Introduction
Pulmonary arterial hypertension (PAH) is a severe
and frequently fatal disease. The hallmark of PAH is the
development of gradually increased pulmonary vascular resistance,
which eventually enhances the afterload of the right ventricle and
leads to right heart failure (1).
The etiopathogenesis of PAH is commonly associated with chronic
hypoxemia in disorders, such as chronic obstructive pulmonary
disease and interstitial lung disease (2). Increasing evidence indicates that
apart from vasoconstriction, smooth muscle cell proliferation and
hypertrophy, usually leading to pulmonary vascular remodeling and
increased resistance, play a crucial role in the development of PAH
(3). Therefore, it would be of
great importance to elucidate the molecular mechanisms underlying
hypoxia-induced smooth muscle cell proliferation, which may in turn
lead to the discovery of novel therapeutic agents/targets.
Oxidative stress, characterized by increased
reactive oxygen species (ROS) production, is involved in cell
proliferation induced by a number of stimuli in a variety of cell
models (4–7). Reportedly, in human pulmonary artery
smooth muscle cells (HPASMCs), NADPH oxidase (NOX)4 mediates cell
proliferation triggered by transforming growth factor-β1 (TGF-β1)
(3). The inhibition of oxidative
stress with nitrite or superoxide dismutase (SOD) has been shown to
ameliorate PAH (8,9). Of note, prostacyclin (also known as
prostaglandin I2 or PGI2), an effective but expensive
clinical drug, has been used for the treatment of PAH via
vasodilatation (10). The
endogenous formation of PGI2 is mainly attributed to
normal cyclooxygenase-2 (COX-2) expression; however, COX-2
expression is reportedly downregulated in hypoxia-induced PAH
(2).
Hydrogen sulfide (H2S) has been
recognized as an important cellular signaling molecule, alongside
nitric oxide (NO) and carbon monoxide (CO), playing a number of
physiological and pathological roles in mammals (11–13). H2S has been shown to
exert various protective effects on the cardiovascular system,
including myocardial preservation, the improvement of endothelial
function, inhibition of proliferation and/or induction of the
apoptosis of smooth muscle cells (14). Many of these effects elicited by
H2S are mediated by the upregulation of COX-2 (15). However, plasma H2S
levels in Wistar rats have been shown to be reduced in
hypoxia-induced pulmonary vascular structural remodeling (16). We therefore hypothesized that
H2S can abolish the hypoxia-induced proliferation of
pulmonary artery smooth muscle cells (PASMCs) and may thus
consequently ameliorate PAH through the upregulation of
COX-2/PGI2.
To confirm our hypothesis, in the present study, we
carried out the following experiments: The chemical hypoxia agent,
cobalt chloride (CoCl2), was employed to treat HPASMCs
in order to establish a cellular model of hypoxic PAH.
Hypoxia-induced changes, such as oxidative stress, a decrease in
COX-2/PGI2 expression, as well as endogenous
H2S levels were observed. In addition, we aimed to
determined whether the exogenous administration of H2S
in the form of sodium hydrosulfide (NaHS) affects the chemical
hypoxia-induced proliferation of HPASMCs, as well as to elucidate
the mechanisms involved.
Materials and methods
Materials
CoCl2, NaHS, N-acetyl-L-cysteine (NAC),
PGI2 and 2′,7′-dichlorofluorescein-diacetate (DCFH-DA)
were all purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). The
Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Laboratories
(Kyushu, Japan). Specific monoclonal antibodies against COX-2,
hypoxia-inducible factor-1α (HIF-1α) or β-actin were obtained from
Epitomics, an Abcam Company (San Francisco, CA, USA). Dulbecco’s
Modified Eagle’s Medium (DMEM) high glucose medium and fetal bovine
serum (FBS) were both supplied by Gibco-BRL (Grand Island, NY,
USA).
Cell culture
HPASMCs, derived from human pulmonary arterial
tissue, were supplied by ScienCell Research Laboratories (Carlsbad,
CA, USA), and maintained in DMEM high glucose medium supplemented
with 15% FBS at 37°C under an atmosphere of 5% CO2 and
95% air. The cells were passaged approximately every 2 days.
Determination of cell proliferation
Cell viability was assessed according to the
instructions provided with the CCK-8. A viability >100%
indicated cell proliferation, whereas a viability of <100%
indicated cell damage, as previously described (17). The HPASMCs were plated in 96-well
plates at a density of 5,000 cells/well. When the cells were grown
to approximately 60–70% confluence, the indicated treatments were
applied. CCK-8 solution (10 μl) at a 1:10 dilution with FBS-free
DMEM high glucose medium (100 μl) was added to each well followed
by another 3 h of incubation at 37°C. The absorbance (A) was
measured at 450 nm on a microplate reader (Molecular Devices, LLC,
Silicon Valley, CA USA). The experiments were performed 4 times, as
previously described (18).
Observation of intracellular ROS
content
Intracellular ROS was determined by the oxidative
conversion of cell permeable DCFH-DA to fluorescent
2′,7′-dichlorofluorescein (DCF) (18). At the end of the indicated
treatments, the HPASMCs were washed and incubated with 10 μmol/l
DCFH-DA solution at 37°C for 20 min in the dark. Intercellular DCF
fluorescence was observed over the entire field of vision using a
fluorescence microscope connected to an imaging system (BX50-FLA;
Olympus, Tokyo, Japan). The mean fluorescence intensity (MFI) of
DCF from 4 random fields was analyzed using ImageJ 1.47
software.
Measurement of intracellular
H2S content
Intracellular free H2S levels were
determined using the H2S fluorescent probe (WSP-1;
kindly provided by Professor Ming Xian at the Department of
Chemistry, Washington State University, Pullman, WA, USA), as
previously described (19,20)
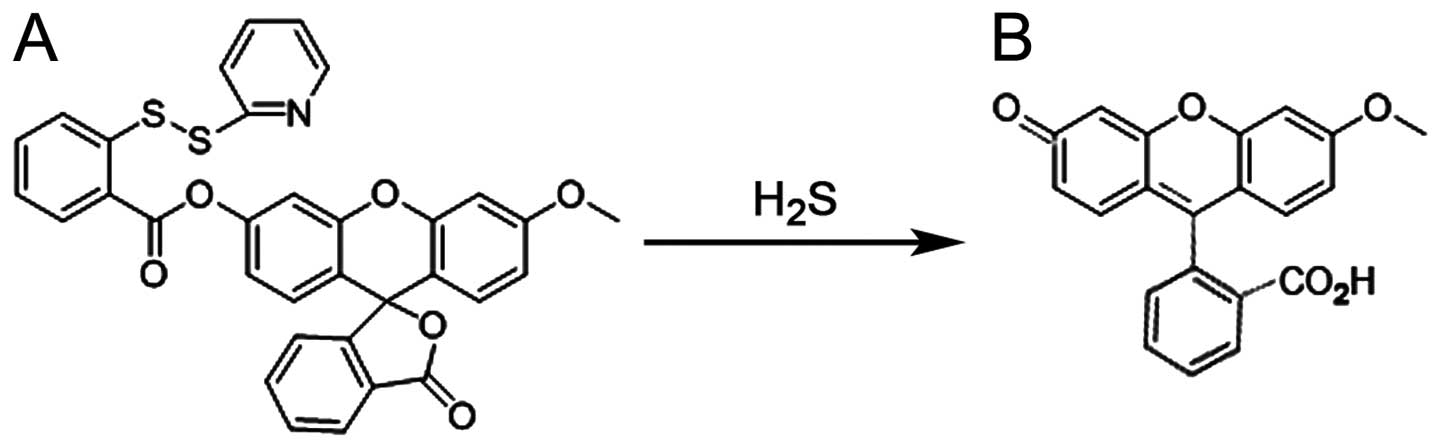
and the chemical equation is presented in Fig. 1. After the indicated treatments,
the HPASMCs were washed with phosphate-buffered saline (PBS) twice
and incubated with 100 μmol/l WSP-1 combined with the surfactant,
cetyltrimethylammonium bromide (CTAB; 50 μmol/l), at 37°C for 20
min in the dark. H2S-derived fluorescence was measured
over the entire field of vision under a fluorescent microscope
connected to an imaging system (BX50-FLA; Olympus). The MFI of 4
random fields was analyzed using ImageJ 1.47 software.
Western blot analysis of protein
expression
Following heat-induced denaturation at 100°C for 5
min, equal amounts of protein from the indicated groups were
loaded. The total proteins were separated in 12% SDS-PAGE by
electrophoresis, and then transferred into PVDF membranes. After
blocking with 5% BSA in TBS-T, the membranes were incubated with
primary antibodies against HIF-1α, COX-2 or β-actin at 4°C
overnight. After 3 washes with TBS-T, the membranes were incubated
with horseradish peroxidase-conjugated secondary antibodies at room
temperature for 2 h. The membranes were washed again and developed
with an enhanced chemiluminescence system (Applygen Technologies,
Beijing, China), and the signals were then exposed to X-ray film.
The integrated optical density of the protein bands was calculated
using ImageJ 1.47 software.
Measurement of PGI2 by
enzyme-linked immunosorbent assay (ELISA)
The HPASMCs were plated in 96-well plates. After the
indicated treatments, the levels of PGI2 in the culture
medium were determined by ELISA according to the manufacturer’s
instructions (Boster Biotechnology Co., Ltd., Wuhan, China). The
amount of PGI2 in the culture medium was normalized to
the cell number measured using the CCK-8. The experiment was
performed at least 4 times with similar outcomes.
Statistical analysis
All the data are expressed as the means ± SD, and
analyzed using SPSS 13.0 software. The significance of inter-group
differences was evaluated by one-way analyses of variance (ANOVA).
Differences were considered to be significant if the two-sided
probability was P<0.05.
Results
Chemically-induced hypoxia enhances the
proliferation of HPASMCs
In order to determine whether chemical hypoxia
induces cell proliferation, we first exposed the HPASMCs to the
chemical hypoxia agent, CoCl2, at the indicated
concentrations and exposure times. We then assessed the cell
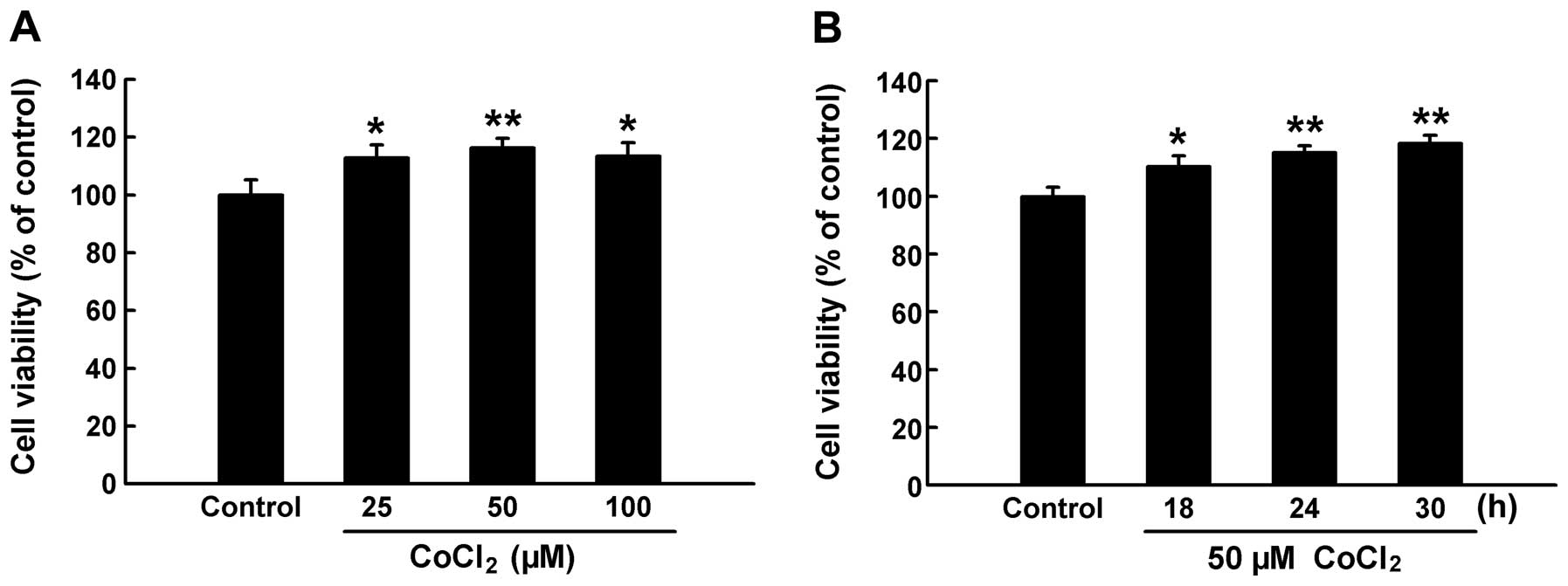
viability using the CCK-8. As shown in Fig. 2A, treatment with CoCl2
at concentrations ranging from 25 to 100 μM significantly increased
cell viability to >100% (P<0.05 and P<0.01 compared with
the control group), indicating the induction of cell proliferation
by chemical hypoxia. In addition, our results indicated that 50 μM
CoCl2 had the most prominent effect on the proliferation
of the HPASMCs. Subsequently, we exposed the HPASMCs to 50 μM
CoCl2 for the indicated periods of time in order to
observe the time course of chemical hypoxia-induced proliferation.
As shown in Fig. 2B, during the
time period of 18–30 h, exposure of the cells to 50 μM
CoCl2 enhanced the proliferation of the HPASMCs in a
time-dependent manner. In addition, treatment with 50 μM
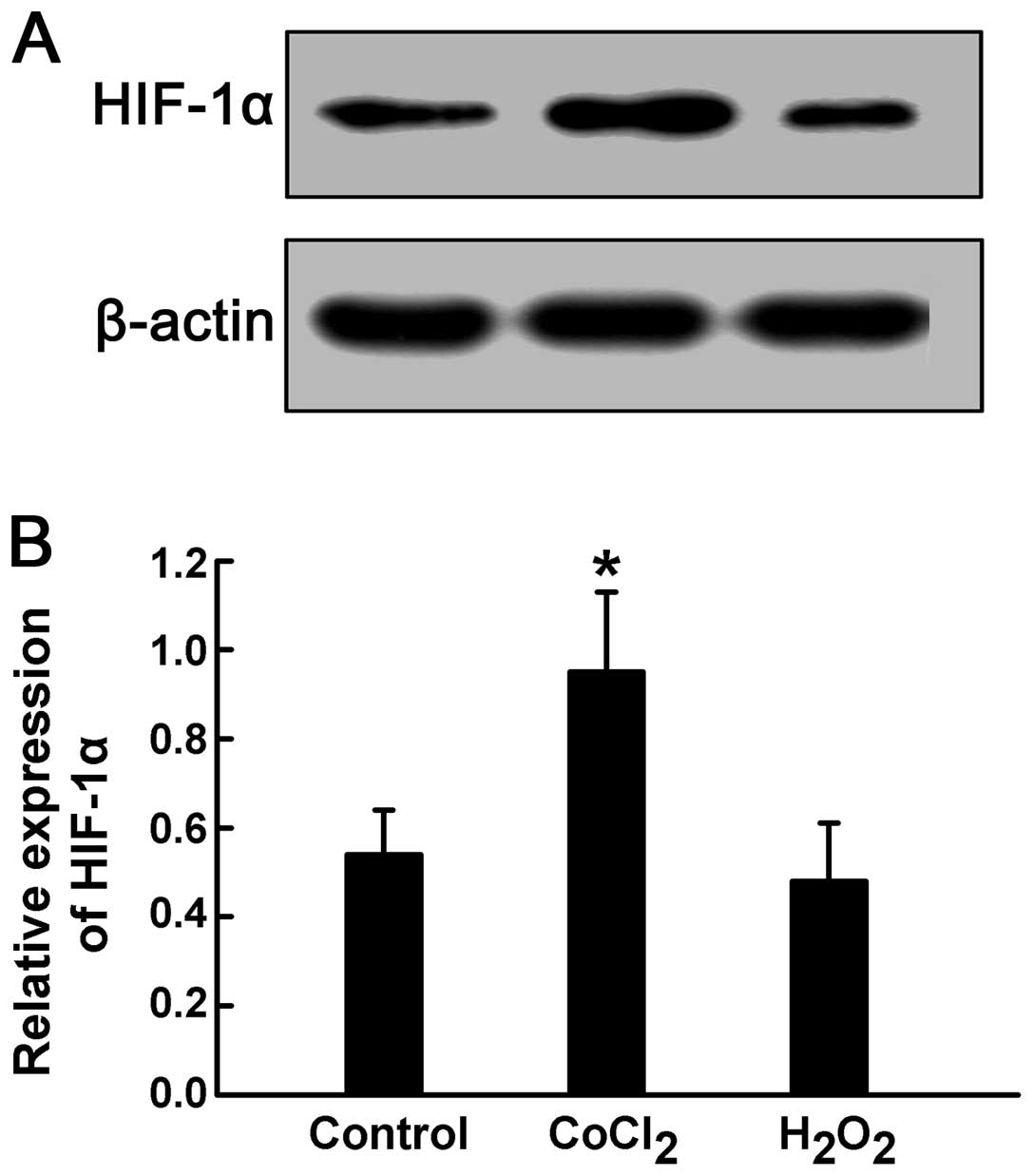
CoCl2 for 24 h significantly increased HIF-1α expression
in the HPASMCs, indicating a hypoxic state (P<0.05) (Fig. 3). These results suggest that
CoCl2 promotes the proliferation of HPASMCs through the
induction of hypoxia.
ROS may not be involved in chemical
hypoxia-induced proliferation of HPASMCs
A growing body of evidence indicates that ROS play a
critical role in hypoxia-induced cell proliferation (3,9).
In this study, to examine the role of ROS in the proliferation of
HPASMCs, the cells were exposed to the exogenous ROS donor,
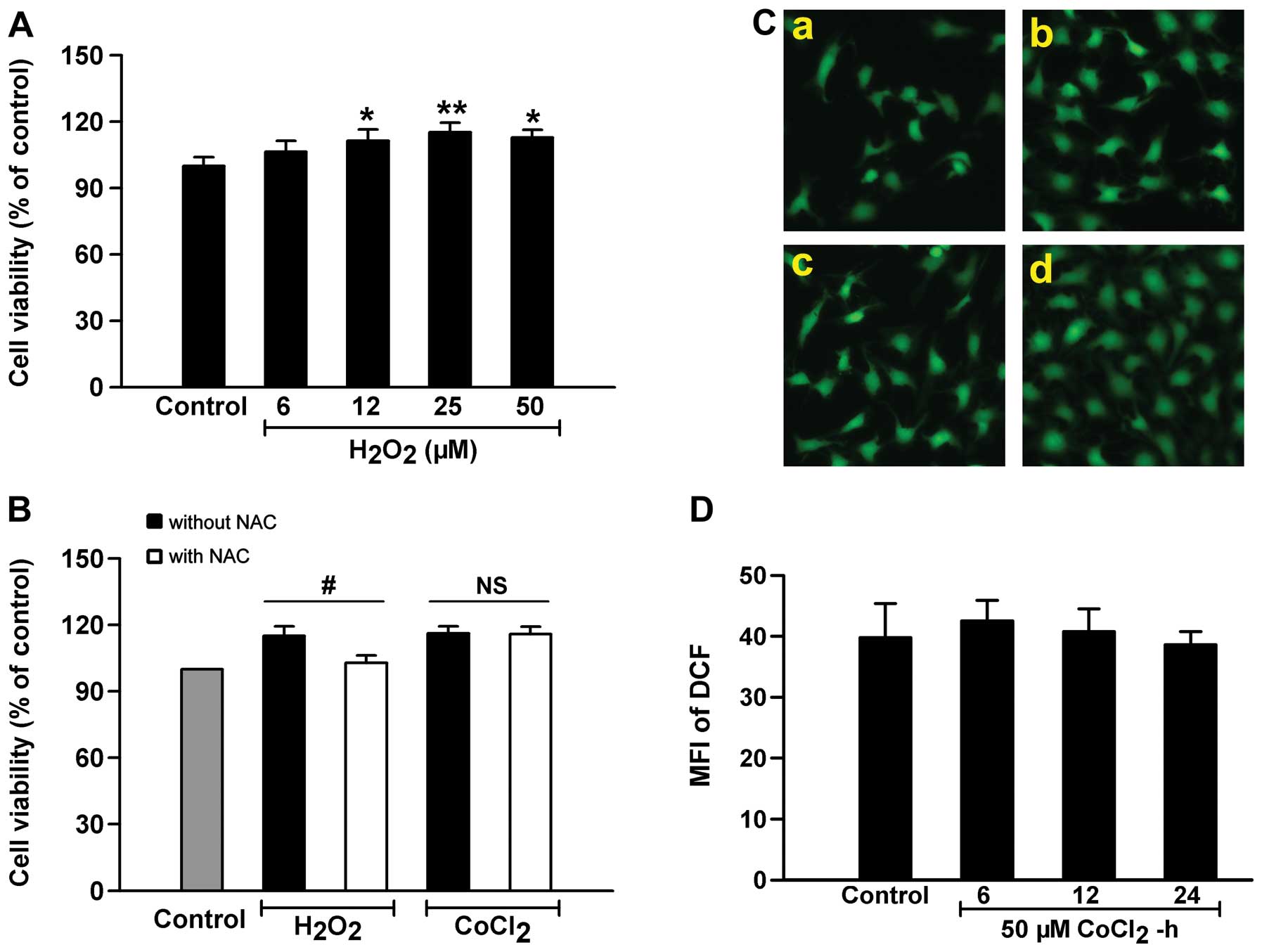
H2O2. As shown in Fig. 4A, H2O2 had
similar effects to CoCl2 in the induction of cell
proliferation; i.e., at concentrations ranging from 6 to 50 μM,
exposure of the HPASMCs to H2O2 for 24 h
induced marked proliferation, and the most prominent effects on
cell proliferation were induced at 25 μM; however, treatment with
25 μM H2O2 did not alter HIF-1α expression in
the HPASMCs (Fig. 3). Of note,
prior to exposure to 50 μM CoCl2 or 25 μM
H2O2, the cells were pre-conditioned with the
ROS scavenger, NAC. The results revealed that pre-treatment with
NAC markedly blocked the H2O2-induced cell
proliferation, but did not alter the effects of CoCl2
(Fig. 4B). Moreover, we observed
the effects of exposure to CoCl2 on the intercellular
ROS content and found that treatment with 50 μM CoCl2
for 6–24 h did not affect the levels of ROS (Fig. 4C and D). These data suggest that
chemical hypoxia-induced cell proliferation may not be dependent on
ROS, and that other mechanisms are involved.
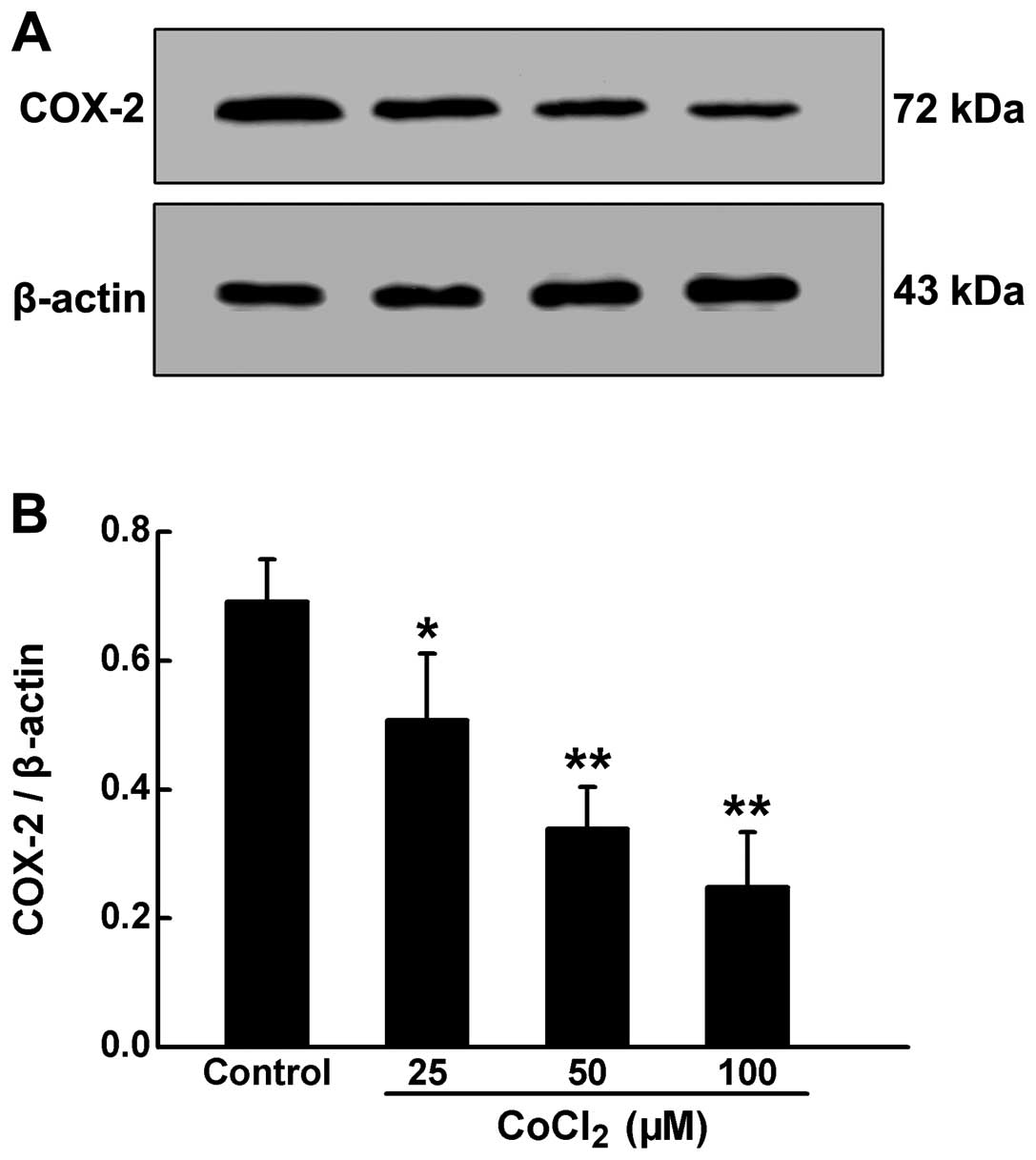
Chemically-induced hypoxia inhibits COX-2
expression in HPASMCs
COX-2 is responsible for the regulation of
proliferation and the hypertrophy of PASMCs. In order to elucidate
the roles of COX-2 in the proliferation induced by chemical
hypoxia, we measured COX-2 protein expression by western blot
analysis. As shown in Fig. 5,
following exposure of the HPASMCs to CoCl2 at increasing
concentrations for 24 h, the expression of COX-2 was significantly
reduced in a dose-dependent manner.
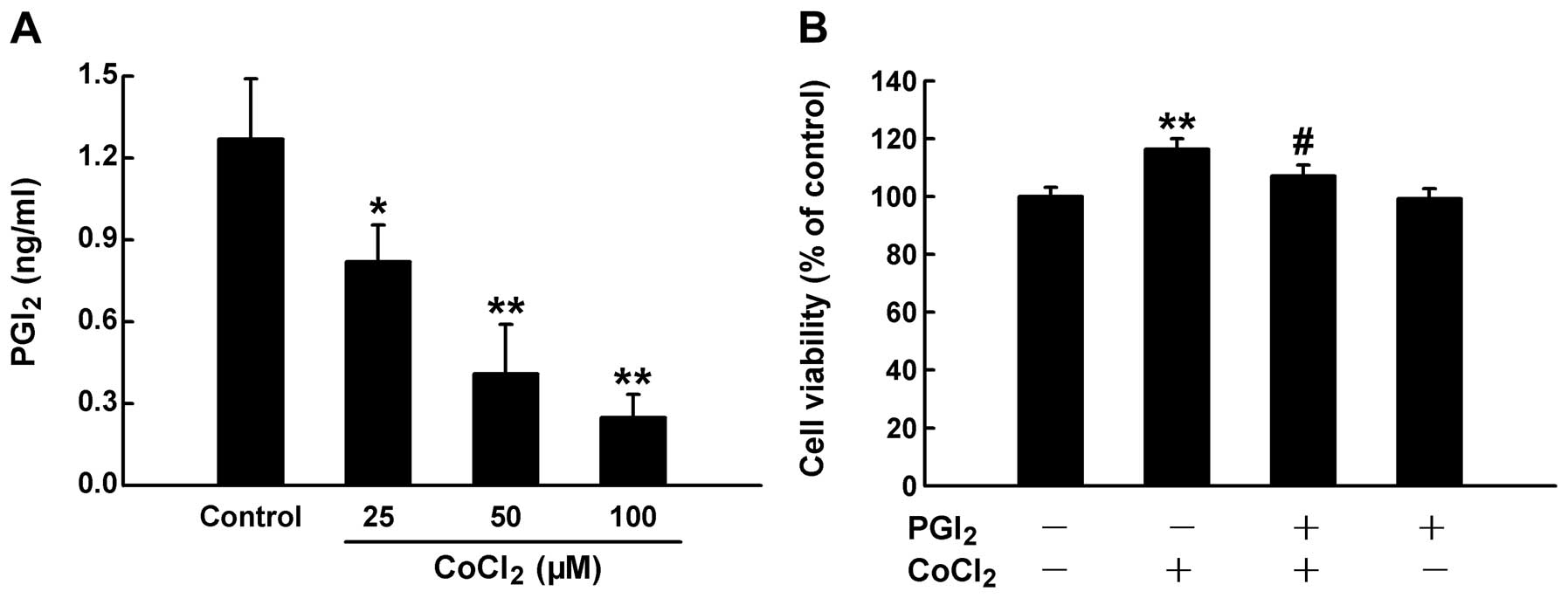
PGI2 is involved in chemical
hypoxia-induced proliferation of HPASMCs
PGI2 is produced by the catalytic action
of COX-2. To determine the downstream pathway of COX-2, we
investigated the effects of PGI2 on the chemical
hypoxia-induced proliferation of HPASMCs. Following exposure of the
cells to CoCl2 at increasing concentrations for 24 h, we
measured the levels of PGI2 in medium using ELISA. As
shown in Fig. 6A, the exposure to
CoCl2 markedly suppressed the release of PGI2
from the HPASMCs. In addition, the exogenous administration of
PGI2 partially abolished the CoCl2-induced
proliferation of HPASMCs (Fig.
6B).
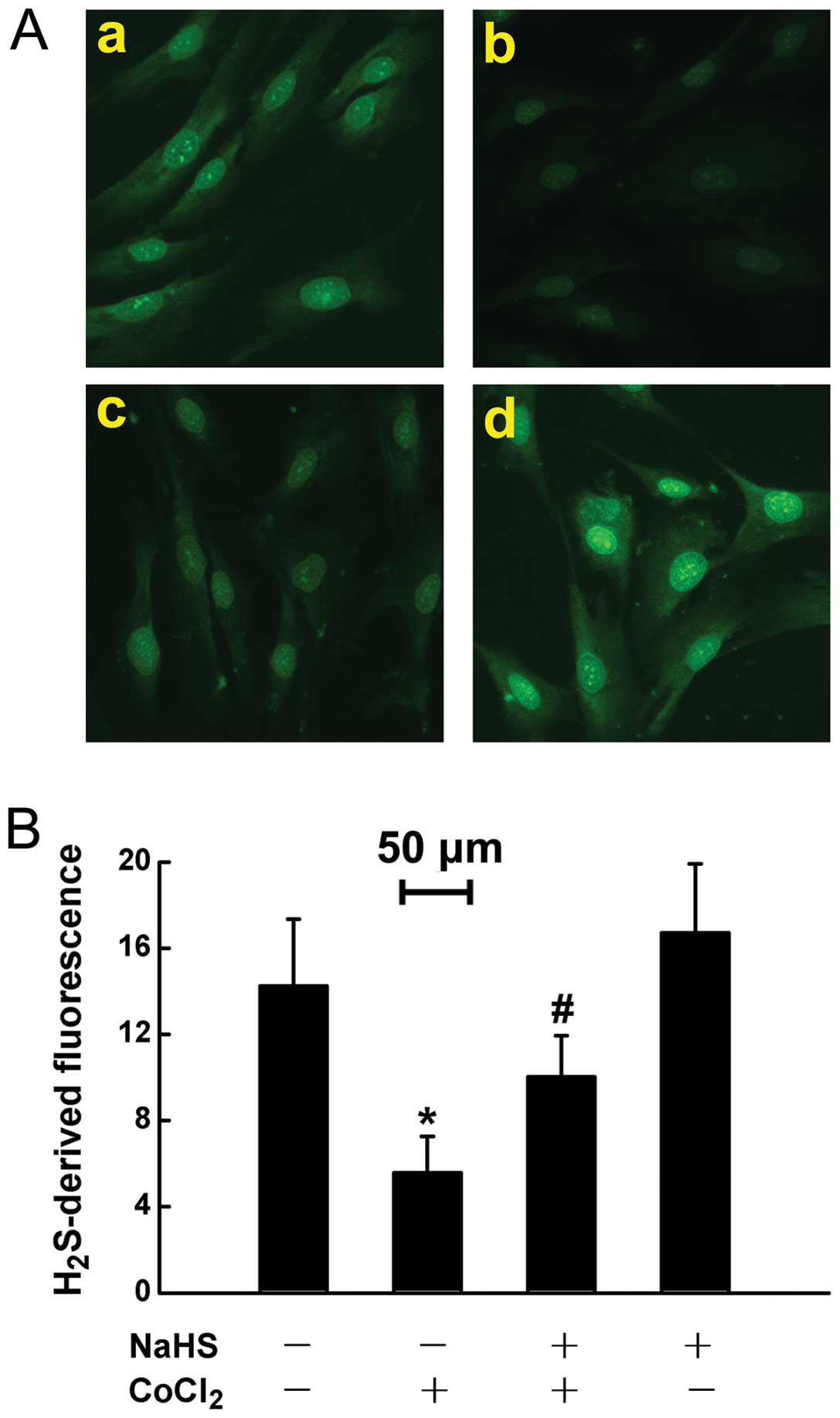
Endogenous H2S deficiency in
HPASMCs exposed to chemically-induced hypoxia
In a number of tissues, normal COX-2 expression
attributes to basic H2S levels in cardiomyocytes and
gastrointestinal mucosa (11,21). Thus, we hypothesized that the
chemical hypoxia-induced COX-2/PGI2 downregulation may
be due to endogenous H2S deficiency. To confirm our
hypothesis, experiments were carried out to detect intercellular
H2S levels using the fluorescent probe, WSP-1, followed
by photofluorography. The results revealed that exposure to 50 μM
CoCl2 for 24 h markedly suppressed the generation of
H2S in the HPASMCs (Fig.
7A–b). This inhibitory effect of CoCl2 was
significantly reversed, in part by the exogenous administration of
NaHS (a donor of H2S) (Fig. 7A–c), which alone did not alter
intercellular H2S levels (Fig. 7A–d).
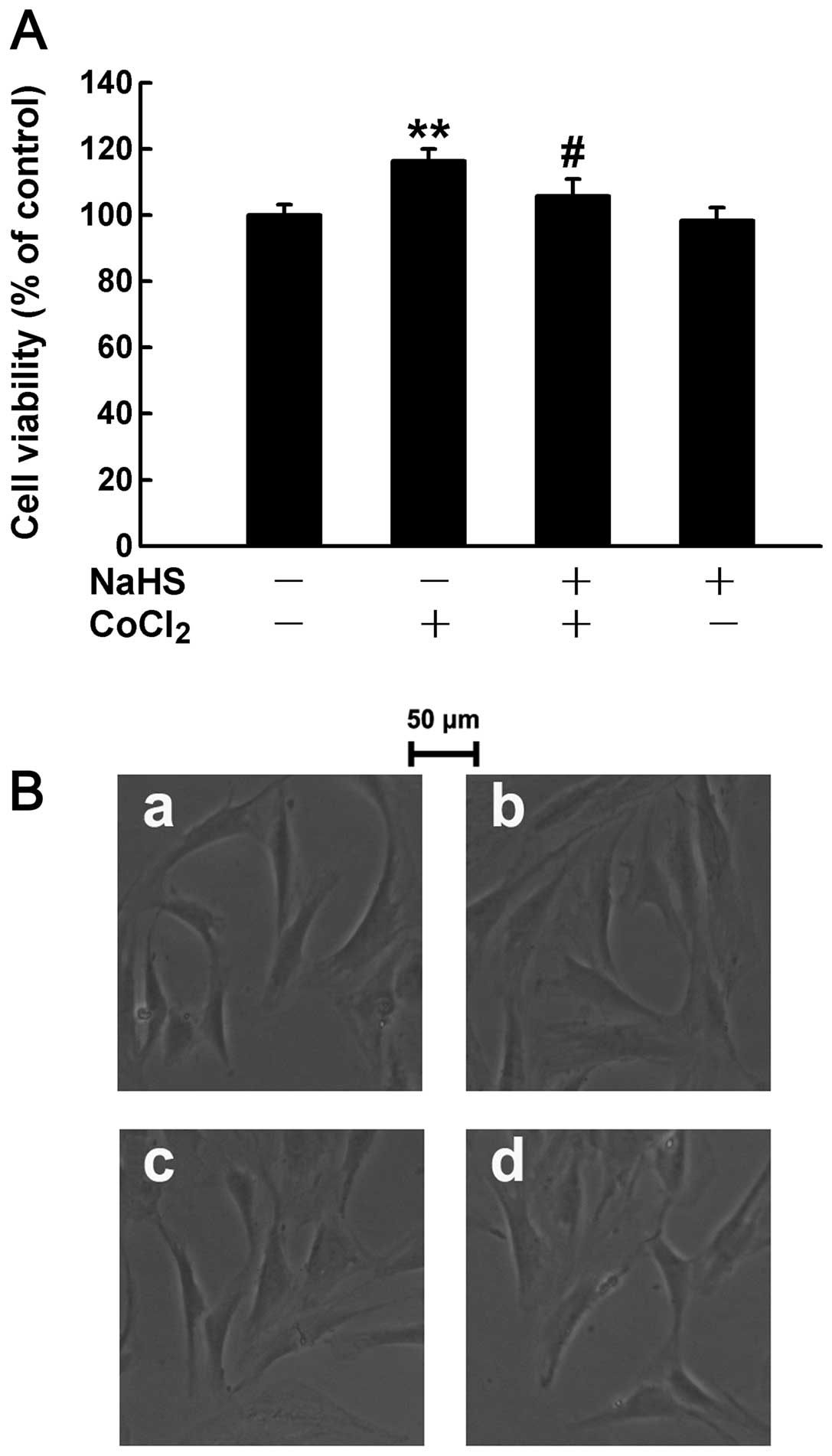
Administration of H2S partly
abolishes the chemical hypoxia-induced proliferation of
HPASMCs
Although the exogenous administration of
H2S prevented the CoCl2-induced
H2S deficiency to a certain extent, we hypothesized that
H2S may affect the chemical hypoxia-induced
proliferation of HPASMCs. The cells were therefore treated with 400
μM NaHS (a donor of H2S) for 30 min prior to treatment
with 50 μM CoCl2 for 24 h followed by the measurement of
cell proliferation. The results revealed that pre-treatment with
NaHS markedly eliminated the CoCl2-induced proliferation
of HPASMCs (Fig. 8A). In
addition, cell growth was observed by acquring images using a
microscope. The images depicted that the chemical hypoxia-induced
changes were mainly characterized by an increased cell number
(proliferation), rather than by the cell size (hypertrophy)
(Fig. 8B–b). Of note, this effect
was suppressed by pre-treatment with H2S (Fig. 8B–c). These results indicate that
insufficient H2S levels may contribute to the chemical
hypoxia-induced proliferation of HPASMCs.
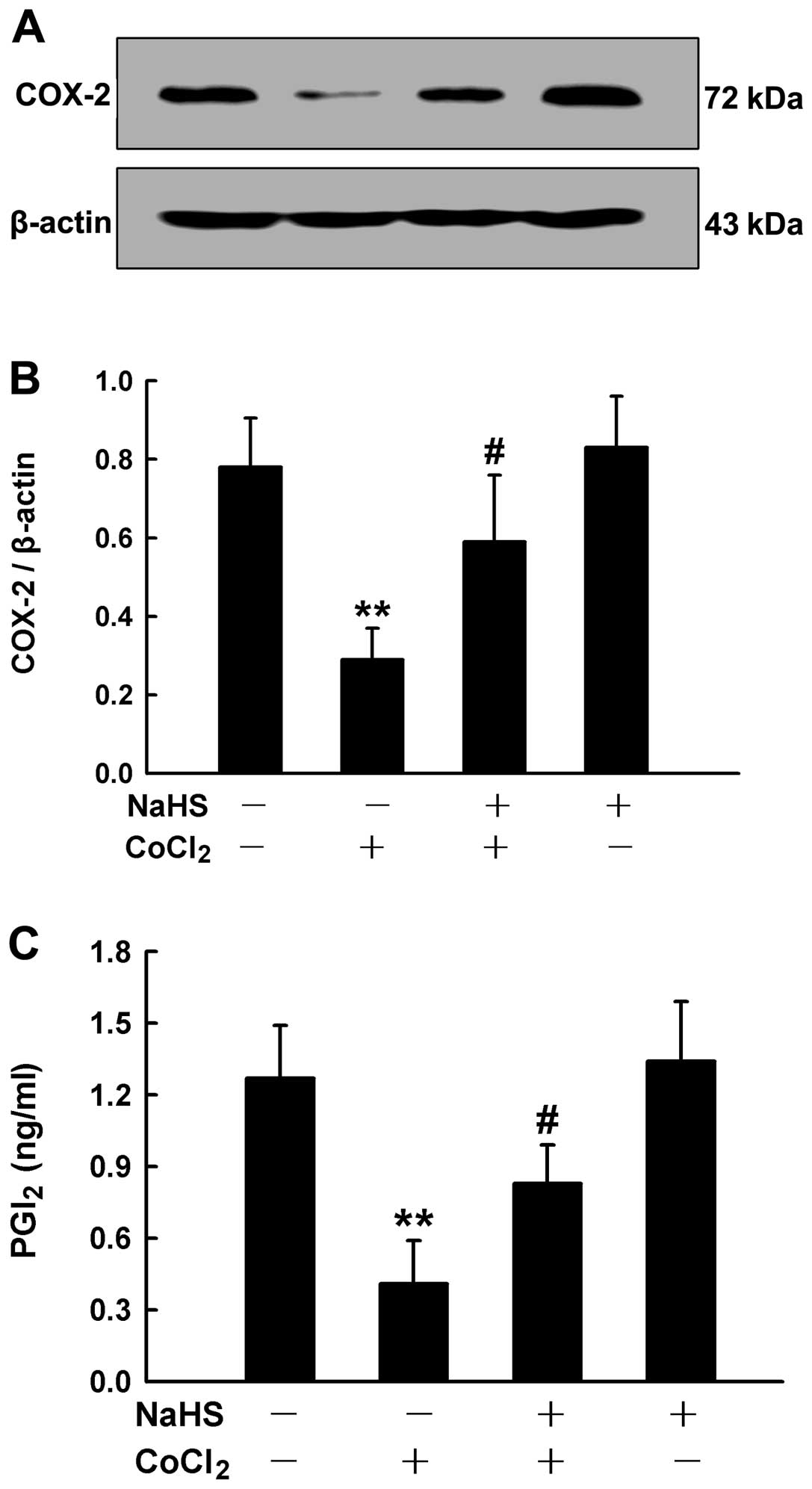
Involvement of COX-2/PGI2 in
the inhibition of cell proliferation by H2S
Since the chemical hypoxia-induced proliferation was
associated with the downregulation of COX-2/PGI2 and
insufficient levels of H2S in the HPASMCs, we wished to
determine the association between endogenous H2S and
COX-2/PGI2. Thus, prior to the treatment of HPASMCs with
50 μM CoCl2 for 24 h, the cells were pre-conditioned
with 400 μM NaHS for 30 min, followed by the measurement of COX-2
expression and PGI2 secretion. We found that exogenously
applied H2S (NaHS) partially abrogated the
downregulation in the expression of COX-2 (Fig. 9A and B) and the reduced secretion
of PGI2, which was induced by CoCl2 (Fig. 9C).
Discussion
The present study demonstrates that
chemically-induced hypoxia enhances the proliferation of HPASMCs.
During this process, there a marked decrease in COX-2 expression
and PGI2 secretion from the cells occurs; however, as
shown by our results, the levels of oxidative stress were not
significantly altered. Of note, our findings also indicated that
the production of H2S, a novel endogenous gaseous
molecule, was reduced, whose donor, NaHS, blocked the chemical
hypoxia-induced proliferation of HPASMCs. Our results also revealed
that the inhibition of cell proliferation by H2S was
attributed to the upregulation of COX-2/PGI2.
In this study, we first created a simple cellular
model of hypoxic PAH. PAH is a disease of the pulmonary
circulation, which can be defined as a mean pulmonary arterial
pressure of >25 mmHg at rest or >30 mmHg during exercise,
accompanied by a pulmonary capillary wedge pressure of <15 mmHg
(22). Sustained PAH usually
leads to an increase in pulmonary vascular resistance, which in
turn enhances right ventricular load, and ultimately induces right
ventricular failure and death (23). On the other hand, pulmonary
vascular remodeling and the resultant increases in pulmonary
vascular resistance play important roles in the development of PAH.
Through the inhibition of cell proliferation or the hypertrophy of
PASMCs and the protection of endothelial function, the development
of PAH can be effectively attenuated. Since hypoxia is one of the
main mechanisms involved in the proliferation of PASMCs (24), we created a cellular model of
hypoxia-induced PAH by the exposure of HPASMCs to the chemical
hypoxia agent, CoCl2. Hypoxic conditions can be induced
either by physical hypoxia, such as 1% O2, 5%
CO2 and 94% N2 in a modular incubator
chamber, or by chemical hypoxia, using hypoxia-mimicking agents,
such as CoCl2, manganese chloride (MnCl2) and
sodium thiosulfate (18). Due to
the advantages of simple operation and stable hypoxic effects, we
selected the latter in the current study. As shown by our results,
the exposure of HPASMCs to CoCl2 markedly elicited the
upregulation of HIF-1α expression, indicating the hypoxic condition
of the cells. Of note, exposure to CoCl2 induced marked
cell proliferation characterized by an increase in cell number
without an obvious increase in cell size. To measure cell
proliferation, we used the CCK-8 assay, as previously used in the
study by Wu et al to examine the proliferation of T
lymphocytes (17). We therefore
believed that our cellular model of hypoxic PAH was functional.
A novel finding of the present study was that
oxidative stress may not play a critical role in the chemical
hypoxia-induced proliferation of HPASMCs. Oxidative stress is
considered a key regulator of the balance between cell
proliferation and the onset of differentiation through ROS
(7). Abnormal ROS accumulation
often leads to delayed differentiation and/or uncontrollable cell
growth, namely proliferation. The development of hypertension and
atherosclerosis is closely associated with the overproduction of
ROS (6,25,26). Similar studies on hypoxia-induced
PAH are also available (3) and,
moreover, the ROS scavenger, SOD, has been shown to ameliorate PAH
(8). In the current study, we
also found that the ROS donor, H2O2, promoted
HPASMC proliferation, which was suppressed by another ROS
scavenger, NAC. However, NAC did not suppress
CoCl2-induced proliferation and treatment with
CoCl2 did not enhance the levels of ROS in the HPASMCs,
indicating that ROS may not be involved in the chemical
hypoxia-induced proliferation of HPASMCs. The differences in the
results between some of the above mentioned studies and our
findings, may be due to the different experimental models used; for
instance, differences between in vivo and in vitro
studies, or between physically- and chemically-induced hypoxia.
Another important finding of the current study was
that the downregulation of COX-2/PGI2 participated in
the chemical hypoxia-induced proliferation of HPASMCs. COX-2 is a
multifunctional protein, whose roles are complex and ambivalent in
different models, mediating either neuronal toxicity or cardiac
protection (27,28). For example, in the cardiovascular
system, COX-2 usually exerts beneficial effects. As previously
demonstrated, the cardioprotective effects of estrogen, zileuton
and atorvastatin are mediated by COX-2 (27,29,30). In accordance with a previous study
(2), our data demonstrated that
exposure to CoCl2 markedly reduced COX-2 expression in
the HPASMCs. Our data further indicated that PGI2, a
product of COX-2 from cells was also markedly reduced. The
exogenous administration of PGI2 markedly suppressed the
CoCl2-induced proliferation of HPASMCs, suggesting that
COX-2/PGI2 are not only involved in classical
vasodilatation, but also in the proliferation of smooth muscle
cells; this fact enhances our knowledge of the mechanisms of action
of PGI2. Currently, PGI2 and its analogues
have been widely used in the clinical management of patients with
PAH through effective vasodilatation. However, the mortality rate
of patients with this disorder has not been significantly reduced
over the past years. In addition, treatment costs, namely the
inhalation of aerosolized PGI2 are extremely high,
approximately several hundred dollars per day (10).
In chemical hypoxia-induced proliferation, the
deficiency of H2S was observed in the HPASMCs. Similar
to NO and CO, H2S, as a gaseous signaling molecule, has
a number of physiological and pathological roles, such as
cardioprotection (15,31), vasodilatation (32–34) and dermal protection (13,35). The endogenous formation of
H2S is attributed to enzymes, such as cystathionine
β-synthase (CBS), cystathionine γ-lyase (CSE), and
3-mercaptopyruvate sulfurtransferase (MPST) (12,19). These enzymes convert L-cysteine or
its derivatives to H2S in various tissues and organs.
However, in human pulmonary arterial tissues, the function of
H2S has not yet been fully elucidated. In the present
study, we found that the levels of H2S were markedly
decreased by treatment with CoCl2, whcn compared to the
control (untreated) HPASMCs. These findings are in accordance with
those presented in the study by Zhang et al on lung tissue
and pulmonary arteries of Wistar rats exposed to physical hypoxia
(36). We also found that
exogenously applied NaHS (donor of H2S) partially
recovered the deficiency in H2S in the HPASMCs exposed
to CoCl2 and, more importantly, NaHS suppressed the
CoCl2-induced cell proliferation. This was perhaps one
of the mechanisms underlying the H2S inhibition of
pulmonary arterial remodeling in vivo. As previously
demonstrated, H2S can also inhibit arterial remodeling
by endothelial protection (37)
and the induction of the apoptosis of smooth muscle cells (38). In the current study, we mainly
investigated the effects of H2S on cell proliferation
and the underlying mechanisms. Certain studies have suggested that
H2S reduces the proliferation of smooth muscle cells
through the downregulation of the mitrogen-activated protein kinase
(MAPK) pathway (39). Others
studies have indicated that the inhibition of cell proliferation by
H2S is involved in the stabilization of p53 coupled with
the induction of downstream molecules, including p21 and Bax
(40). Notably, a recent study
demonstrated that H2S inhibits both HIF-1α translation
by enhancing the phosphorylation of eukaryotic translation
initiation factor 2α (41);
another study demonstrated that H2S inhibits the
activation of HIF-1α in a von Hippel-Lindau protein- and
mitochondrial-dependent manner (42). In this study, treatment with
H2S markedly upregulated COX-2 expression and enhanced
PGI2 secretion from CoCl2-stimulated HPASMCs,
which may be associated with the inhibition of HIF-1α. Taking the
above mentioned data into consideration, we therefore suggested
that the H2S inhibition of the proliferation of HPASMCs
induced by chemical hypoxia may be associated with the upregulation
of COX-2/PGI2. In the future, the H2S donor
or its precursor, L-cysteine, may be applied to the treatment of
patients with PAH through the enhancement of PGI2
production, thus effectively reducing the treatment costs.
In conclusion, the present study demonstrates that
during the chemical hypoxia-induced proliferation of HPASMCs, the
expression of COX-2/PGI2 and the levels of
H2S are markedly suppressed. The exogenous
administration of PGI2 or H2S markedly
eliminated the chemical hypoxia-induced cell proliferation. In
addition, our data demonstrated that the H2S-induced
inhibition of cell proliferation was mediated by the upregulation
of COX-2/PGI2. The data from the present study provide
novel insight into the role of H2S in ameliorating the
chemical hypoxia-induced proliferation of HPASMCs. The modulation
of endogenous H2S production or the exogenous
administration of the H2S donor, NaHS, may be a novel
therapeutic strategy for PAH.
Acknowledgements
The present study was supported by the Science and
Technology Planning Project of Guangdong Province in China (no.
2012B031800298). We would like to thank Professor Ming Xian
(Department of Chemistry, Washington State University, Pullman, WA,
USA) for providing the H2S fluorescent probe
(WSP-1).
References
|
1
|
Schannwell CM, Steiner S and Strauer BE:
Diagnostics in pulmonary hypertension. J Physiol Pharmacol.
58(Suppl 5): 591–602. 2007.
|
|
2
|
Fredenburgh LE, Liang OD, Macias AA, et
al: Absence of cyclooxygenase-2 exacerbates hypoxia-induced
pulmonary hypertension and enhances contractility of vascular
smooth muscle cells. Circulation. 117:2114–2122. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ismail S, Sturrock A, Wu P, et al: NOX4
mediates hypoxia-induced proliferation of human pulmonary artery
smooth muscle cells: the role of autocrine production of
transforming growth factor-β1 and insulin-like growth factor
binding protein-3. Am J Physiol Lung Cell Mol Physiol.
296:L489–L499. 2009.PubMed/NCBI
|
|
4
|
Burdon RH, Gill V and Rice-Evans C:
Oxidative stress and tumour cell proliferation. Free Radic Res
Commun. 11:65–76. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Immenschuh S and Baumgart-Vogt E:
Peroxiredoxins, oxidative stress, and cell proliferation. Antioxid
Redox Signal. 7:768–777. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shimizu H, Hirose Y, Nishijima F,
Tsubakihara Y and Miyazaki H: ROS and PDGF-β receptors are
critically involved in indoxyl sulfate actions that promote
vascular smooth muscle cell proliferation and migration. Am J
Physiol Cell Physiol. 297:C389–C396. 2009.
|
|
7
|
Tsukagoshi H, Busch W and Benfey PN:
Transcriptional regulation of ROS controls transition from
proliferation to differentiation in the root. Cell. 143:606–616.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lakshminrusimha S, Russell JA, Wedgwood S,
et al: Superoxide dismutase improves oxygenation and reduces
oxidation in neonatal pulmonary hypertension. Am J Respir Crit Care
Med. 174:1370–1377. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zuckerbraun BS, Shiva S, Ifedigbo E, et
al: Nitrite potently inhibits hypoxic and inflammatory pulmonary
arterial hypertension and smooth muscle proliferation via xanthine
oxidoreductase-dependent nitric oxide generation. Circulation.
121:98–109. 2010. View Article : Google Scholar
|
|
10
|
Ruan CH, Dixon RA, Willerson JT and Ruan
KH: Prostacyclin therapy for pulmonary arterial hypertension. Tex
Heart Inst J. 37:391–399. 2010.PubMed/NCBI
|
|
11
|
Pan TT, Feng ZN, Lee SW, Moore PK and Bian
JS: Endogenous hydrogen sulfide contributes to the cardioprotection
by metabolic inhibition preconditioning in the rat ventricular
myocytes. J Mol Cell Cardiol. 40:119–130. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang XY, Yang CT, Zheng DD, et al:
Hydrogen sulfide protects H9c2 cells against doxorubicin-induced
cardiotoxicity through inhibition of endoplasmic reticulum stress.
Mol Cell Biochem. 363:419–426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang C, Yang Z, Zhang M, et al: Hydrogen
sulfide protects against chemical hypoxia-induced cytotoxicity and
inflammation in HaCaT cells through inhibition of
ROS/NF-kappaB/COX-2 pathway. PLoS One. 6:e219712011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang R: Physiological implications of
hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev.
92:791–896. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu LF, Pan TT, Neo KL, Yong QC and Bian
JS: Cyclooxygenase-2 mediates the delayed cardioprotection induced
by hydrogen sulfide preconditioning in isolated rat cardiomyocytes.
Pflugers Arch. 455:971–978. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin Hongfang J, Bailin Cong, Bin Zhao, et
al: Effects of hydrogen sulfide on hypoxic pulmonary vascular
structural remodeling. Life Sci. 78:1299–1309. 2006.PubMed/NCBI
|
|
17
|
Wu C, Liu F, Zhou X, et al: Effect of
protein kinase C on proliferation and apoptosis of T lymphocytes in
idiopathic thrombocytopenic purpura children. Cell Mol Immunol.
2:197–203. 2005.PubMed/NCBI
|
|
18
|
Yang C, Ling H, Zhang M, et al: Oxidative
stress mediates chemical hypoxia-induced injury and inflammation by
activating NF-kappab-COX-2 pathway in HaCaT cells. Mol Cells.
31:531–538. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao Y, Bhushan S, Yang C, et al:
Controllable hydrogen sulfide donors and the activity against
myocardial ischemia-reperfusion injury. ACS Chem Biol. 8:1283–1290.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu C, Pan J, Li S, et al: Capture and
visualization of hydrogen sulfide by a fluorescent probe. Angew
Chem Int Ed Engl. 50:10327–10329. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Coruzzi G, Venturi N and Spaggiari S:
Gastrointestinal safety of novel nonsteroidal antiinflammatory
drugs: selective COX-2 inhibitors and beyond. Acta Biomed.
78:96–110. 2007.PubMed/NCBI
|
|
22
|
Badesch DB, Abman SH, Simonneau G, Rubin
LJ and McLaughlin VV: Medical therapy for pulmonary arterial
hypertension: updated ACCP evidence-based clinical practice
guidelines. Chest. 131:1917–1928. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Simonneau G, Galie N, Rubin LJ, et al:
Clinical classification of pulmonary hypertension. J Am Coll
Cardiol. 43:S5–S12. 2004. View Article : Google Scholar
|
|
24
|
Yu M, Gong D, Lim M, Arutyunyan A, Groffen
J and Heisterkamp N: Lack of bcr and abr promotes hypoxia-induced
pulmonary hypertension in mice. PLoS One. 7:e497562012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Touyz RM: Reactive oxygen species,
vascular oxidative stress, and redox signaling in hypertension:
what is the clinical significance? Hypertension. 44:248–252. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Landmesser U, Dikalov S, Price SR, et al:
Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial
cell nitric oxide synthase in hypertension. J Clin Invest.
111:1201–1209. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Booth EA, Flint RR, Lucas KL, Knittel AK
and Lucchesi BR: Estrogen protects the heart from
ischemia-reperfusion injury via COX-2-derived PGI2. J
Cardiovasc Pharmacol. 52:228–235. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dore S, Otsuka T, Mito T, et al: Neuronal
overexpression of cyclooxygenase-2 increases cerebral infarction.
Ann Neurol. 54:155–162. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kwak HJ, Park KM, Choi HE, Lim HJ, Park JH
and Park HY: The cardioprotective effects of zileuton, a
5-lipoxygenase inhibitor, are mediated by COX-2 via activation of
PKC delta. Cell Signal. 22:80–87. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Birnbaum Y, Ye Y, Rosanio S, et al:
Prostaglandins mediate the cardioprotective effects of atorvastatin
against ischemia-reperfusion injury. Cardiovasc Res. 65:345–355.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang Z, Yang C, Xiao L, et al: Novel
insights into the role of HSP90 in cytoprotection of H2S
against chemical hypoxia-induced injury in H9c2 cardiac myocytes.
Int J Mol Med. 28:397–403. 2011.PubMed/NCBI
|
|
32
|
Zhao W, Zhang J, Lu Y and Wang R: The
vasorelaxant effect of H2S as a novel endogenous gaseous
KATP channel opener. EMBO J. 20:6008–6016. 2001.PubMed/NCBI
|
|
33
|
Bhatia M: Hydrogen sulfide as a
vasodilator. IUBMB Life. 57:603–606. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng Y, Ndisang JF, Tang G, Cao K and
Wang R: Hydrogen sulfide-induced relaxation of resistance
mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol.
287:H2316–H2323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shimizu K, Ogawa F, Hara T, et al:
Exogenous application of hydrogen sulfide donor attenuates
inflammatory reactions through the L-selectin-involved pathway in
the cutaneous reverse passive Arthus reaction. J Leukoc Biol.
93:573–584. 2013. View Article : Google Scholar
|
|
36
|
Zhang C, Du J, Bu D, Yan H, Tang X and
Tang C: The regulatory effect of hydrogen sulfide on hypoxic
pulmonary hypertension in rats. Biochem Biophys Res Commun.
302:810–816. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bearden SE, Beard RS Jr and Pfau JC:
Extracellular transsulfuration generates hydrogen sulfide from
homocysteine and protects endothelium from redox stress. Am J
Physiol Heart Circ Physiol. 299:H1568–H1576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang G, Sun X and Wang R: Hydrogen
sulfide-induced apoptosis of human aorta smooth muscle cells via
the activation of mitogen-activated protein kinases and caspase-3.
FASEB J. 18:1782–1784. 2004.PubMed/NCBI
|
|
39
|
Du J, Hui Y, Cheung Y, et al: The possible
role of hydrogen sulfide as a smooth muscle cell proliferation
inhibitor in rat cultured cells. Heart Vessels. 19:75–80. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Baskar R, Sparatore A, Del Soldato P and
Moore PK: Effect of S-diclofenac, a novel hydrogen sulfide
releasing derivative inhibit rat vascular smooth muscle cell
proliferation. Eur J Pharmacol. 594:1–8. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu B, Teng H, Yang G, Wu L and Wang R:
Hydrogen sulfide inhibits the translational expression of
hypoxia-inducible factor-1alpha. Br J Pharmacol. 167:1492–1505.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kai S, Tanaka T, Daijo H, et al: Hydrogen
sulfide inhibits hypoxia- but not anoxia-induced hypoxia-inducible
factor 1 activation in a von hippel-lindau- and
mitochondria-dependent manner. Antioxid Redox Signal. 16:203–216.
2012. View Article : Google Scholar : PubMed/NCBI
|