Introduction
Sinodielide A (SA) is a naturally occurring
guaianolide, which is isolated from the root of Sinodielsia
yunnanensis (1). This root is
used in traditional Chinese medicine as an antipyretic, analgesic
and diaphoretic agent. There are a number of studies on the
anticancer effects of agents isolated from a species of
Umbelliferae (Apiaceae). The major component of Angelica
japonica roots, 3′-O-acetylhamaudol, has dual functions:
it has anti-angiogenic functions and can activate intestinal
intraepithelial lymphocytes (2).
Furthermore, xanthoangelol D, isolated from the roots of
Angelica keiskei, inhibits endothelin-1 production by
suppressing nuclear factor-κB (NF-κB) (3). When cancer cells are subjected to
hyperthermia, they typically acquire thermotolerance during or
after the heating process, similar to other treatments, such as
radiotherapy and chemotherapy; this is one of the problems
associated with thermotherapy. In an attempt to overcome the
problems of thermotolerance and drug resistance associated with
thermo- and chemotherapy, combined treatment with hyperthermia and
chemotherapeutic agents has become widely adopted as a cancer
treatment strategy to gain greater therapeutic effects by reducing
the cytotoxicity of the administered chemotherapeutic drug. SA,
extracted from plants of the genus Oenanthe of the Umbelliferae
family, is an original crude medicinal substance developed by Wang
et al (Osaka University of Pharmaceutical Sciences, Osaka,
Japan) (1); however, to our
knowledge, no studies on its potential roles in cancer therapy have
been published to date.
We previously reported the in vitro
thermosensitization of human cancer cell lines by combined therapy
with hyperthermia and the sesquiterpene lactone, parthenolide, an
inhibitor of the transcription factor, NF-κB and further
investigated the kinetics of apoptosis induction and the cell cycle
distribution with regards to the transcription factor, NF-κB, and
the proto-oncogene mitogen-activated protein (MAP) kinase (MAPK)
signaling pathways (4–6). Cellular thermosensitivity was
observed by combined treatment with parthenolide, an NF-κB
inhibitor, and subsequent exposure to hyperthermia in A549 human
non-small cell lung adenocarcinoma cells bearing the wild-type
p53 gene. Apoptosis was induced through the direct
suppression of NF-κB in a p53-independent and heat-induced
hsp72-independent manner via the NF-κB signaling pathway (4). We also reported that combination
therapy with parthenolide administered prior to exposure to
hyperthermia caused lethal damage to A549 cells by targeting the S
phase; this effect correlated with the induction of apoptosis and
the G2/M arrest via the NF-κB cascade, and possibly
occurred due to the blockade of NF-κB activation by the
heat-induced hsp72 protein. It was concluded that parthenolide
contributes to thermosensitization through other pathways related
to NF-κB signaling or by crosstalk with other mediator genes
(5). In another study,
combination therapy with parthenolide and heating was carried out
in the human androgen-independent prostate cancer cell lines, PC3
and DU145. We reported a higher number of apoptotic PC3 cells than
DU145 cells, which was in agreement with the finding that the
amount of damage to the cells (assessed by survival curves) was
greater in the PC3 than in the DU145 cells (6).
Ras activates a number of pathways, of which MAPK
has been the most studied. This cascade transmits signals
downstream and results in the transcription of genes involved in
cell growth or mitosis (7). The
unusual activation of the Ras/Raf/MAPK signaling pathway is
characteristic of human cancer; thus, the pathway has attracted
attention as a potential target in anticancer therapy. Similar to
hsp family members, known to be thermotolerance inducers,
ras is known as a gene that causes thermoresistance in
hyperthermia through various signaling cascades. The association of
thermotolerance with the ras gene has been demonstrated,
since thermotolerance or thermoresistance developed following the
blockade of the activation of the Ras-cAMP or MAPK cascades
(8–10). Among the variety of intracellular
signaling pathways, the most relevant to the development of cell
malignancy are the MAPK growth signaling cascade and the
phosphoinositide 3-kinase (PI3K)-Akt survival signaling cascade.
The MAPK pathway has been reported to be activated in various types
of cancer, such as breast cancer, colon cancer, melanoma, lung
cancer and prostate cancer, which indicates its involvement in
tumor progression and metastasis (11–15). Three distinct groups of MAPKs have
been identified in mammalian cells: the extracellular-regulated
kinase (ERK), the stress-activated protein kinase/c-Jun N-terminal
kinase (SAPK/JNK) and p38 (16–19); they all act as mediators of
signals from the cell surface to the nucleus (20). The SAPK/JNK signaling pathway also
regulates cellular proliferation, apoptosis and tissue
morphogenesis (21). Xia et
al (22) reported opposing
effects of the ERK and SAPK/JNK-p38 MAPKs on apoptosis; thus, the
dynamic balance between the growth factor-activated ERK and the
stress-activated SAPK/JNK-p38 pathways may be important for
determining whether a cell survives or undergoes apoptosis
(22). We previously reported
that parthenolide in combination with hyperthermia activated the
p-SAPK/JNK protein mainly through the abovementioned MAPK family
signaling pathways (ERK1/2, SAPK/JNK and p38) in DU145 cells.
However, our results further indicated that the induction of
apoptosis and cell cycle arrest at the G2/M phase mostly
occurred via the SAPK/JNK pathway (6).
The MAPK pathway is activated by different
extracellular stimuli and has distinct downstream targets;
therefore its disruption can halt cancer progression by inhibiting
tumor angiogenesis, proliferation, invasion and metastasis
(23). In prostate cancer, a
number of other pathways have been shown to be activated, including
the PI3K/Akt and NF-κB signal transduction pathways, which have
been associated with tumor development and progression (24,25). In the present study, we confirmed
that SA and hyperthermia exert antitumor effects by inducing
apoptosis and cell cycle arrest. The related mechanisms, involving
the Ras/MAPK cascade in the development of thermotolerance through
the regulation of ERK and SAPK/JNK, and the involvement of the
PI3K/Akt signal transduction pathway in the inhibition of
apoptosis, were also examined. Furthermore, we investigated the
effects of combined therapy using SA and heating on the
thermosensitization of DU145 human prostate cancer cells, an
androgen-independent cell line, as well as the relevant
mechanisms.
Materials and methods
Cells and culture medium
DU145 cells from a human androgen-independent
prostate cancer cell line were provided by the Department of
Urology of Kitasato University (Tokyo, Japan), courtesy of Dr
Takefumi Sato. The cells were cultured in RPMI-1640 medium
(Invitrogen Life Technologies, Grand Island, NY, USA) under
standard conditions at 37°C in a humidified incubator with 5%
CO2 in 95% air (26,27). RPMI-1640 medium was supplemented
with 10% fetal bovine serum (ICN Biomedicas Inc., Aurora, OH, USA),
1% minimum essential medium (MEM) non-essential amino acid (NEAA)
solution, 1% MEM vitamin solution (both from Invitrogen Life
Technologies), 1% sodium pyruvate, and 1% of a penicillin and
streptomycin mix (both from Nacalai Tesque Inc., Kyoto, Japan).
SA, hyperthermia and combined
treatment
SA was dissolved in culture medium to a final
concentration of 20.0 μM prior to use. Cells that adhered to the
inner side of the bottom of culture flasks were exposed to SA by
replacement with 6 ml of SA solution for various periods of time.
The cells were then treated with 20.0 μM of SA at 37°C for 4 h,
which resulted in 50% lethal damage (LD50). The SA
solution was removed and the adhered cells were gently rinsed twice
with culture medium containing 3% serum, then resupplied with 6 ml
of RPMI-1640 medium at 37°C. Hyperthermia was established by
immersing culture flasks equipped with tightened screw tops in a
temperature-regulated water bath (Advantec, model LF-480; Toyo
Seisakusho Co., Ltd., Tokyo, Japan) preset to the desired
temperature, which was maintained within ±0.05°C, as measured by a
thermistor (model D116-1251; Takara Thermistor Instructions Co.,
Yokohama, Japan). For the combined treatment, applications of SA
and heating were sequentially performed, with cells first exposed
to SA for 4 h, then rinsed twice with culture medium containing 3%
serum, placed in RPMI-1640 medium, and finally subjected to
hyperthermia. Kinetic assessments of the sensitivity of DU145 cells
to SA and hyperthermia were carried out by colony formation assays,
and the results were corrected based on the plating efficiency of
the control cells (i.e., 80–90%). The average colony multiplicity
was <1.1.
Cells exposed to hyperthermia produce linear
survival curvers. The relationship between the survival fraction
S and the heating period T is then calculated as:
S = e−αT, where S
is the number of surviving cells, −α is the slope and
T is the heating period. This relationship is more commonly
represented as: S =
e−T/T0
by defining T0 as 1/α. When T = T0,
S = e−1 = 0.037.
S0, T0 show the
heating period required to reduce the experimental survival rate by
1/e. The paramater T0 can then be used to characterize
the thermosensitivity in this region of the curve.
The T0 value, which was adopted as the
criterion for cellular thermosensitivity and sensitivity to SA, was
defined as the treatment period required to reduce survival by 1/e
in the exponentially regressing portion of the survival curve,
i.e., the linear portions of the treatment period shown in the
semilogarithmic survival curves.
Apoptosis assay and cell cycle
distribution
The kinetics of apoptosis induction, as well as the
G1 and G2/M cell cycle arrest of DU145 cells
following treatment with SA, hyperthermia and their combination
were analyzed by flow cytometry. After 0, 12, 24 and 48 h of
incubation at 37°C following treatments, cells (1×105)
were harvested by trypsinization, resuspended in culture medium,
rinsed twice with ice-cold PBS(−), and fixed in ice-cold 70%
ethanol, following the addition of PBS into the culture tubes at a
rate of 3. The cells were stored at 4°C for at least 24 h, then
collected by centrifugation, rinsed twice with ice-cold PBS, and
treated with 1 mg/ml of RNase A (type II-A; Sigma-Aldrich Corp.,
St. Louis, MO, USA) at room temperature for 30 min. The cells were
then stained with 100 μg/ml of propidium iodide (PI) (Sigma-Aldrich
Corp.) for at least 30 min on ice in the dark. The cell cycle
distribution was analyzed using a flow cytometer (Beckman Coulter,
Inc., Fullerton, CA, USA). Immediately prior to analysis, cell
suspensions were filtered through 40-μm diameter nylon meshes to
remove cell aggregates and debris. Ten thousand events per
determination were analyzed for each sample and the quantification
of the cell cycle distribution was performed using software
provided by the manufacturer. Cell cycle distribution based on DNA
content is represented by a histogram.
Examination of ERK1/2, SAPK/JNK and Akt
cascades by western blot analysis
Intracellular protein and the phosphorylation of
ERK1/2, SAPK/JNK MAPK and Akt following SA application, heating,
and their combination were examined in DU145 cells
(1×106) by western blot analysis. Cells were harvested
by trypsinization and resuspended in RPMI-1640 medium. After
rinsing with ice-cold PBS twice, they were dissolved in RIPA lysis
buffer containing a protease and a phosphatase inhibitor and were
treated by freezing at −20°C and thawing on ice 3 times. Cell
lysates were centrifuged at 14,000 rpm at 4°C for 10 min to remove
cell debris. The supernatants were then diluted 2-fold with
SDS-PAGE, and subjected to a block incubator at 95°C for 3 min
following stirring and vortex mixing and transformation. The
protein contents of the supernatants were quantified using a
protein assay kit (Bio-Rad Laboratories, Richmond, CA, USA).
Aliquots of protein (10 μg) were subjected to western blot analysis
using ERK1/2, SAPK/JNK, Akt and their phosphorylated antibodies
(Cell Signaling Technology, Japan KK). Following electrophoresis on
10% polyacrylamide gels containing 0.1% solution dodecyl sulfate
(SDS) and electrophoretic transfer onto Immobilon-P PVDF-membranes
(Millipore Corp., Medford, MA, USA), the membranes were incubated
with phosphorylated (p-)ERK1/2, p-SAPK/JNK and p-Akt antibodies.
GAPDH antibody (Cell Signaling Technology) served as the loading
control, as previously described (28).
Results
Thermosensitizing effects of SA with
hyperthermia at 40–44°C
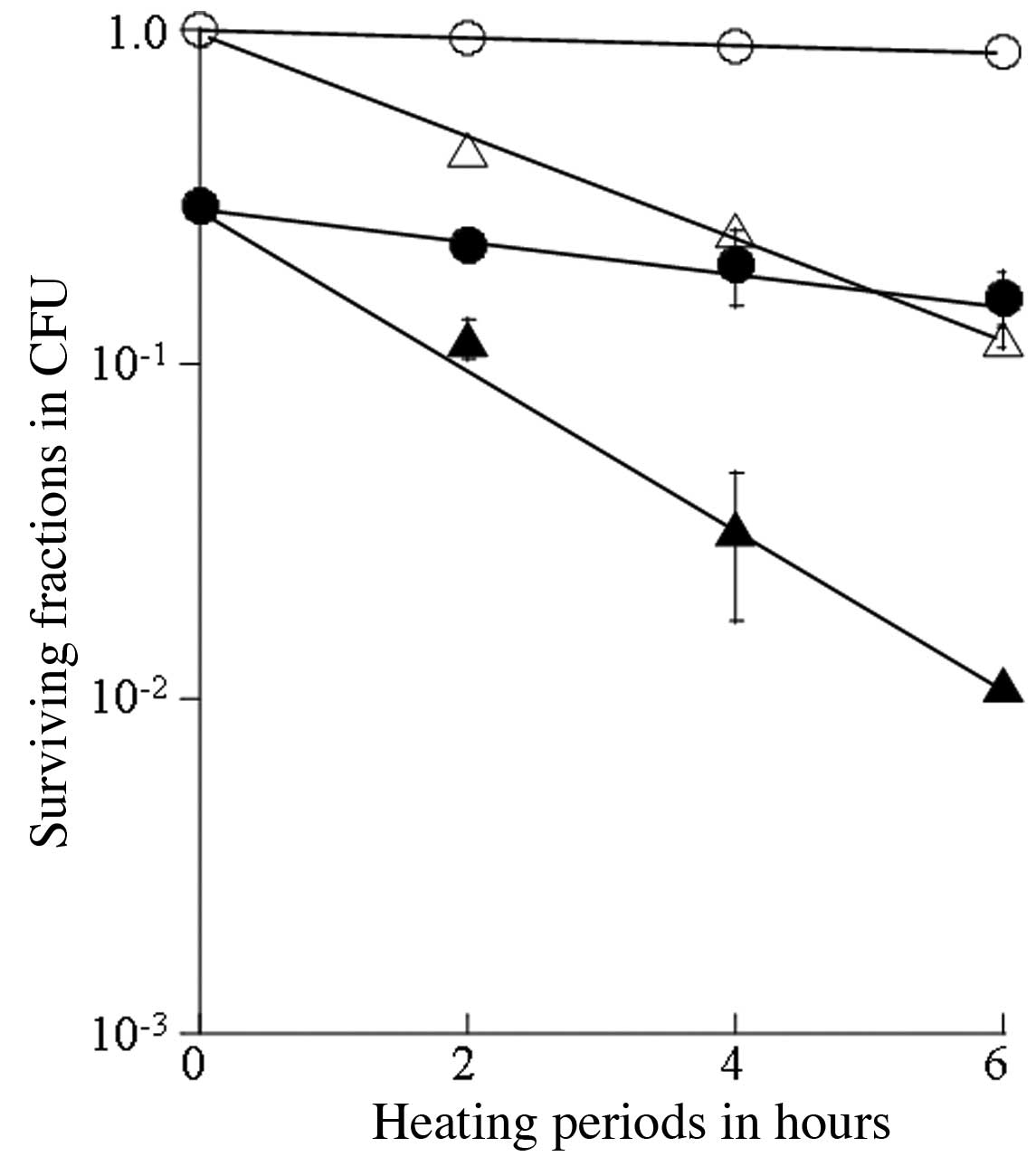
The thermosensitizing effects of SA were
investigated in DU145 cells based on semilogarithmic survival
curves produced from the results of the single and the combined
therapy with 20.0 μM of SA for 4 h, and hyperthermia at 40, 42, 43
and 44°C (Figs. 1 and 2). The survival curve following the
heating treatment at 40°C for up to 6 h indicated a slight
cytotoxicity, while synergistic antitumor effects were observed for
the treatment with 20.0 μM SA for 4 h prior to heating at 40°C
(Fig. 1). Cellular lethal
thermosensitivity was estimated based on the T0 value
(heating time required to reduce survival by 1/e), which was the
reciprocal of the slope of the survival curve in the exponential
phase. This value was 39.8 h for heating at 40°C, and 9.55 h for
sequential treatment with SA and heating at 40°C (Table I). Similarly, the
thermosensitivity of the cells increased with heating at 42°C, and
the T0 value was 2.12 h for heating at 42°C, and 1.29 h
for sequential treatment with SA and heating at 42°C. The survival
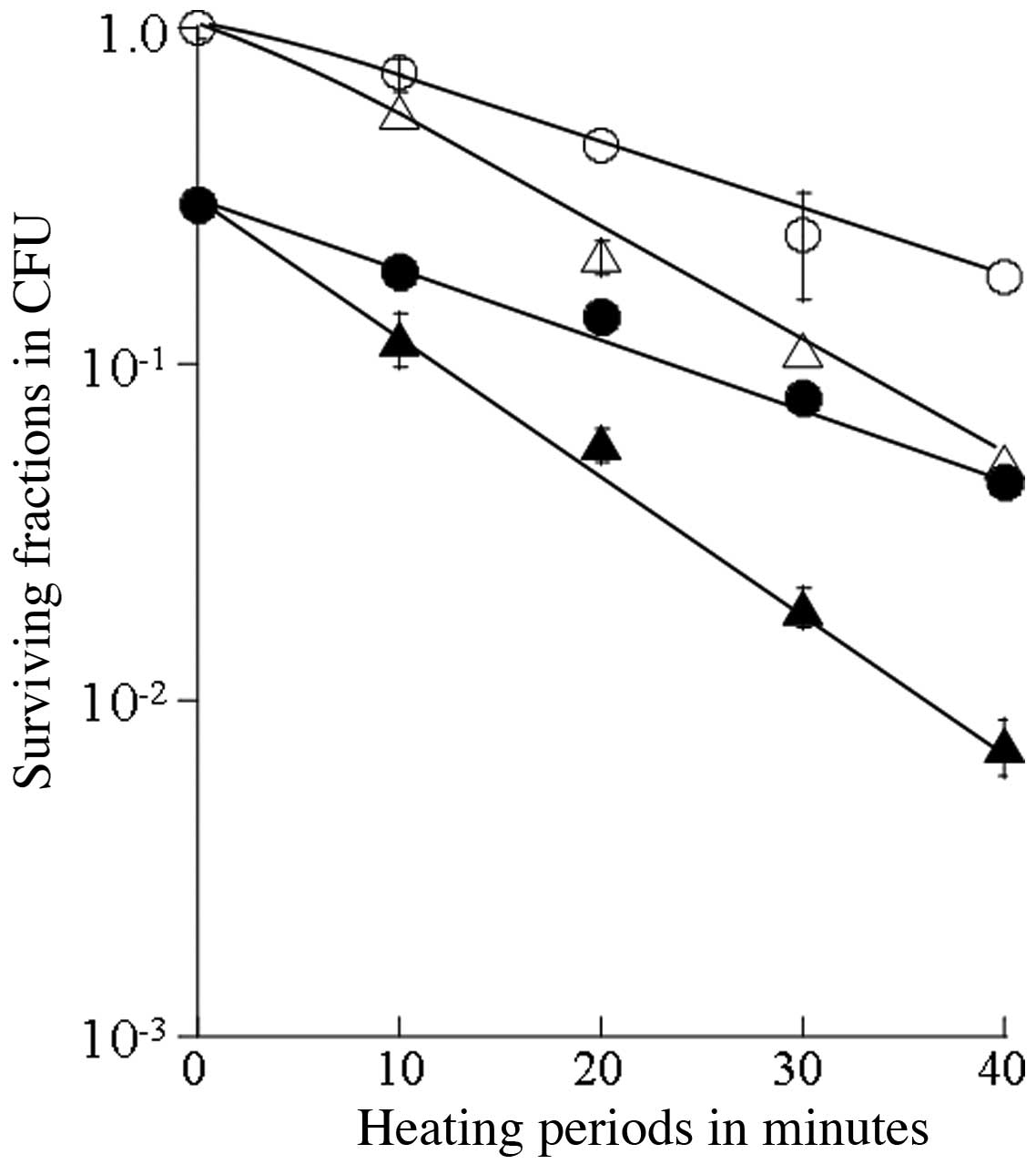
of cells heated at 43°C or 44°C and of those receiving combined
treatment with SA was estimated for up to a 40-min heating period
(Fig. 2). The T0 value
was 20.2 min for heating at 43°C and 12.4 min for the combined
treatment with SA and heating at 43°C, 14.4 min for heating at
44°C, and 10.6 min for the combined treatment with SA and heating
at 44°C (Table I). Thus, the
synergistic thermosensitizing effects were observed with the
combination of thermotherapy at 40–44°C and SA.
 | Table IEnhancement ratio based on
T0 values for survival of DU145 cells after subjection
to hyperthermia and the combination of hyperthermia and SA. |
Table I
Enhancement ratio based on
T0 values for survival of DU145 cells after subjection
to hyperthermia and the combination of hyperthermia and SA.
| Temp. | Hyperthermia
T01 | SA and hyperthermia
T02 | Enhancement ratio
T01/T02 |
|---|
| 40°C | 39.81 h | 9.55 h | 4.16 |
| 42°C | 2.12 h | 1.29 h | 1.64 |
| 43°C | 20.2 min | 12.4 min | 1.63 |
| 44°C | 14.4 min | 10.6 min | 1.36 |
Apoptosis assay and cell cycle
distribution
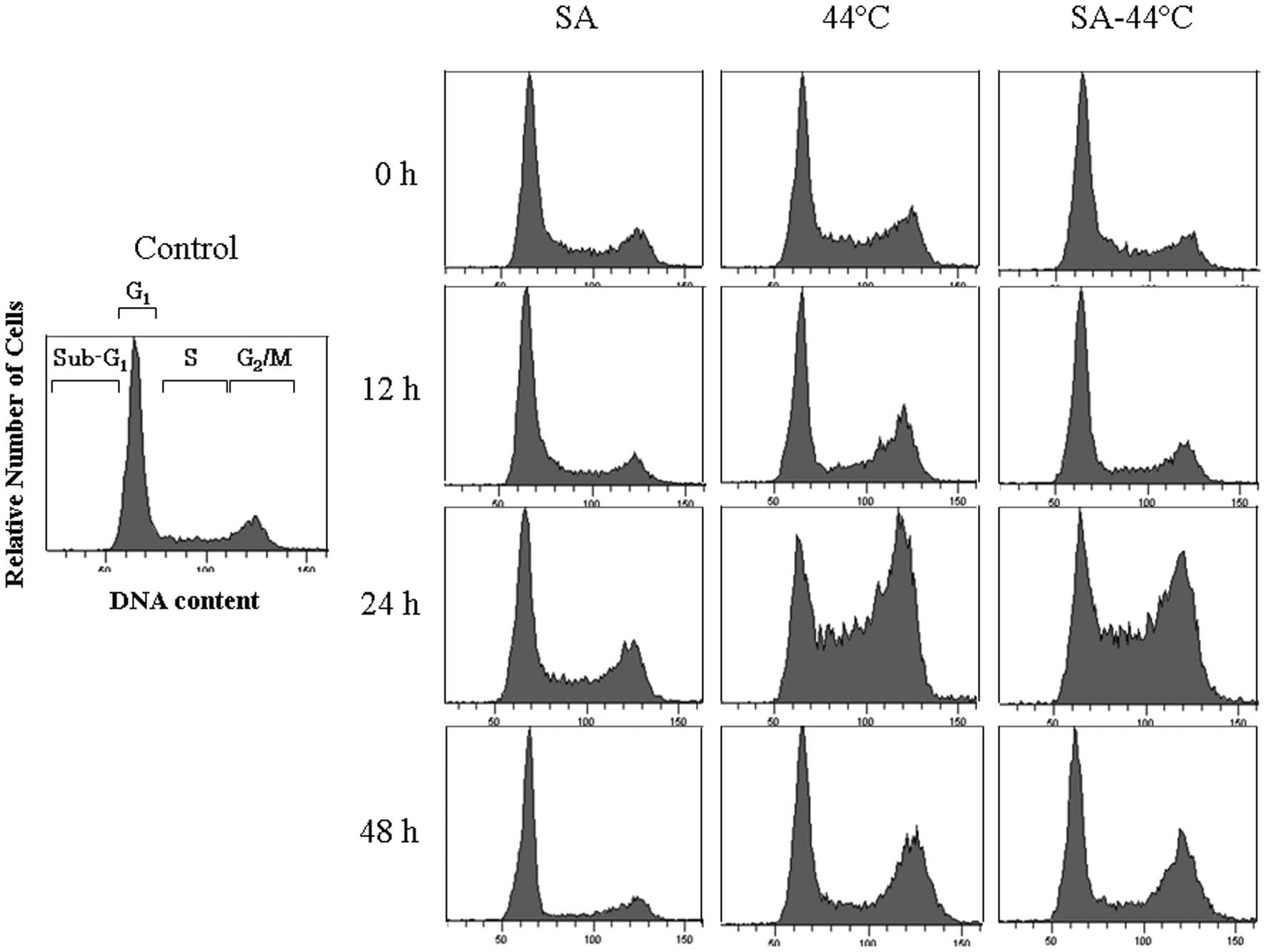
Cell cycle distribution at 0, 12, 24 and 48 h after
treatment with 20.0 μM SA for 4 h, 44°C hyperthermia for 30 min,
and their combination was examined by flow cytometry, to examine
the induction of apoptosis and cell cycle arrest in relation to the
thermosensitivity of DU145 cells. Representative histograms showing
the population distribution (based on DNA content as measured by
PI) for the cell cycle phases (sub-G1, G1 and
G2/M) are shown in Fig.
3. The distribution of cells in the sub-G1
(apoptotic cell population), the G1 and G2/M
phases are expressed as a percentage (%) of the coefficient of
variation (CV) values (Table
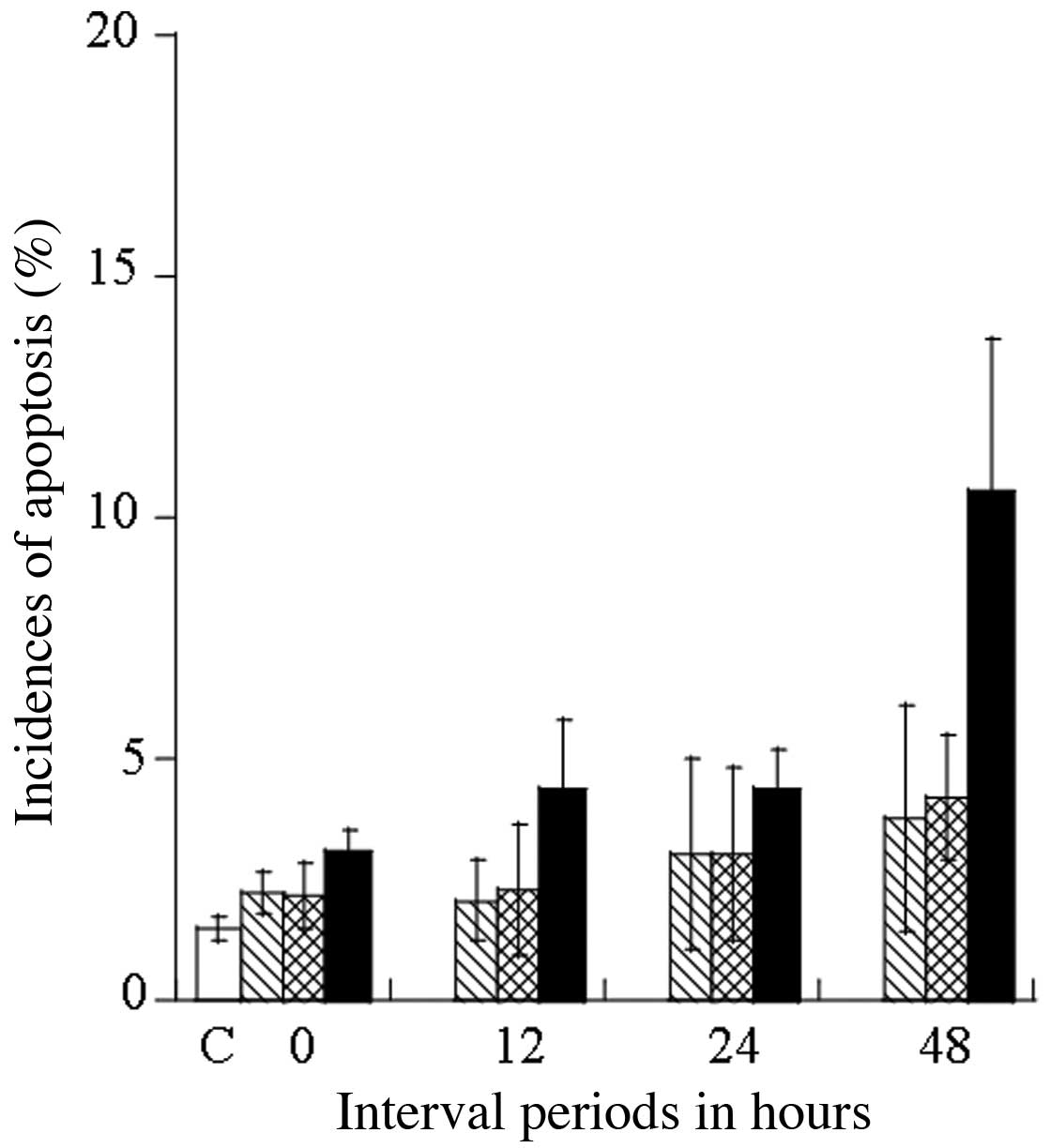
II). The induction of apoptosis in the DU145 cells was
estimated after 48 h by determining the number of cells in the
sub-G1 phase: 1.50±0.25% for the control, at 3.75±2.35%
for SA treatment alone, at 4.20±1.30% for 44°C heating treatment
alone, and at 10.53±5.02% for the treatment combining SA and
heating at 44°C (Table II). With
the combined treatment, SA significantly enhanced heat-induced
apoptosis in the DU145 cells, which was estimated to be
approximately 2.0-fold greater than when heating alone was applied.
Fig. 4 shows the kinetics of
apoptosis induction up to 48 h after treatment expressed as a
percentage (%) of apoptotic cells at the sub-G1 phase.
The distribution (%) of DU145 cells at the G2/M phase
was significantly increased following heating at 44°C (25.9±6.86%),
and following the combined treatment of heating at 44°C and SA
(24.4±4.78%), as compared with the control (19.6±6.36%) (Table II). The distribution (%) of DU145
cells at the G2/M phase was markedly increased at 24 h
after heating at 44°C, as well as after the combined treatment with
SA (Fig. 3). Taken together,
these results indicate that treatment with heating at 44°C and the
combination of heating at 44°C and SA exert thermosensitizing
effects by inducing cell cycle arrest at the G2/M phase
and apoptosis.
| Table IIDistribution (%) of DU145 cell
population in different phases of the cell cycle following
treatment with SA, hyperthermia at 44°C, and the combination of
both. |
Table II
Distribution (%) of DU145 cell
population in different phases of the cell cycle following
treatment with SA, hyperthermia at 44°C, and the combination of
both.
| Cell cycle
phase | Interval periods
(h) | Control | SA | Hyperthermia at
44°C | SA + hyperthermia
at 44°C |
|---|
|
Sub-G1 | 0 | 1.5±0.25 | 2.23±0.45 | 2.17±0.66 | 3.10±0.40 |
| 12 | | 2.05±0.83 | 2.30±1.37 | 4.37±1.46 |
| 24 | | 3.03±1.97 | 3.02±1.77 | 4.40±0.80 |
| 48 | | 3.75±2.35 | 4.20±1.30 | 10.53±5.02 |
| Average | | 2.77±1.40 | 2.92±1.27 | 5.60±1.92 |
| G1 | 0 | 47.5±9.89 | 44.65±9.64 | 38.60±2.00 | 43.00±8.60 |
| 12 | | 48.72±3.46 | 41.47±5.20 | 45.77±5.24 |
| 24 | | 49.37±10.4 | 22.2±7.15 | 23.10±3.46 |
| 48 | | 57.60±8.74 | 30.02±3.00 | 30.82±6.70 |
| Average | | 50.08±8.06 | 33.07±4.34 | 35.67±6.00 |
|
G2/M |
| 0 | 19.6±6.36 | 23.7±6.82 | 22.2±5.32 | 21.6±6.47 |
| 12 | | 17.8±4.40 | 22.25±4.55 | 20.0±3.94 |
| 24 | | 19.4±3.99 | 34.6±9.70 | 28.6±3.34 |
| 48 | | 15.2±1.82 | 24.7±7.89 | 27.42±5.37 |
| Average | | 19.0±4.26 | 25.9±6.86 | 24.4±4.78 |
Western blot analysis of the MAPK cascade
in response to treatment with SA and heating
We investigated the potential involvement of the
Ras/Raf/MAPK and PI3K/Akt pathways in SA- and/or heat-induced
apoptosis and cell cycle arrest in DU145 cells. The levels of
constitutively activated proteins, as well as those of p-ERK1/2,
p-SAPK/p-JNK and p-Akt in the MAPK cascade were assayed at 0, 12,
24 and 48 h following treatment with 20.0 μM of SA for 4 h,
hyperthermia at 44°C for 30 min, and their combination by western
blot analysis (Fig. 5). ERK1/2
levels were not increased in the DU145 cells by any of the
treatments, while the phosphorylation of ERK1/2 was suppressed
following combined treatment with SA and heating, since it appeared
to localize in the cell nuclei in smaller quantities (Fig. 5). Furthermore, the SAPK/JNK levels
were not increased after any of the treatments. However, the level
of p-SAPK/p-JNK was markedly increased immediately after treatment
with heating at 44°C and after the combined treatment (Fig. 5). In addition, the levels of Akt
markedly increased at 12, 24 and 48 h after treatment with SA and
increased even further at 0, 12 and 24 h following treatment with
heating at 44°C, whereas the combination of SA and heating resulted
in a slight increase at 48 h after treatment; this increase was
less pronounced than the one observed with heating alone. Finally,
the level of p-Akt/p-PKB was increased only immediately after
treatment with heating at 44°C.
 | Figure 5Western blot analysis of cellular
amounts of extracellular-regulated kinase (ERK)1/2, phosphorylated
(p-)ERK1/2, stress-activated protein kinase/c-Jun N-terminal kinase
(SAPK/JNK), p-SAPK/JNK, Akt and p-Akt in DU145 cells following
treatment with 20.0 μM sinodielide A (SA) for 4 h, hyperthermia at
44°C for 30 min, and the combination of these treatments. The
numbers 0, 12, 24 and 48 on each well denote intervals of
incubation at 37°C, expressed in h after treatment, while ‘C’
represents the untreated control. |
Discussion
If thermal sensitizers are found to be effective in
clinical anticancer thermotherapy by enhancing thermosensitivity or
by significantly reducing thermotolerance without any side-effects,
then they can be used in anticancer therapy for certain patients.
We previously studied various chemicals, such as parthenolide,
adriamycin (doxorubicin), bleomycin, cisplatin and amrubicin and
its metabolite, amrubicinol, with regards to their ability to
modify the effects of hyperthermia at the kinetic and molecular
level (4–6,26–29). Cellular thermosensitivity was
acquired through the combined treatment with parthenolide, an NF-κB
inhibitor, and hyperthermia in A549 human non-small cell lung
adenocarcinoma cells bearing the wild-type p53 gene. The
mechanisms underlying this effect appeared to involve the induction
of apoptosis by the direct suppression of NF-κB in a
p53-independent and heat-induced hsp72-independent manner via the
NF-κB signaling pathway (4).
Combined therapy using parthenolide and hyperthermia blocked the
re-activation of NF-κB by the heat-induced hsp72 protein, and we
suggested that parthenolide contributed to thermosensitization via
another pathway related to NF-κB signaling or by crosstalk with
another mediator gene (5). In PC3
cells, parthenolide in combination with hyperthermia gradually
activated all phosphorylation cascades, with the most prominent
increase observed for p-ERK1/2 and p-p38, which were associated
with the induction of apoptosis or G2/M cell cycle
arrest mainly via the ERK1/2 and p38 cascades (6). Parthenolide in combination with
hyperthermia activated p-p38 and p-SAPK/p-JNK in DU145 cells, an
effect associated with the induction of apoptosis and
G2/M cell cycle arrest mainly via the SAPK/JNK cascades.
The contrasting results between the two cell lines may be due to
the fact that PC3 cells are null for the p53 gene, while
DU145 cells possess a mutant copy of p53. Furthermore,
parthenolide- and heat-induced apoptosis and G2/M cell
cycle arrest in PC3 cells that do not have the p53 gene
occurred via the ERK1/2 and p38 MAPK signaling cascades with the
upregulation of NF-κB, irrespective of p53, while the same effects
in DU145 cells possessing a mutant p53 copy may have been
caused by a slight p53-dependent thermoresistance. In the
present study, we investigated the medicinal herbal compound, SA,
which has been shown to have few side-effects (1), for use in thermo-chemotherapy; the
long-term aim was to identify molecules exerting therapeutic
effects, while overcoming problems associated with thermotolerance
and/or drug resistance. In addition, we examined the mechanisms
involved in the in vitro interaction of hyperthermia and
chemotherapy at the kinetic and molecular level, using DU145 human
prostate cancer cells.
Thermosensitizing effects of SA combined
with heating at 40–44°C
The survival curve for heating at 40°C indicated a
slight heat-induced cytotoxicity for up to 6 h, while treatment
with 20.0 μM SA for 4 h prior to heating at 40°C enhanced the
antitumor effects in a synergistic manner. The T0 value
was 39.8 h for heating treatment at 40°C and 9.55 h for the
sequential combination of SA and heating at 40°C (Table I). Similarly, the
thermosensitizing effects became more prominent with heating at
42°C, showing a T0 value of 2.12 h, while T0
was 1.29 h for the sequential treatment with SA and heating at
42°C. Moreover, the T0 values for heating at higher
temperatures were 20.2 min for 43°C and 14.4 min for 44°C, while
they were 12.4 min for the combination of SA and heating at 43°C,
and 10.6 min for SA and heating at 44°C. The thermal enhancement
ratios for SA at 40, 42, 43 and 44°C were 4.16, 1.64, 1.63 and
1.36, respectively. These results demonstrated that the
thermosensitizing effects were more pronounced with the combination
therapy with SA and lower temperatures; in particular, the use of
mild hyperthermia and SA reduced the heating time required to
obtain effects equal to those induced by hyperthermia alone.
Apoptosis assay and cell cycle
distribution
A number of studies on gene and signaling pathways
related to the induction of apoptosis have been presented. Mayo
et al (30) reported that
the activation of NF-κB suppressed the induction of p53-independent
apoptosis, while Kalra et al (31) found that apoptosis in prostate
cancer cells was mediated through two related pathways; the
upregulation of p53 and the downregulation of NF-κB.
Hyperthermia-induced apoptosis has also been found to be mediated
by caspase-3 (32). Furthermore,
other studies have demonstrated that the inhibition of NF-κB
induces apoptosis and cell cycle arrest in the G2/M
phase through various signaling pathways. In addition, the
inhibition of the radiation-induced activation of NF-κB in prostate
cancer cells has been shown to promote apoptosis and
G2/M cell cycle arrest, which correlates with increased
p21/WAF1/Cip1 and decreased cyclin B1 expression (33). In another study, the viability of
HeLa cells was reduced by inducing cell cycle arrest at the
G2/M phase and mitochondrial apoptosis through the
p53-dependent expression of proteins of the Bcl-2 family
(34). In addition, the pathway
for NF-κB/caspase activation was shown to be independent of the
NF-κB/cell cycle pathway, and the events downstream of the
NF-κB/caspase-9 cascade have been shown to lead to apoptosis
(35). In the present study,
using flow cytometry, we examined the kinetics of apoptosis at
various periods of time following exposure to SA (20.0 μM) for 4 h,
hyperthermia at 44°C for 30 min, and the combination of these
treatments using DU145 cells (Fig.
3). The distribution of the cell populations at the
sub-G1 (apoptotic cell population), G1, and
G2/M phases after these treatments is shown in Table II. The induction of apoptosis is
indicated by the average percentage of cells at the
sub-G1 phase at 48 h and was estimated at 1.50±0.25% for
the control, 2.77±1.40% for the SA-treated cells, 2.92±1.27% for
the 44°C heat-treated cells, and 5.60±1.92% for the combination of
these treatments (Table II).
When used in combination with heating, SA markedly enhanced
heat-induced apoptosis in DU145 cells by approximately 1.9-fold, as
compared with heating treatment alone. The thermosensitizing
effects, estimated by examining the induction of apoptosis in DU145
cells (percentage of cells in the sub-G1, G1
and G2/M phase) became more prominent with time. The DNA
content at the G2/M and G1 fractions in the
flow cytometry distributions indicated the amount of cells in cell
cycle arrest. The greatest increase was observed for cells in
G1 arrest following treatment with SA (50.08±8.06%),
while the percentage of cells in G2/M arrest was
increased by both heating at 44°C and the combined treatment to
25.9±6.86 and 24.4±4.78%, respectively, as compared with the
control (Table II). In addition,
the relative amount of DNA content in the G2/M cell
fraction after heating at 44°C and the combined treatment with SA
was relatively high at 24 h (Fig.
3).
Examination of MAPK and Akt cascades by
western blot analysis
Ras regulates multiple downstream effector
pathways, such as Raf/MAP/ERK, PI3K/Akt and JNK/p38 (36–41). The Ras/Raf/MAPK cascade promotes
mitogen activation and cell growth via the EGFR signaling pathway,
which mainly participates in proliferation, inhibition of
apoptosis, infiltration and metastasis (42,43). Among a number of intracellular
signal transduction pathways, the most relevant to cancer
development are the MAPK/ERK (growth signaling) and PI3K/Akt
(survival signaling) cascades regulating the downstream gene,
ras. A previous study demonstrated that DU145 cells possess
mutant the ras gene (44).
In this study, we examined the constitutional and phosphorylated
activation of ERK1/2, JNK/SAPK and Akt in cascades related to the
ras signaling pathway, regulating cell cycle check points
and transcription factors, in order to investigate the mechanisms
underlying the thermosensitizing effects of the combination of SA
and hyperthermia in DU145 cells possessing mutant ras
copies. The dimerization product of p-ERK1 and p-ERK2, ERK1/2,
translocates from the cytoplasm to the nucleus to activate the
transcription factor, NF-κB (45,46). However, another study suggested
that ERK induces neuronal apoptosis (47). ERK1/2 in DU145 cells was not
increased following treatment with SA alone, heating at 44°C, or
the combination of both, while ERK1/2 phosphorylation was
suppressed by treatment with the combination of SA and heating and
was localized in small quantities in the nucleus. By contrast, the
SAPK/JNK level was increased by each of these treatments, with the
most prominent increase of its phosphorylated form occurring
immediately after treatment with heating at 44°C and after the
combined treatment with SA (Fig.
5). The JNK signaling pathway induces apoptosis through diverse
mediators (48–51). We found that the Akt level was
markedly increased at 12, 24 and 48 h following treatment with SA,
and at 0, 12 and 24 h following heating at 44°C. By contrast, the
combination of these treatments only slightly increased the Akt
level for up to 48 h after treatment. Furthermore, p-Akt levels
were increased only immediately after heating at 44°C. Thus, SA
further contributed to thermosensitization by inducing apoptosis
and G2/M arrest by inhibiting the phosphorylation of
Akt. The PI3K/Akt pathway regulates apoptosis, a process where
kinetics are either pro- or anti-apoptotic, depending on the type
of stimuli and circumstances (52,53). Numerous studies on the PI3K/Akt
pathway have indicated that its inhibition can inhibit the
proliferation of cancer cells (54–57). We conclude that SA exerts its
thermosensitizing effects on DU145 cells by inhibiting the
activation of the ERK1/2 and PI3K/Akt signaling pathways and
promoting apoptosis via the JNK signaling cascade.
Acknowledgements
The present study was supported in part by a
Grant-in-Aid for Scientific Research (C) (no. 23591854) from the
Ministry of Education, Science and Culture of Japan, for
elucidation of the mechanism of proton beam-specific cellular
responses in radiotherapy, 2011–2013. Support was also provided by
an incorporation grant from the Suzuki Urology Promotion Research.
We express our gratitude to Ms. Junko Yamamoto, Division of
Bioresearch, Faculty of Medical Science, University of Fukui,
Japan, for her assistance with the measurements and processing of
the flow cytometry data, as well as with the western blot analysis
procedures.
References
|
1
|
Wang NH, Taniguchi M, Tsuji D, Doi M,
Ohishi H, Yoza K and Baba K: Four guaianolides from Sinodielsia
yunnanensis. Chem Pharm Bull (Tokyo). 51:68–70. 2003.
View Article : Google Scholar
|
|
2
|
Kimura Y, Sumiyoshi M and Baba K:
Anti-tumor actions of major component 3′-O-acetylhamaudol of
Angelica japonica roots through dual actions,
anti-angiogenesis and intestinal intraepithelial lymphocyte
activation. Cancer Lett. 265:84–97. 2008.PubMed/NCBI
|
|
3
|
Sugii M, Ohkita M, Taniguchi M, Baba K,
Kawai Y, Tahara C, Takaoka M and Matsumura Y: Xanthoangelol D
isolated from the roots of Angelica keiskei inhibits
endothelin-1 production through the suppression of nuclear
factor-kappaB. Biol Pharm Bull. 28:607–610. 2005.
|
|
4
|
Hayashi S, Hatashita M, Hayashi A,
Matsumoto H, Shioura H and Kitai R: Thermosensitization by
parthenolide in human lung adenocarcinoma A549 cells and p53- and
hsp72-independent apoptosis induction via the nuclear factor-κB
signal pathway. Int J Mol Med. 21:585–592. 2008.PubMed/NCBI
|
|
5
|
Hayashi S, Sakurai H, Hayashi A, Tanaka Y,
Hatashita M and Shioura H: Inhibition of NF-κB by combination
therapy with parthenolide and hyperthermia and kinetics of
apoptosis induction and cell cycle arrest in human lung
adenocarcinoma cells. Int J Mol Med. 25:81–87. 2010.
|
|
6
|
Hayashi S, Koshiba K, Hatashita M, Sato T,
Jujo Y, Suzuki R, Tanaka Y and Shioura H: Thermosensitization and
induction of apoptosis or cell-cycle arrest via the MAPK cascade by
parthenolide, an NF-κB inhibitor, in human prostate cancer
androgen-independent cell lines. Int J Mol Med. 28:1033–1042.
2011.
|
|
7
|
Storer RD, Stein RB, Sina JF, DeLuca JG,
Allen HL and Bradley MO: Malignant transformation of preneoplastic
hamster epidermal cell line by the EJ c-Ha-ras-oncogene. Cancer
Res. 46:1458–1464. 1986.PubMed/NCBI
|
|
8
|
Cameron S, Levin L, Zoller M and Wigler M:
cAMP-independent control of sporulation, glycogen metabolism, and
heat shock resistance in S. cerevisiae. Cell. 53:555–566.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shirayama M, Kawakami K, Matsui Y, Tanaka
K and Toh-e A: MSI3, a multicopy suppressor of mutants
hyperactivated in the RAS-cAMP pathway, encodes a novel HSP70
protein of Saccharomyces cerevisiae. Mol Gen Genet.
240:323–332. 1993.PubMed/NCBI
|
|
10
|
Mivechi NF and Giaccia AJ:
Mitogen-activated protein kinase acts as a negative regulator of
the heat shock response in NIH3T3 cells. Cancer Res. 55:5512–5519.
1995.PubMed/NCBI
|
|
11
|
Oh AS, Lorant LA, Holloway JN, Miller DL,
Kern FG and El-Ashry D: Hyperactivation of MAPK induces loss of
ERalpha expression in breast cancer cells. Mol Endocrinol.
15:1344–1359. 2001.PubMed/NCBI
|
|
12
|
Barault L, Veyrie N, Jooste V, et al:
Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH
kinase) signaling network correlate with poor survival in a
population-based series of colon cancers. Int J Cancer.
122:2255–2259. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fecher LA, Amaravadi RK and Flaherty KT:
The MAPK pathway in melanoma. Curr Opin Oncol. 20:183–189. 2008.
View Article : Google Scholar
|
|
14
|
Chen KH, Weng MS and Lin JK: Tangeretin
suppresses IL-1beta-induced cyclooxygenase (COX)-2 expression
through inhibition of p38 MAPK, JNK, and AKT activation in human
lung carcinoma cells. Biochem Pharmacol. 73:215–227. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang YQ, Jaganath I, Manikam R and Sekaran
SD: Phyllanthus suppresses prostate cancer cell, PC-3,
proliferation and induces apoptosis through multiple signalling
pathways (MAPKs, PI3K/Akt, NFκB, and hypoxia). Evid Based
Complement Alternat Med. 2013:6095812013.PubMed/NCBI
|
|
16
|
Cobb MH and Goldsmith EJ: How MAP kinases
are regulated. J Biol Chem. 270:14843–14846. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Robinson MJ and Cobb MH: Mitogen-activated
protein kinase pathways. Curr Opin Cell Biol. 9:180–186. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Widmann C, Gibson S, Jarpe MB and Johnson
GL: Mitogen-activated protein kinase: conservation of a
three-kinase module from yeast to human. Physiol Rev. 79:143–180.
1999.PubMed/NCBI
|
|
19
|
Mielke K and Herdegen T: JNK and p38
stresskinases - degenerative effectors of
signal-transduction-cascades in the nervous system. Prog Neurobiol.
61:45–60. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Whitmarsh AJ and Davis RJ: Transcription
factor AP-1 regulation by mitogen-activated protein kinase signal
transduction pathways. J Mol Med (Berl). 74:589–607. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ip YT and Davis RJ: Signal transduction by
the c-Jun N-terminal kinase (JNK) - from inflammation to
development. Curr Opin Cell Biol. 10:205–219. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ahn KS, Sethi G and Aggarwal BB: Nuclear
factor-kappaB: from clone to clinic. Curr Mol Med. 7:619–637. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Majumder PK and Sellers WR: Akt-regulated
pathways in prostate cancer. Oncogene. 24:7465–7474. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hayashi S, Kano E, Tsuji K,
Furukawa-Furuya M, Yoshikawa S, Hatashita M, Matsumoto H, Jin ZH,
Ohtsubo T and Kitai R: Modification of thermosensitivity and
chemosensitivity induced by combined treatments with hyperthermia
and adriamycin. Int J Mol Med. 8:417–422. 2001.PubMed/NCBI
|
|
27
|
Shioura H, Hayashi S, Matsumoto H, Kitai
R, Ohtsubo T, Nishida T, Zhang SW, Yoshida M, Ishii Y and Kano E:
The effects of combined treatments with low hyperthermia and
bleomycin on survivals of murine L cells. J Exp Clin Cancer Res.
16:147–152. 1997.
|
|
28
|
Ohtsubo T, Saito H, Matsumoto H, Hayashi
S, Shioura H, Kitai R, Saito T and Kano E: In vitro effects of
hyperthermia combined with cisplatin or peplomycin on the human
maxillary carcinoma cell line IMC-2. Int J Hyperthermia. 13:59–67.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hayashi S, Hatashita M, Matsumoto H, Jin
ZH, Shioura H and Kano E: Modification of thermosensitivity by
amrubicin or amrubicinol in human lung adenocarcinoma A549 cells
and the kinetics of apoptosis and necrosis induction. Int J Mol
Med. 16:381–387. 2005.PubMed/NCBI
|
|
30
|
Mayo MW, Wang CY, Cogswell PC,
Rogers-Graham KS, Lowe SW, Der CJ and Baldwin AS Jr: Requirement of
NF-kappaB activation to suppress p53-independent apoptosis induced
by oncogenic Ras. Science. 278:1812–1815. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kalra N, Seth K, Prasad S, Singh M, Pant
AB and Shukla Y: Theaflavins induced apoptosis of LNCaP cells is
mediated through induction of p53, down-regulation of NF-kappa B
and mitogen-activated protein kinases pathways. Life Sci.
80:2137–2146. 2007. View Article : Google Scholar
|
|
32
|
Vertrees RA, Das GC, Coscio AM, Xie J,
Zwischenberger JB and Boor PJ: A mechanism of hyperthermia-induced
apoptosis in ras-transformed lung cells. Mol Carcinog. 44:111–121.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Raffoul JJ, Wang Y, Kucuk O, Forman JD,
Sarkar FH and Hillman GG: Genistein inhibits radiation-induced
activation of NF-kappaB in prostate cancer cells promoting
apoptosis and G2/M cell cycle arrest. BMC Cancer. 6:1072006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vidya Priyadarsini R, Senthil Murugan R,
Maitreyi S, Ramalingam K, Karunagaran D and Nagini S: The flavonoid
quercetin induces cell cycle arrest and mitochondria-mediated
apoptosis in human cervical cancer (HeLa) cells through p53
induction and NF-κB inhibition. Eur J Pharmacol. 649:84–91.
2010.PubMed/NCBI
|
|
35
|
Mogi M, Ozeki N, Nakamura H and Togari A:
Dual roles for NF-kappaB activation in osteoblastic cells by serum
deprivation: osteoblastic apoptosis and cell-cycle arrest. Bone.
35:507–516. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Barbacid M: ras genes (Review). Annu Rev
Biochem. 56:779–827. 1987. View Article : Google Scholar
|
|
37
|
Bos JL: ras oncogenes in human cancer: a
review (Review). Cancer Res. 49:4682–4689. 1989.PubMed/NCBI
|
|
38
|
Serrano M, Lin AW, McCurrach ME, Beach D
and Lowe SW: Oncogenic ras provokes premature cell senescence
associated with accumulation of p53 and p16INK4a. Cell. 88:593–602.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang W, Chen JX, Liao R, Deng Q, Zhou JJ,
Huang S and Sun P: Sequential activation of the MEK-extracellular
signal-regulated kinase and MKK3/6-p38 mitogen-activated protein
kinase pathways mediates oncogenic ras-induced premature
senescence. Mol Cell Biol. 22:3389–3403. 2002. View Article : Google Scholar
|
|
40
|
Hingorani SR and Tuveson DA: Ras redux:
rethinking how and where Ras acts. Curr Opin Genet Dev. 13:6–13.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Boguski MS and McCormick F: Proteins
regulating Ras and its relatives. Nature. 366:643–654. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Benvenuti S, Sartore-Bianchi A, Di
Nicolantonio F, Zanon C, Moroni M, Veronese S, Siena S and Bardelli
A: Oncogenic activation of the RAS/RAF signaling pathway impairs
the response of metastatic colorectal cancers to anti-epidermal
growth factor receptor antibody therapies. Cancer Res.
67:2643–2648. 2007. View Article : Google Scholar
|
|
43
|
Russo A, Rizzo S, Bronte G, Silvestris N,
Colucci G, Gebbia N, Bazan V and Fulfaro F: The long and winding
road to useful predictive factors for anti-EGFR therapy in
metastatic colorectal carcinoma: the KRAS/BRAF pathway. Oncology.
77(Suppl 1): 57–68. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pergolizzi RG, Kreis W, Rottach C, Susin M
and Broome JD: Mutational status of codons 12 and 13 of the N- and
K-ras genes in tissue and cell lines derived from primary and
metastatic prostate carcinomas. Cancer Invest. 11:25–32. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim-Kaneyama, Nose K and Shibanuma M:
Significance of nuclear relocalization of ERK1/2 in reactivation of
c-fos transcription and DNA synthesis in senescent fibroblasts. J
Biol Chem. 275:20685–20692. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Godeny MD and Sayeski PP: ANG II-induced
cell proliferation is dually mediated by c-Src/Yes/Fyn-regulated
ERK1/2 activation in the cytoplasm and PKCzeta-controlled ERK1/2
activity within the nucleus. Am J Physiol Cell Physiol.
291:C1297–C1307. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cheung EC and Slack RS: Emerging role for
ERK as a key regulator of neuronal apoptosis. Sci STKE.
2004:PE452004.PubMed/NCBI
|
|
48
|
Aloyz RS, Bamji SX, Pozniak CD, Toma JG,
Atwal J, Kaplan DR and Miller FD: p53 is essential for
developmental neuron death as regulated by the TrkA and p75
neurotrophin receptors. J Cell Biol. 143:1691–1703. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Leppä S and Bohmann D: Diverse functions
of JNK signaling and c-Jun in stress response and apoptosis.
Oncogene. 18:6158–6162. 1999.PubMed/NCBI
|
|
50
|
Shen HM and Liu ZG: JNK signaling pathway
is a key modulator in cell death mediated by reactive oxygen and
nitrogen species. Free Radic Biol Med. 40:928–939. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim HJ, Chakravarti N, Oridate N, Choe C,
Claret FX and Lotan R: N-(4-hydroxyphenyl)retinamide-induced
apoptosis triggered by reactive oxygen species is mediated by
activation of MAPKs in head and neck squamous carcinoma cells.
Oncogene. 25:2785–2794. 2006. View Article : Google Scholar
|
|
52
|
Rosner D, Stoneman V, Littlewood T,
McCarthy N, Figg N, Wang Y, Tellides G and Bennett M:
Interferon-gamma induces Fas trafficking and sensitization to
apoptosis in vascular smooth muscle cells via a PI3K- and
Akt-dependent mechanism. Am J Pathol. 168:2054–2063. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lu B, Wang L, Stehlik C, Medan D, Huang C,
Hu S, Chen F, Shi X and Rojanasakul Y: Phosphatidylinositol
3-kinase/Akt positively regulates Fas (CD95)-mediated apoptosis in
epidermal Cl41 cells. J Immunol. 176:6785–6793. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Aksamitiene E, Kiyatkin A and Kholodenko
BN: Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt
pathways: a fine balance. Biochem Soc Trans. 40:139–146. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ho AL and Sherman E: Clinical development
of kinase inhibitors for the treatment of differentiated thyroid
cancer. Clin Adv Hematol Oncol. 9:32–41. 2011.PubMed/NCBI
|
|
56
|
Liu D and Xing M: Potent inhibition of
thyroid cancer cells by the MEK inhibitor PD0325901 and its
potentiation by suppression of the PI3K and NF-kappaB pathways.
Thyroid. 18:853–864. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Brzezianska E and Pastuszak-Lewandoska D:
A minireview: the role of MAPK/ERK and PI3K/Akt pathways in thyroid
follicular cell-derived neoplasm (Review). Front Biosci (Landmark
Ed). 16:422–439. 2011. View
Article : Google Scholar : PubMed/NCBI
|