Introduction
Cell dysfunction induced by lipid accumulation or
lipotoxicity in pancreatic β-cells may contribute to the
pathogenesis of type 2 diabetes. In recent years, autophagy has
been identified as a novel mechanism that regulates β-cell function
(1–3) and death (4–6).
Autophagy is a conserved self-digestion process among eukaryotes
that regulates cellular component degradation through lysosomes.
Autophagy plays an important role in maintaining cell homeostasis
by regulating the synthesis, degradation and recycling of cellular
components (7). A low level of
constitutive autophagy exists in order to control the quality of
proteins and organelles. Autophagy is important for survival as it
reallocates nutrients to essential processes from less important
ones (8). In addition, autophagy
can also be induced under stress conditions to maintain the balance
of the cell. Growing evidence (4,9)
indicates that autophagy in β-cells is activated by free fatty
acids, and suggests that addressing the underlying mechanisms
involved in lipid-induced autophagy may provide clues for treating
or preventing β-cell lipotoxicity.
Free fatty acids are known as inducers of
endoplasmic reticulum (ER) stress. Previous evidence has revealed
that saturated fatty acid induces β-cell dysfunction and death via
ER stress activation (10–13).
Meanwhile, autophagy can also be activated by ER stress (14), and ER stress was reported to
enhance autophagy by regulating the Akt/TSC/mTOR pathway (15). Thus, the possibility that
lipid-induced autophagy acts as a downstream response to lipotoxic
ER stress in β-cells is conceivable. The aim of this study was to
verify saturated fatty acid-induced autophagy and to investigate
the relationship between autophagy and ER stress.
Materials and methods
Reagents and antibodies
The following reagents were used. Palmitic acid,
bovine serum albumin (BSA), bafilomycin A1, SP600125, LY294002,
3-methyladenine (3-MA),
3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and thapsigargin (TG) were purchased from Sigma-Aldrich Inc. (St.
Louis, MO, USA). Tauroursodeoxycholic acid dihydrate (TUDCA) was
obtained from Tokyo Chemical Industry Co. (Tokyo, Japan), and fetal
bovine serum (FBS) and PageRuler™ Prestained ProteinLadder were
both from Thermo Scientific (Rockford, IL, USA). The rat insulin
radioimmunoassay (RIA) kit was from Linco Research, Inc. (St.
Charles, MO, USA), and RIPA lysis buffer, complete protease
inhibitor mixture, protein phosphatase inhibitor, and Super
Enhanced chemiluminescence detection reagents were all from
Applygen Technologies Inc. (Beijing, China). PVDF membranes were
obtained from Millipore (Billerica, MA, USA), and the BCA protein
assay kit was from KW Biotech (Beijing, China). The following
antibodies were used: light chain 3 (LC3)A/B, BiP, CHOP,
phospho-SAPK/JNK (Thr183/Tyr185), SAPK/JNK, phospho-Akt (Ser473)
and Akt (all from Cell Signaling Technology, Danvers, MA, USA).
Actin, insulin receptor β (IRβ), and the horseradish
peroxidase-conjugated secondary antibody were purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell culture and palmitate
preparation
Mouse insulinoma-derived MIN6 cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM, containing 25 mM glucose)
supplemented with 15% FBS, 100 U/ml penicillin, 100 μg/ml
streptomycin, 10 mM HEPES, 2 mM L-glutamine, 1 mM sodium pyruvate
and 5 μl/l β-mercaptoethanol at 37°C in a 5% CO2/95% air
atmosphere and 100% humidity. For detection of insulin secretion,
the culture medium was replaced with DMEM containing 5.6 mM
glucose. Palmitic acid was dissolved in 0.1 N NaOH at a
concentration of 100 mM and adjusted to 10 mM palmitate with fatty
acid-free BSA solution. The palmitate/BSA complex was then filtered
and added to serum-free medium to achieve a final concentration of
0.5 mM palmitate/1% BSA.
Western blot analysis
The expression and phosphorylation levels of target
proteins were detected by western blot analysis. Cells were lysed
with RIPA lysis buffer containing complete protease inhibitor
mixture and protein phosphatase inhibitor. After protein
extraction, cell lysates were separated by sodium dodecyl sulfate
polyacrylamide gel electrophoresis and transferred to PVDF
membranes. Molecular weights were estimated by comparison with a
prestained protein ladder. Nonspecific binding was blocked using 5%
skim milk. The membranes were then incubated with specific primary
antibodies overnight at 4°C. Subsequently, secondary antibody
conjugated to horseradish peroxidase and Super Enhanced
chemiluminescence detection reagents were used to detect the
specific bands. Immunoblots were quantified by densitometric
analysis using ImageTool 3.0 software. Quantification of protein
phosphorylation was normalized with the corresponding total protein
expression, and the relative expression level of a certain protein
was normalized with actin. To visualize the accumulation of LC3-II,
20 nM bafilomycin A1 was added to the culture medium during the
final 4-h period. Bafilomycin A1, an inhibitor of vacuolar type
H+-ATPase, inhibits the fusion of antophagosomes with
lysosomes (16,17).
Cell viability and morphological
examination
Cell viability was determined using MTT assays
following the experimental treatments. Briefly, cells were seeded
in 96-well plates and incubated under different conditions. Then
culture medium was removed, and 200 μl/well diluted MTT solution
(0.5 mg/ml) was added to the culture. After a 4-h incubation at
37°C, MTT solution in each well was replaced with 150 μl
dimethylsulfoxide (DMSO) to ensure that the crystals dissolved.
After mixing, the absorbance was measured at 490 nm using a
microplate reader. Cell viability was calculated as a percentage of
absorbance. For morphological examination, cells were cultured in 6
cm plates and examined using light microscopy (×100).
Insulin secretion
Cells were seeded in 24-well plates at a density of
5×105 cells/well and incubated in different treatment
media. After the indicated duration of culture, cells were washed
with PBS and equilibrated in Kerbs-Ringer bicarbonate buffer (KRBB,
pH 7.4) containing 118.5 mM NaCl, 2.54 mM CaCl2, 1.19 mM
KH2PO4, 4.74 mM KCl, 1.19 mM
MgSO4, 25 mM NaHCO3, 10 mM HEPES, 0.5% BSA
(fatty acid free) and 5.6 mM glucose at 37°C for 30 min. Then the
buffer was removed and replaced with fresh KRBB containing 2.8 or
27.8 mM glucose for 60 min. The supernatants from cell cultures
were collected, and the insulin concentration was measured using
the insulin RIA kit after appropriate dilutions. The total protein
was extracted with RIPA lysis buffer supplemented with 1 mM
phenylmethyl sulfonylfluoride, and then the concentration of the
protein was determined using the BCA protein assay kit. The levels
of insulin secretion were normalized with the respective protein
content. Insulin secretion of the cells following stimulation with
2.8 and 27.8 mM glucose was defined as basal insulin secretion
(BIS) and glucose-stimulated insulin secretion (GSIS),
respectively.
Statistical analysis
Data are expressed as means ± SD. Statistical
differences were determined using the Student’s two-sample t-test
or one-way ANOVA followed by the Student-Newman-Keuls test where
appropriate, using SPSS 12.0 (SPSS Inc., Chicago, IL, USA). A
P-value <0.05 was considered to indicate a statistically
significant result.
Results
Palmitate upregulates autophagy via the
ER stress-dependent JNK pathway
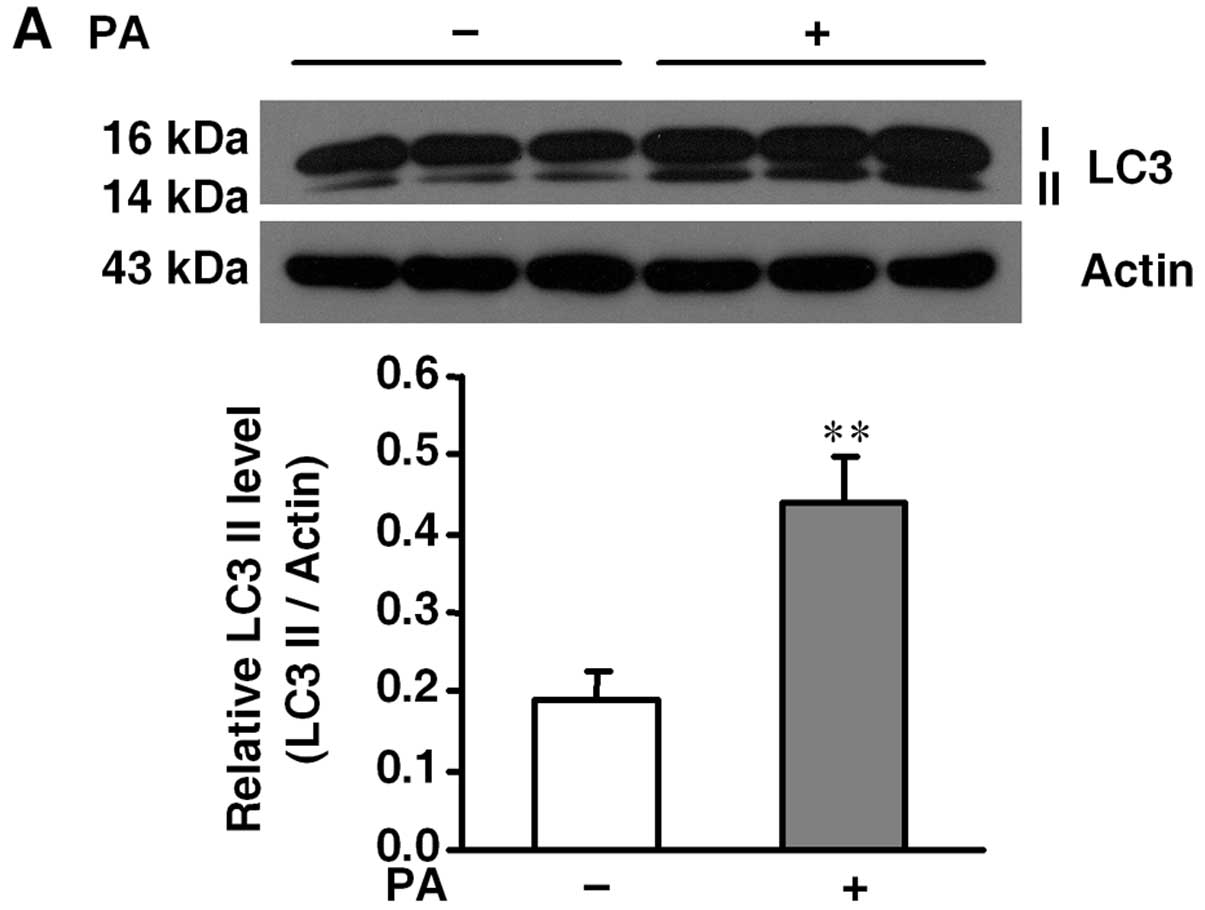
To confirm palmitate-induced autophagy in β-cells,
we detected the expression of LC3-II, a hallmark of autophagy
activation (16), with or without
0.5 mM palmitate for 36 h. Western blot analysis showed that
palmitate treatment significantly enhanced the LC3-II level when
compared to the level in the control group (Fig. 1A). Meanwhile, MIN6 cells were
cultured in 0.5 mM palmitate for different times to determine
whether ER stress and JNK phosphorylation were activated by
palmitate. We found that the expression of ER stress markers, BiP
and CHOP, and JNK phosphorylation were gradually increased
following palmitate treatment (Fig.
1B). ER stress inhibitor TUDCA (500 μM) and JNK inhibitor
SP600125 (20 μM) both exhibited an inhibitory effect on the
palmitate-induced increase in LC3-II (Fig. 1C and D).
Involvement of Akt phosphorylation in
palmitate-stimulated autophagy
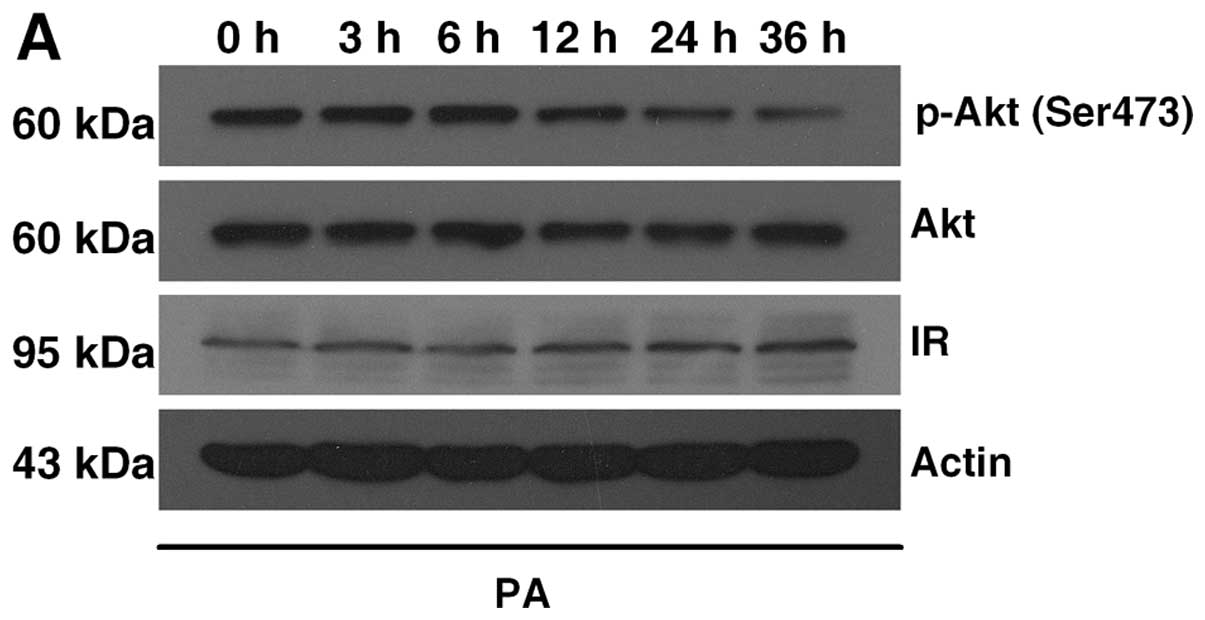
We assessed the possible involvement of Akt
phosphorylation in autophagy induction. First, we found that the
insulin-induced p-Akt (Ser473) gradually decreased following
palmitate treatment, while the expression of IRβ did not change
within the 36-h treatment (Fig.
2A), indicating that palmitate induced a reduction in p-Akt
independent of the insulin receptors. Next, to investigate the role
of p-Akt reduction, we suppressed the PI3K/Akt pathway using
LY294002. LY294002 (50 μM) was found to facilitate
palmitate-induced autophagy by increasing LC3-II expression
(Fig. 2B). We also investigated
the upstream reduction in p-Akt and found that both TUDCA (500 μM)
and SP600125 (20 μM) inhibited the palmitate-induced reduction of
p-Akt (Fig. 2C and D). Our
results suggest an involvement of decreased Akt phosphorylation in
palmitate-induced autophagy through ER stress and JNK
activation.
Inhibition of autophagy aggravates
palmitate-induced ER stress
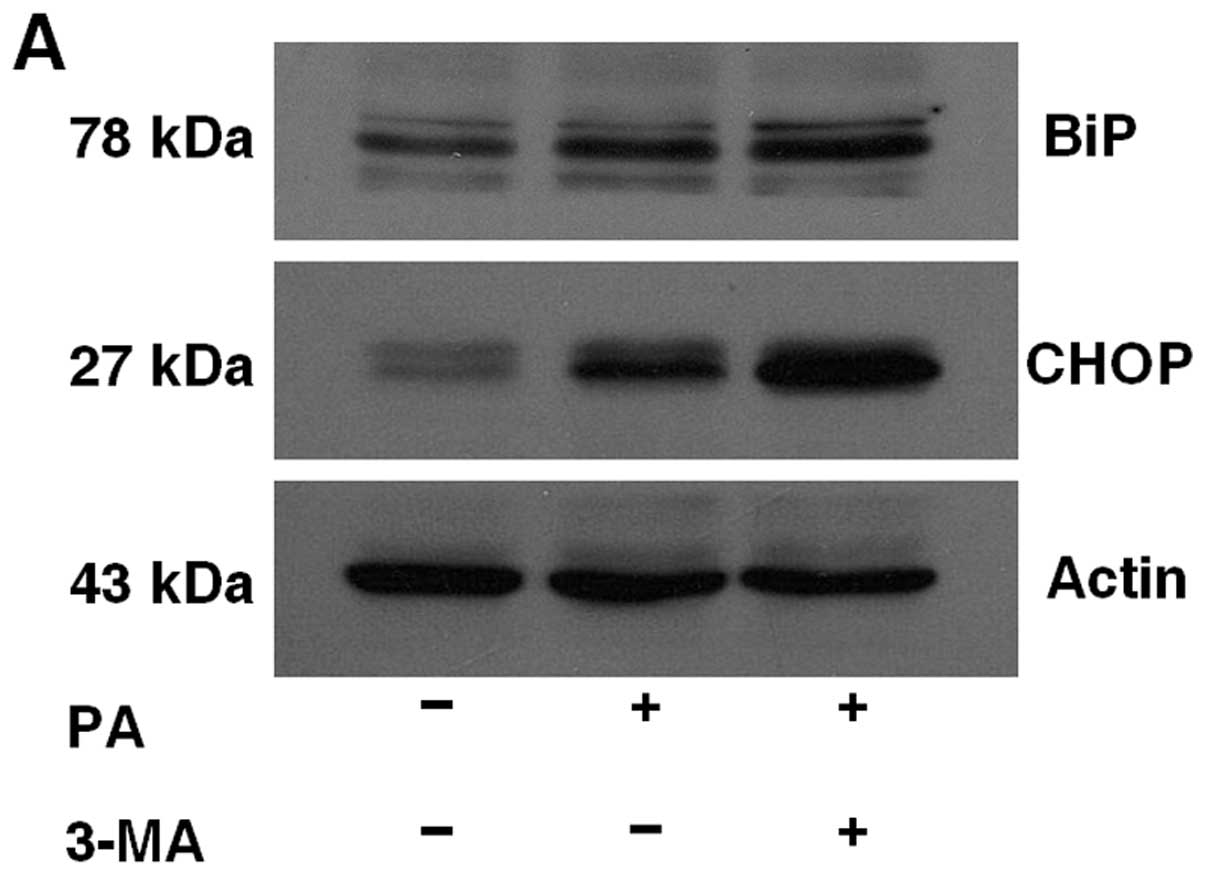
To examine the effect of autophagy inhibition on ER
stress, MIN6 cells were pretreated with or without 5 mM 3-MA for 1
h and then treated with 0.5 mM plamitate for 36 h. As shown in
Fig. 3, the expression of ER
stress markers, BiP and CHOP, was increased by 3-MA
pretreatment.
Inhibition of autophagy aggravates cell
injuries caused by ER stress
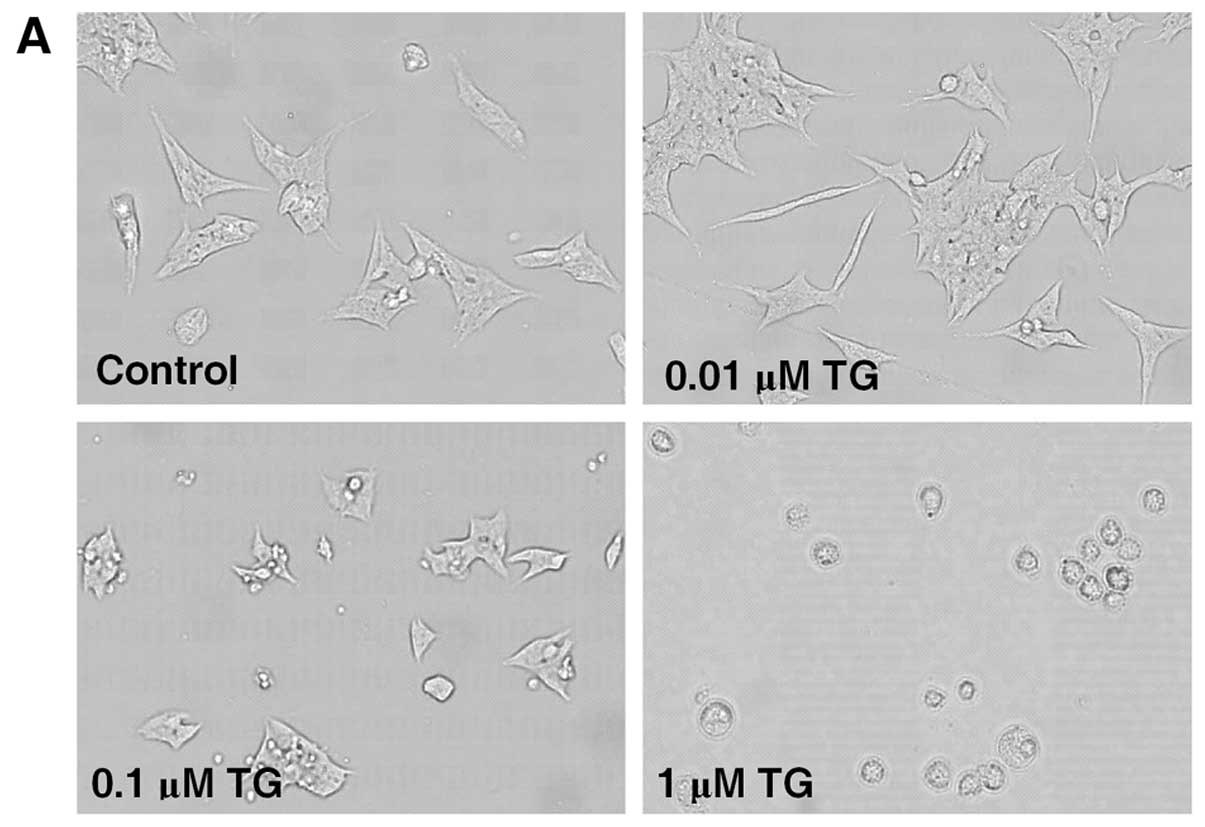
As shown in Fig. 4A
and B, TG caused cell injury in MIN6 cells in a dose-dependent
manner. A concentration of 0.1 μM TG was chosen for use in the
subsequent experiments. MTT assay revealed that treatment with 0.1
μM TG for 36 h decreased the cell viability of MIN6 cells from
100.0±4.1 to 81.4±6.0%, while pretreatment with 3-MA followed by
0.1 μM TG caused a further decrease to 68.2±6.1% (Fig. 4C). TG also inhibited insulin
secretion, including BIS from 145.96±25.65 to 48.38±25.47 ng/mg
protein/h and GSIS from 285.14±28.03 to 65.62±22.81 ng/mg
protein/h. Compared with TG alone, TG exposure following 3-MA
pretreatment significantly reduced insulin secretion to a greater
extent, with a BIS value of 21.26±10.66 ng/mg protein/h and a GSIS
value of 19.42±9.32 ng/mg protein/h, respectively (Fig. 4D).
Discussion
In the present study, we examined the molecular
mechanisms of lipid-induced autophagy in β-cells. In accordance
with previous studies (4,9), enhancement of LC3-II levels, a
marker of autophagy, was observed following palmitate treatment.
Palmitate-induced autophagy is considered to be a ubiquitous
process, as autophagy was also confirmed to be induced in several
other cell lines (9). The
possible involvement of ER stress and the JNK pathway was examined
to further understand the mechanisms of palmitate-induced
autophagy. The results showed that the expression of ER stress
markers and p-JNK was gradually increased in conjunction with
prolonged treatment time of palmitate. In addition, TUDCA and
SP600125 were used to further verify whether ER stress and JNK are
associated with palmitate-induced autophagy. TUDCA is a type of
chemical chaperone which can effectively alleviate ER stress
(18), and SP600125 is widely
used as a JNK-specific inhibitor. Results showed that
palmitate-induced autophagy was blocked both by TUDCA and by
SP600125, suggesting that ER stress and JNK do participate in the
induction of autophagy. The concept that the JNK pathway plays a
role in palmitate-induced autophagy was also proven in another
study (9), and it was shown that
ER stress-related JNK activation was mediated by ER stress marker
IRE1 (14,19).
Since JNK activation was reported to take part in
the suppression of insulin signaling by inducing serine
phosphorylation of insulin receptor substrate-1 (20), we also examined the effect of
palmitate on insulin-induced phosphorylation of Akt, a downstream
target of insulin signaling. As shown in Fig. 2A, insulin-induced Akt
phosphorylation was reduced following palmitate treatment, and this
reduction was independent of the change in IRβ expression.
Moreover, the downregulation of Akt phosphorylation was associated
with ER stress and JNK phosphorylation, as TUDCA and SP600125
treatment blocked the decrease in Akt phosphorylation level caused
by palmitate. Previously, the concept of β-cell insulin resistance
has been discussed (21,22). The reduction in insulin-induced
Akt phosphorylation here was a result of an alteration in the
insulin signaling cascade, and may be considered as a sign of
β-cell insulin resistance. LY294002 was found to be a specific PI3K
inhibitor for suppressing the activation of the PI3K-Akt-mTOR axis
(23). Following the use of
LY294002, the level of autophagy induced by palmitate was further
increased, indicating the involvement of Akt in the formation of
palmitate-induced autophagy. Notably, palmitate is the precursor of
ceramide, and ceramide was shown to stimulate autophagy by
inhibiting Akt phosphorylation (24). Furthermore, the control of
autophagy is characterized by the necessity of class-III PI3K for
autophagosome formation or by being inhibited by class-I
PI3K/Akt/mTOR (25,26). Although reduced insulin signaling
was reported to accompany pancreatic β-cell dysfunction (27), we believe that autophagy and its
upstream reduction in insulin signaling act as adaptive and
protective responses to ER stress.
Inhibition of autophagy by 3-MA, a widely used
inhibitor of autophagic flux, facilitated the induction of ER
stress by palmitate. TG is known as a chemical which can induce ER
stress by inhibiting ER Ca2+-ATPase (28). We also found that cell injuries
caused by TG were further aggravated by 3-MA pretreatment,
including a decrease in cell viability and insulin secretion.
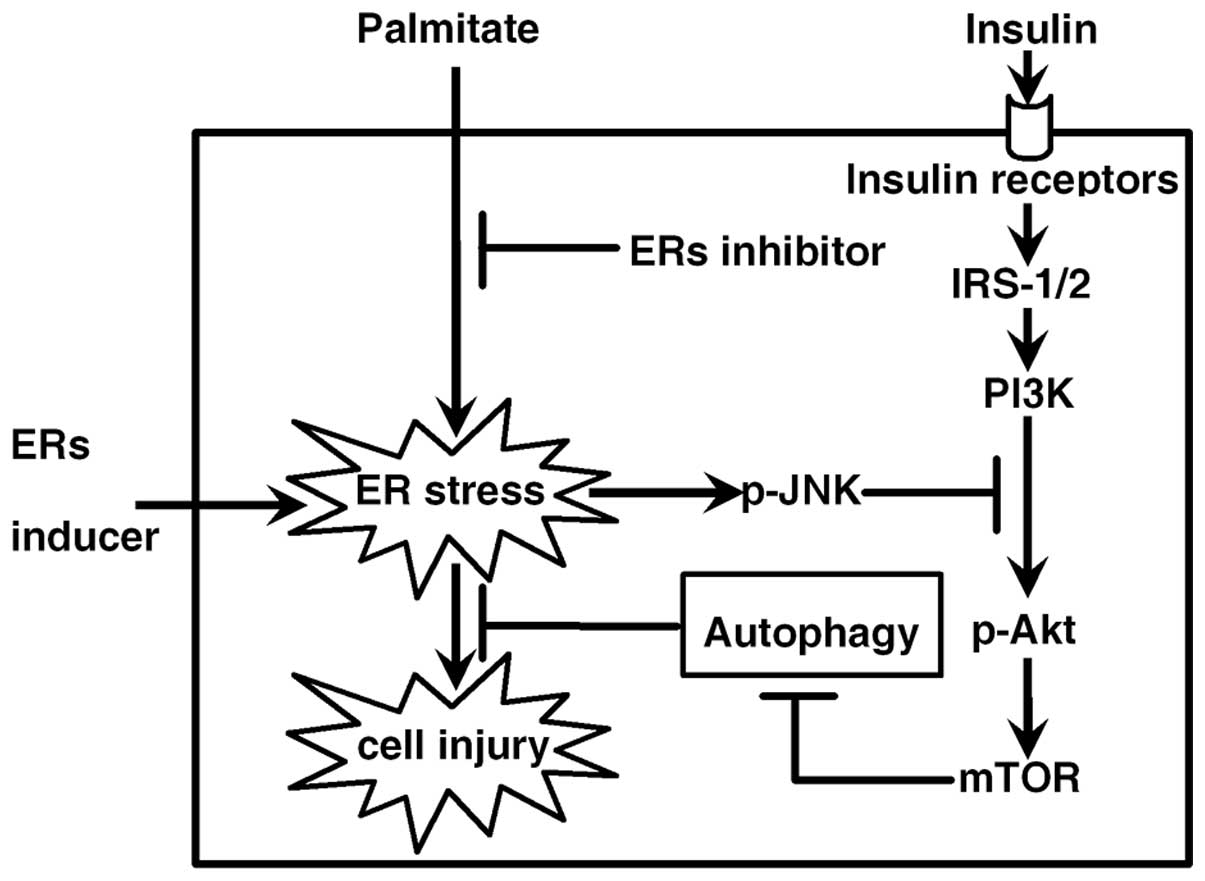
In summary, our results revealed that ER stress, its
downstream JNK pathway and the inhibition of insulin signaling
contributed to palmitate-induced autophagy. A model of the pathway
is proposed in Fig. 5. A
reduction in autophagy aggravated ER stress and related cell
injuries. If autophagy is elevated in type 2 diabetes, one could
speculate that the onset of autophagy under stress conditions may
be an adaptive response intended to protect β-cells against
damage.
References
|
1
|
Fujitani Y, Ebato C, Uchida T, Kawamori R
and Watada H: β-cell autophagy: a novel mechanism regulating β-cell
function and mass: lessons from β-cell-specific Atg7-deficient
mice. Islets. 1:151–153. 2009.
|
|
2
|
Ebato C, Uchida T, Arakawa M, Komatsu M,
Ueno T, Komiya K, Azuma K, et al: Autophagy is important in islet
homeostasis and compensatory increase of beta cell mass in response
to high-fat diet. Cell Metab. 8:325–332. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jung HS, Chung KW, Won Kim J, Kim J,
Komatsu M, Tanaka K, Nguyen YH, et al: Loss of autophagy diminishes
pancreatic beta cell mass and function with resultant
hyperglycemia. Cell Metab. 8:318–324. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choi SE, Lee SM, Lee YJ, Li LJ, Lee SJ,
Lee JH, Kim Y, et al: Protective role of autophagy in
palmitate-induced INS-1 beta-cell death. Endocrinology.
150:126–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fujimoto K, Hanson PT, Tran H, Ford EL,
Han Z, Johnson JD, Schmidt RE, et al: Autophagy regulates
pancreatic beta cell death in response to Pdx1 deficiency and
nutrient deprivation. J Biol Chem. 284:27664–27673. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Masini M, Bugliani M, Lupi R, del Guerra
S, Boggi U, Filipponi F, Marselli L, et al: Autophagy in human type
2 diabetes pancreatic beta cells. Diabetologia. 52:1083–1086. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kuma A, Hatano M, Matsui M, Yamamoto A,
Nakaya H, Yoshimori T, Ohsumi Y, et al: The role of autophagy
during the early neonatal starvation period. Nature. 432:1032–1036.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Komiya K, Uchida T, Ueno T, Koike M, Abe
H, Hirose T, Kawamori R, et al: Free fatty acids stimulate
autophagy in pancreatic β-cells via JNK pathway. Biochem Biophys
Res Commun. 401:561–567. 2010.PubMed/NCBI
|
|
10
|
Karaskov E, Scott C, Zhang L, Teodoro T,
Ravazzola M and Volchuk A: Chronic palmitate but not oleate
exposure induces endoplasmic reticulum stress, which may contribute
to INS-1 pancreatic beta-cell apoptosis. Endocrinology.
147:3398–3407. 2006. View Article : Google Scholar
|
|
11
|
Cnop M, Igoillo-Esteve M, Cunha DA,
Ladrière L and Eizirik DL: An update on lipotoxic endoplasmic
reticulum stress in pancreatic beta-cells. Biochem Soc Trans.
36:909–915. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lai E, Bikopoulos G, Wheeler MB,
Rozakis-Adcock M and Volchuk A: Differential activation of ER
stress and apoptosis in response to chronically elevated free fatty
acids in pancreatic beta-cells. Am J Physiol Endocrinol Metab.
294:E540–E550. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cnop M, Ladrière L, Igoillo-Esteve M,
Moura RF and Cunha DA: Causes and cures for endoplasmic reticulum
stress in lipotoxic β-cell dysfunction. Diabetes Obes Metab.
12:76–82. 2010.PubMed/NCBI
|
|
14
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, et al: Autophagy is activated for
cell survival after endoplasmic reticulum stress. Mol Cell Biol.
26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qin L, Wang Z, Tao L and Wang Y: ER stress
negatively regulates AKT/TSC/mTOR pathway to enhance autophagy.
Autophagy. 6:239–247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Klionsky DJ, Elazar Z, Seglen PO and
Rubinsztein DC: Does bafilomycin A1 block the fusion of
autophagosomes with lysosomes? Autophagy. 4:849–950. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ozcan U, Yilmaz E, Ozcan L, Furuhashi M,
Vaillancourt E, Smith RO, Görgün CZ, et al: Chemical chaperones
reduce ER stress and restore glucose homeostasis in a mouse model
of type 2 diabetes. Science. 313:1137–1140. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hirosumi J, Tuncman G, Chang L, Görgün CZ,
Uysal KT, Maeda K, Karin M, et al: A central role for JNK in
obesity and insulin resistance. Nature. 420:333–336. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leibiger IB, Leibiger B and Berggren PO:
Insulin signaling in the pancreatic beta-cell. Annu Rev Nutr.
28:233–251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kahn CR, Brüning JC, Michael MD and
Kulkarni RN: Knockout mice challenge our concepts of glucose
homeostasis and the pathogenesis of diabetes mellitus. J Pediatr
Endocrinol Metab. 13:1377–1384. 2000.PubMed/NCBI
|
|
23
|
Wu YT, Tan HL, Huang Q, Ong CN and Shen
HM: Activation of the PI3K-Akt-mTOR signaling pathway promotes
necrotic cell death via suppression of autophagy. Autophagy.
5:824–834. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Scarlatti F, Bauvy C, Ventruti A, Sala G,
Cluzeaud F, Vandewalle A, Ghidoni R, et al: Ceramide-mediated
macroautophagy involves inhibition of protein kinase B and
up-regulation of beclin 1. J Biol Chem. 279:18384–18391. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meijer AJ and Codogno P: Macroautophagy:
protector in the diabetes drama? Autophagy. 3:523–526. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Leibowitz G, Cerasi E and Ketzinel-Gilad
M: The role of mTOR in the adaptation and failure of beta-cells in
type 2 diabetes. Diabetes Obes Metab. 10:157–169. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Matsuda T, Kido Y, Uchida T and Kasuga M:
Reduced insulin signaling and endoplasmic reticulum stress act
synergistically to deteriorate pancreatic beta cell function. Kobe
J Med Sci. 54:E114–E121. 2008.PubMed/NCBI
|
|
28
|
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi
NN, Ozdelen E, Tuncman G, et al: Endoplasmic reticulum stress links
obesity, insulin action, and type 2 diabetes. Science. 306:457–461.
2004. View Article : Google Scholar : PubMed/NCBI
|