single heart cells. Am J Physiol. 261:H1115–H1122.
1991.PubMed/NCBI
|
|
23
|
Palojoki E, Saraste A, Eriksson A, Pulkki
K, Kallajoki M, Voipio-Pulkki LM and Tikkanen I: Cardiomyocyte
apoptosis and ventricular remodeling after myocardial infarction in
rats. Am J Physiol Heart Circ Physiol. 280:H2726–H2731.
2001.PubMed/NCBI
|
|
24
|
Fliss H and Gattinger D: Apoptosis in
ischemic and reperfused rat myocardium. Circ Res. 79:949–956. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Danial NN and Korsmeyer SJ: Cell death:
critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arumugam TV, Chan SL, Jo DG, Yilmaz G,
Tang SC, Cheng A, Gleichmann M, Okun E, Dixit VD, Chigurupati S, et
al: Gamma secretase-mediated Notch signaling worsens brain damage
and functional outcome in ischemic stroke. Nat Med. 12:621–623.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Adams JM and Cory S: The Bcl-2 protein
family: arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kharbanda S, Pandey P, Schofield L,
Israels S, Roncinske R, Yoshida K, Bharti A, Yuan ZM, Saxena S,
Weichselbaum R, et al: Role for Bcl-xL as an inhibitor of cytosolic
cytochrome C accumulation in DNA damage-induced apoptosis. Proc
Natl Acad Sci USA. 94:6939–6942. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release of cytochrome c from mitochondria: a
primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Simonis G, Wiedemann S, Schwarz K, Christ
T, Sedding DG, Yu X, Marquetant R, Braun-Dullaeus RC, Ravens U and
Strasser RH: Chelerythrine treatment influences the balance of pro-
and anti-apoptotic signaling pathways in the remote myocardium
after infarction. Mol Cell Biochem. 310:119–128. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brocheriou V, Hagège AA, Oubenaïssa A,
Lambert M, Mallet VO, Duriez M, Wassef M, Kahn A, Menasché P and
Gilgenkrantz H: Cardiac functional improvement by a human Bcl-2
transgene in a mouse model of ischemia/reperfusion injury. J Gene
Med. 2:326–333. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hochhauser E, Kivity S, Offen D, Maulik N,
Otani H, Barhum Y, Pannet H, Shneyvays V, Shainberg A, Goldshtaub
V, et al: Bax ablation protects against myocardial
ischemia-reperfusion injury in transgenic mice. Am J Physiol Heart
Circ Physiol. 284:H2351–H2359. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ohtsuka T, Ryu H, Minamishima YA, Macip S,
Sagara J, Nakayama KI, Aaronson SA and Lee SW: ASC is a Bax adaptor
and regulates the p53-Bax mitochondrial apoptosis pathway. Nat Cell
Biol. 6:121–128. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fridman JS and Lowe SW: Control of
apoptosis by p53. Oncogene. 22:9030–9040. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kumar R, Singh VP and Baker KM: The
intracellular renin-angiotensin system in the heart. Curr Hypertens
Rep. 11:104–110. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kumar R and Boim MA: Diversity of pathways
for intracellular angiotensin II synthesis. Curr Opin Nephrol
Hypertens. 18:33–39. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dell’Italia LJ and Husain A: Dissecting
the role of chymase in angiotensin II formation and heart and blood
vessel diseases. Curr Opin Cardiol. 17:374–379. 2002.PubMed/NCBI
|
|
41
|
Singh VP, Le B, Bhat VB, Baker KM and
Kumar R: High-glucose-induced regulation of intracellular ANG II
synthesis and nuclear redistribution in cardiac myocytes. Am J
Physiol Heart Circ Physiol. 293:H939–H948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ferrario CM, Chappell MC, Tallant EA,
Brosnihan KB and Diz DI: Counterregulatory actions of
angiotensin-(1–7). Hypertension. 30:535–541. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Iyer SN, Chappell MC, Averill DB, Diz DI
and Ferrario CM: Vasodepressor actions of angiotensin-(1–7)
unmasked during combined treatment with lisinopril and losartan.
Hypertension. 31:699–705. 1998.PubMed/NCBI
|
|
44
|
Crackower MA, Sarao R, Oudit GY, Yagil C,
Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang
L, Pei Y, et al: Angiotensin-converting enzyme 2 is an essential
regulator of heart function. Nature. 417:822–828. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Thomas MC, Pickering RJ, Tsorotes D,
Koitka A, Sheehy K, Bernardi S, Toffoli B, Nguyen-Huu TP, Head GA,
Fu Y, et al: Genetic Ace2 deficiency accentuates vascular
inflammation and atherosclerosis in the ApoE knockout mouse. Circ
Res. 107:888–897. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tikellis C, Bialkowski K, Pete J, Sheehy
K, Su Q, Johnston C, Cooper ME and Thomas MC: ACE2 deficiency
modifies renoprotection afforded by ACE inhibition in experimental
diabetes. Diabetes. 57:1018–1025. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Burrell LM, Risvanis J, Kubota E, Dean RG,
MacDonald PS, Lu S, Tikellis C, Grant SL, Lew RA, Smith AI, et al:
Myocardial infarction increases ACE2 expression in rat and humans.
Eur Heart J. 26:369–375, Discussion 322–364. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ferrario CM, Jessup J, Chappell MC,
Averill DB, Brosnihan KB, Tallant EA, Diz DI and Gallagher PE:
Effect of angiotensin-converting enzyme inhibition and angiotensin
II receptor blockers on cardiac angiotensin-converting enzyme 2.
Circulation. 111:2605–2610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Harada E, Yoshimura M, Yasue H, Nakagawa
O, Nakagawa M, Harada M, Mizuno Y, Nakayama M, Shimasaki Y, Ito T,
et al: Aldosterone induces angiotensin-converting-enzyme gene
expression in cultured neonatal rat cardiocytes. Circulation.
104:137–139. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yamamuro M, Yoshimura M, Nakayama M, Abe
K, Sumida H, Sugiyama S, Saito Y, Nakao K, Yasue H and Ogawa H:
Aldosterone, but not angiotensin II, reduces angiotensin converting
enzyme 2 gene expression levels in cultured neonatal rat
cardiomyocytes. Circ J. 72:1346–1350. 2008. View Article : Google Scholar : PubMed/NCBI
|