Introduction
Dilated cardiomyopathy (DCM) is the most common form
of primary heart muscle disease characterized by dilation and
systolic dysfunction of the left or both ventricles with normal
ventricular wall thickness, in the absence of underlying conditions
such as coronary artery disease, hypertension and valvular
anomalies (1). It is the most
frequent cause of congestive heart failure and sudden cardiac death
in the young and is the most common indication for cardiac
transplantation in both child and adult patients worldwide
(1,2). DCM may occur as a result of acquired
factors, including infectious agents, toxins, nutritional
deficiencies and autoimmune disorders (1). However, in approximately 50% of DCM
cases, no acquired risk factors can be found, and such DCM is
defined as idiopathic DCM, of which 25–50% are familial with at
least two affected close relatives in each family, hence termed
familial DCM (3). A growing body
of evidence reveals a genetic origin in many patients with
idiopathic DCM, and a long list of mutations in more than 50 genes
has been associated with familial DCM (3). However, these established
DCM-associated genes only account for roughly one-third of cases,
and each gene has a low mutational frequency, with most occurring
in less than 1% of patients with DCM (4). Therefore, continued genetic studies
in other cohorts of DCM patients are necessary in order to gain
novel insight into the molecular basis for HCM.
The zinc finger-containing transcription factor
GATA4 is abundantly expressed in the heart at various developmental
stages and continues to be highly expressed in adult
cardiomyocytes, where it mediates the expression of several crucial
structural and regulatory genes, including those encoding atrial
natriuretic factor (ANF), brain natriuretic factor, carnitine
palmitoyltransferase Iβ, troponin I, troponin C, α- and β-myosin
heavy chain (5–7). In humans, a great number of
mutations in the GATA4 gene have been implicated in a wide
variety of congenital cardiovascular malformations, including
atrial septal defect, ventricular septal defect, tetralogy of
Fallot, endocardial cushion defect, patent ductus arteriosus,
pulmonary stenosis and hypoplastic right ventricle (8–16),
highlighting the pivotal role of GATA4 in human cardiogenesis.
Recently, a novel GATA4 mutation was reported to underlie
familial DCM (17). In mice,
GATA4 has been substantiated to be essential for proper
cardiovascular morphogenesis, and homozygous GATA4 deletion
causes early embryonic lethality due to abnormal embryogenesis and
failure to form heart tube (18,19). Mice expressing 70% less GATA4
protein died between day 13.5 and 16.5 of gestation, and common
atrioventricular canal, double outlet right ventricle and
hypoplastic ventricular myocardium were observed in these embryos
(20). Moreover, transgenic mice
expressing GATA4 mutants suffered from various cardiac
deformations, including septal defects, right ventricular
hypoplasia, endocardial cushion defect, tetralogy of Fallot, double
outlets of the right ventricle and cardiomyopathy, similar to
abnormalities occurring in humans (9). More importantly, mice with
cardiac-specific deletion of GATA4 survived into adulthood
but showed progressive cardiac enlargement and functional
impairment with increased rates of cardiomyocyte apoptosis that was
positively correlated to GATA4 levels (21). These results demonstrate that
GATA4 plays a key role in maintaining physiological homeostatic
remodeling in adult hearts by promoting cell survival and
regeneration and inhibiting programmed cell death (22–26).
GATA4 regulates downstream gene expression by
forming complexes with other transcriptional factors, including
NKX2-5 and TBX5 (7). NKX2-5 is
another regulator required for normal cardiac development and its
expression and functions overlap with those of GATA4 during
embryogenesis (12). Furthermore,
GATA4 and NKX2-5 have been shown to physically interact and
synergistically regulate the expression of multiple important
cardiac target genes, including those coding for ANF, T- and L-type
Ca2+ channels, connexin40, α-actin, ID2 and LRRC10
(7). In mice, targeted disruption
of NKX2-5 led to impaired cardiac growth and chamber
formation, deranged gene regulatory network, and early embryonic
death, while cardiac-specific knockout of NKX2-5 resulted in
progressive cardiomyopathy and complete heart block (27–29). In humans, mutations in the NKX2-5
gene have been linked to diverse congenital heart diseases,
including cardiac septal defects, tetralogy of Fallot,
transposition of the great arteries, hypoplastic left heart,
valvular deformities and left ventricular contractile dysfunction
(30–32), and adult-onset DCM (33). These data suggest that
GATA4 is an alternative candidate gene for DCM.
Materials and methods
Patients and controls
A cohort of 150 unrelated patients with idiopathic
DCM was recruited from the Han Chinese population. The available
relatives of the index patients were also enlisted. A total of 200
ethnically matched unrelated healthy individuals were enrolled as
controls. All participants were evaluated by detailed medical
history, physical examination, chest radiography,
electrocardiogram, echocardiography and exercise performance
testing. Cardiac catheterization, angiography, endomyocardial
biopsy and cardiac magnetic resonance imaging were performed only
if there was a strong clinical indication. Medical records were
also reviewed in the case of deceased or unavailable relatives.
Diagnosis of idiopathic DCM was made in accordance with the
criteria established by the World Health Organization/International
Society and Federation of Cardiology Task Force on the
Classification of Cardiomyopathy: a left ventricular end-diastolic
diameter >27 mm/m2 and an ejection fraction <40%
or fractional shortening <25% in the absence of abnormal loading
conditions, coronary artery disease, congenital heart lesions, and
other systemic diseases (17,34). Individuals were excluded if they
had insufficient echocardiographic image quality, or coexistent
conditions that may lead to contractile dysfunction, such as
uncontrolled systemic hypertension, coronary artery disease or
valvular heart disease. Familial DCM was defined as having two or
more first-degree relatives with idiopathic DCM. Peripheral venous
blood samples from all the participants were prepared. The clinical
studies were performed with investigators blinded to the results of
the genotypes. This study conformed to the principles of the
Declaration of Helsinki, and the study protocol was approved by the
local institutional ethics committee. Written informed consent was
obtained from all participants prior to the study.
Genotyping
Genomic DNA was extracted from blood samples
obtained from all participants with the Wizard Genomic DNA
purification kit (Promega, Madison, WI, USA). The coding exons and
exon-intron boundaries of the GATA4 gene were sequenced in
150 unrelated patients with idiopathic DCM. When a mutation was
found in an index patient, the available relatives of the mutation
carrier and 200 unrelated healthy controls were genotyped for
GATA4. The referential genomic DNA sequence of GATA4
was derived from GenBank (accession no. NC_000008). The primer
pairs used to amplify the entire coding region and flanking splice
junction sites of GATA4 by polymerase chain reaction (PCR)
were designed as described previously (11). The PCR was conducted using HotStar
Taq DNA polymerase (Qiagen, Hilden, Germany) on a PE 9700 thermal
cycler (Applied Biosystems, Foster, CA, USA) with standard
conditions and concentrations of reagents. Amplified products were
purified with the QIAquick Gel Extraction kit (Qiagen). Both
strands of each PCR product were sequenced with a
BigDye® Terminator v3.1 Cycle Sequencing kit under an
ABI PRISM 3130xl DNA analyzer (both from Applied Biosystems). DNA
sequences were viewed and analyzed with the DNA Sequencing Analysis
Software v5.1 (Applied Biosystems). The variant was validated by
re-sequencing of an independent PCR-generated amplicon from the
same subject. In addition, for an identified sequence variant, the
single nucleotide polymorphism (SNP; http://www.ncbi.nlm.nih.gov/SNP) and human gene
mutation (HGM; http://www.hgmd.org) databases were
queried to confirm its novelty.
Alignment of multiple GATA4 protein
sequences
Multiple GATA4 protein sequences among species were
aligned using the online MUSCLE program, version 3.6 (http://www.ncbi.nlm.nih.gov/).
Prediction of the disease-causing
potential of a GATA4 sequence variation
The causative potential of a GATA4 sequence
variation was predicted by MutationTaster (an online program at
http://www.mutationtaster.org), which
automatically provides a probability for the variation to be either
a pathogenic mutation or a benign polymorphism. Of note, the
P-value used here is the probability of the correct prediction
rather than the probability of error as used in t-test statistics
(i.e., a value close to 1 indicates a high accuracy of the
prediction). Additionally, another online program PolyPhen-2
(http://genetics.bwh.harvard.edu/pph2)
was also used to evaluate the pathogenic likeliness of a
variation.
Expression plasmids and site-directed
mutagenesis
The recombinant expression vector GATA4-pSSRa and
the ANF-luciferase (ANF-luc) reporter plasmid, which contains the
2,600-bp 5′-flanking region of the ANF gene, were kindly
provided by Dr Ichiro Shiojima of the Chiba University School of
Medicine, Japan. The identified mutation was introduced into the
wild-type GATA4 using a QuickChange II XL Site-Directed
Mutagenesis kit (Stratagene, La Jolla, CA, USA) with a
complementary pair of primers. The mutant was sequenced to confirm
the desired mutation and to exclude any other sequence
variations.
Reporter gene assays
HeLa cells were cultured in Dulbecco’s modified
Eagle’s medium (DMEM) supplemented with 10% fetal calf serum. The
internal control reporter plasmid pGL4.75 (hRluc/CMV; Promega) was
used in transient transfection analyses to evaluate the
transcriptional activity of the GATA4 mutant. HeLa cells
were transfected with 0.4 μg of wild-type or mutant GATA4-pSSRa,
1.0 μg of ANF-luc, and 0.04 μg of pGL4.75 using PolyFect
transfection reagent (Qiagen). For co-transfection experiments, 0.2
μg of wild-type GATA4-pSSRa, 0.2 μg of mutant GATA4-pSSRa, 1.0 μg
of ANF-luc, and 0.04 μg of pGL4.75 were used. Firefly luciferase
and Renilla luciferase activities were measured with the
Dual-Glo luciferase assay system (Promega) 48 h after transfection.
The activity of the ANF promoter was presented as fold
activation of firefly luciferase relative to Renilla
luciferase. Three independent experiments were performed at minimum
for wild-type and mutant GATA4.
Statistical analysis
Data are expressed as means ± standard deviation
(SD). Continuous variables were tested for normality of
distribution, and the Student’s unpaired t-test was used for
comparison of numeric variables between two groups. Comparison of
the categorical variables between two groups was performed using
Pearson’s χ2 test or Fisher’s exact test when
appropriate. A two-tailed P-value of <0.05 indicated statistical
significance.
Results
Clinical characteristics of the study
subjects
A total of 150 unrelated patients with idiopathic
DCM were clinically evaluated in contrast to 200 control
individuals. None had apparent traditional risk factors for DCM.
All of the patients manifested with a typical DCM phenotype as
described previously (34). The
control individuals had no evidence of structural cardiac diseases,
and their echocardiogram results were normal. The baseline clinical
characteristics of the study subjects are summarized in Table I.
 | Table IBaseline clinical characteristics of
the patients and controls. |
Table I
Baseline clinical characteristics of
the patients and controls.
| Parameters | Patients
(n=150) | Controls
(n=200) | P-value |
|---|
| Mean age
(years) | 45.6±12.5 | 47.2±10.9 | 0.2029 |
| Male, n (%) | 82 (54.7) | 111 (55.5) | 0.8767 |
| Family history of
DCM, n (%) | 65 (43.3) | 0 (0) | <0.0001 |
| SBP (mmHg) | 112.8±15.3 | 120.0±13.6 | 0.0001 |
| DBP (mmHg) | 71.4±9.1 | 82.5±8.6 | <0.0001 |
| HR (bpm) | 110.5±16.4 | 78.3±10.7 | <0.0001 |
| LVEDD (mm) | 72.6±8.3 | 47.9±6.0 | <0.0001 |
| LVESD (mm) | 59.5±9.8 | 35.0±5.6 | <0.0001 |
| LVEF (%) | 34.7±10.0 | 65.2±7.4 | <0.0001 |
| NYHA function class
(%) |
| I | 23 (15.3) | NA | NA |
| II | 58 (38.7) | NA | NA |
| III | 50 (33.3) | NA | NA |
| IV | 19 (12.7) | NA | NA |
GATA4 mutation
By direct sequencing of the GATA4 gene, a
heterozygous mutation was identified in 1 out of 150 unrelated
patients with idiopathic DCM, with a mutational prevalence of
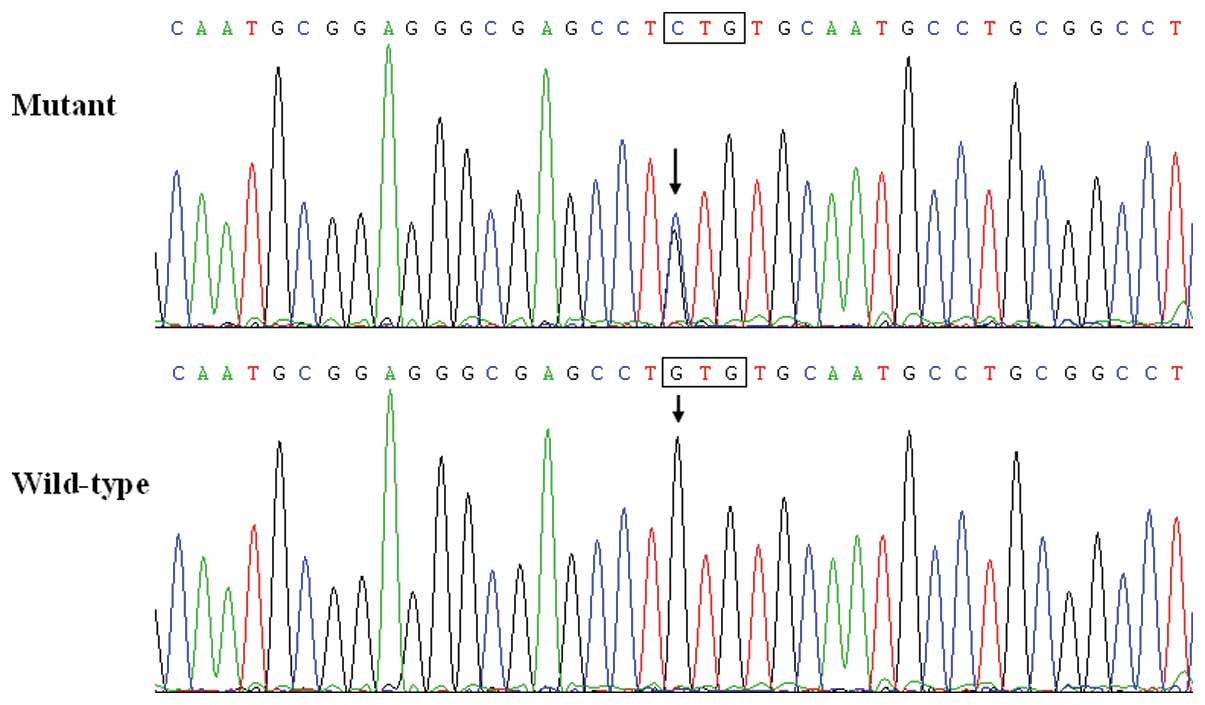
~0.67%. Specifically, a substitution of C for G in the first
nucleotide of codon 291 (c.871G>C), predicting the transition of
valine (V) into leucine (L) at amino acid position 291 (p.V291L)
was identified in the proband from family 1. The sequence
chromatograms showing the detected heterozygous GATA4
mutation of c.871G>C compared with its control sequence are
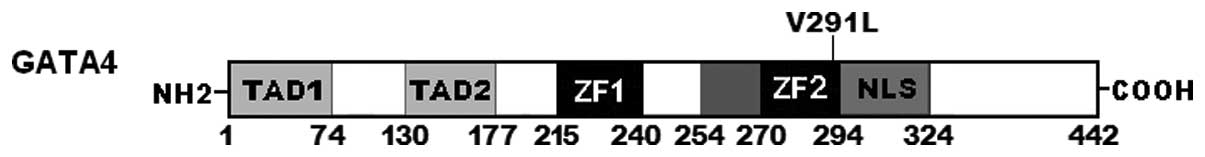
shown in Fig. 1. A schematic
diagram of GATA4 protein depicting the structural domains and
location of the mutation identified in this study is presented in
Fig. 2. The missense mutation was
neither observed in the control population nor reported in the SNP
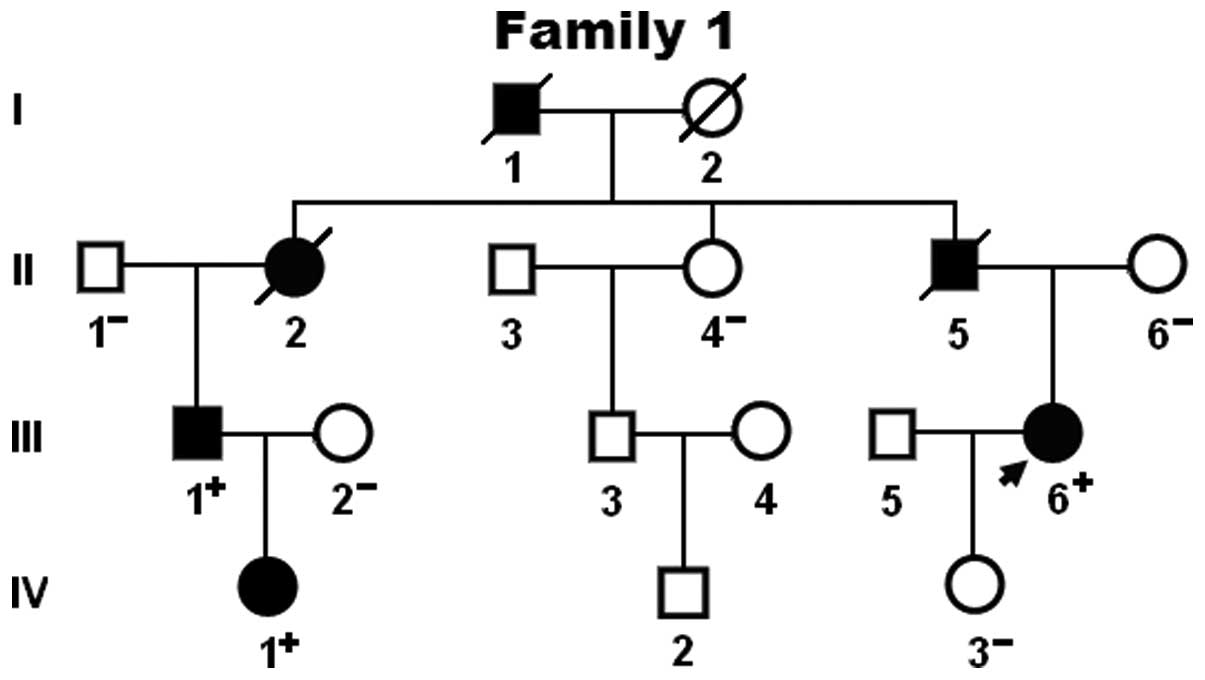
and HGM databases. The genetic scan of the family showed that the
mutation was present in all affected living family members, but
absent in the unaffected family members examined. Analysis of the
pedigree demonstrated that the mutation co-segregated with DCM was
transmitted in an autosomal dominant pattern in the family with
complete penetrance. The pedigree structure of the family is
illustrated in Fig. 3. The
phenotypic characteristics and status of the GATA4 mutation of the
affected living family members are listed in Table II.
| Table IIPhenotypic characteristics and status
of the GATA4 mutation in the affected living pedigree members. |
Table II
Phenotypic characteristics and status
of the GATA4 mutation in the affected living pedigree members.
| Individual | Gender | Age (years) | Cardiac
phenotype | LVEDD (mm) | LVESD (mm) | LVEF (%) | ECG findings | GATA4 mutation |
|---|
| III-1 | M | 53 | DCM, ASD | 75 | 68 | 32 | PAF, AVB, RBBB | +/− |
| III-6 | F | 46 | DCM | 82 | 71 | 27 | | +/− |
| IV-1 | F | 27 | DCM | 60 | 52 | 39 | | +/− |
In addition, individual III-1 and his mother (II-2)
also had documented congenital atrial septal defect and paroxysmal
atrial fibrillation as well as first-degree atrioventricular
conduction block and incomplete right bundle branch block.
Multiple alignments of GATA4 protein
sequences
A cross-species alignment of GATA4 protein sequences
showed that the altered amino acid p.V291 was completely conserved
evolutionarily (Fig. 4).
Disease-causing potential of the GATA4
variation
The GATA4 sequence variation of c.871G>C was
predicted by MutationTaster to be a disease-causing mutation with a
P-value of nearly 1.0. No SNPs in the altered region were found in
the MutationTaster database. This GATA4 sequence variation
was also predicted by PolyPhen-2 to be probably damaging, with a
score of 0.997 (sensitivity 0.41; specificity 0.98).
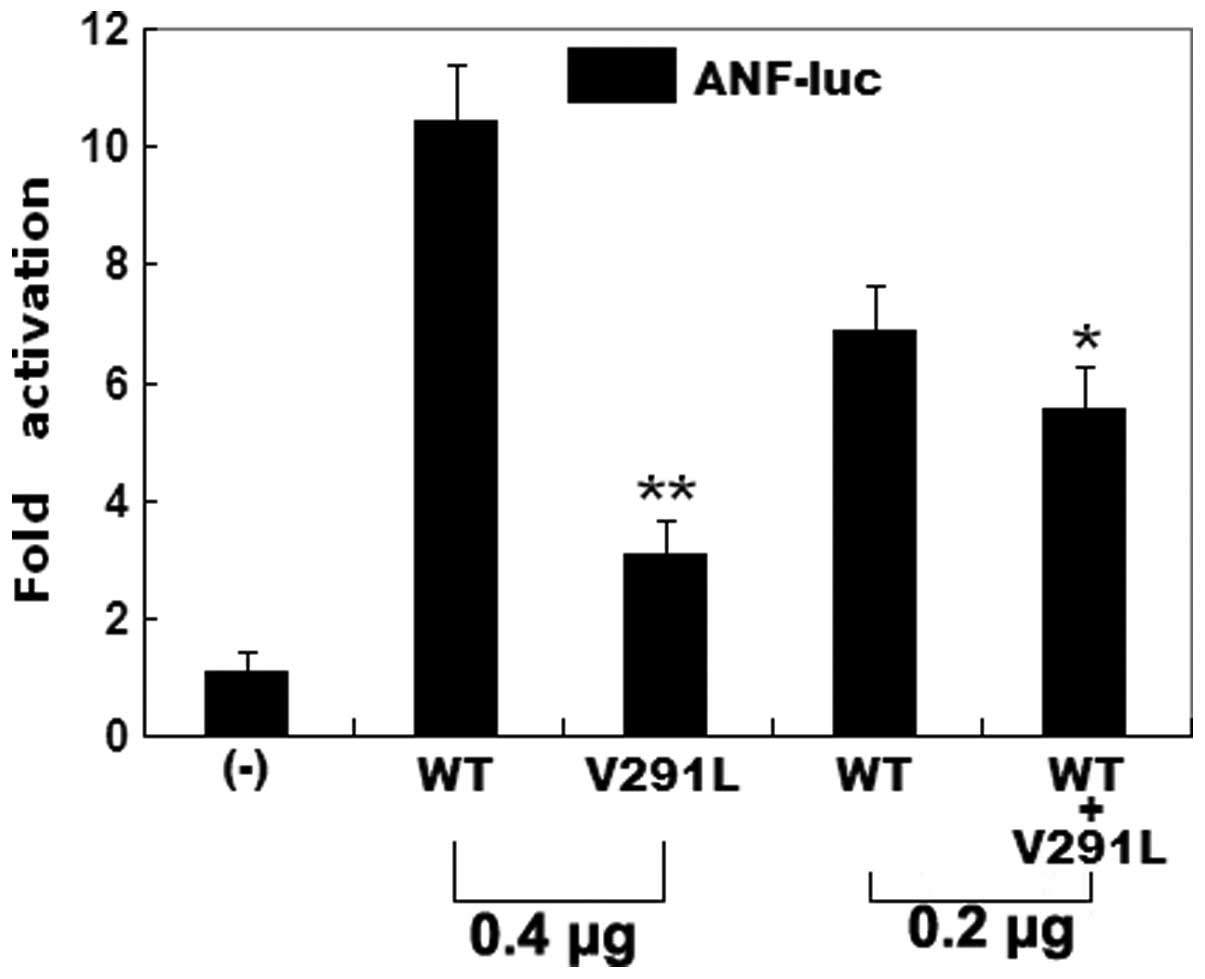
Transcriptional activity of the GATA4
mutant
The same amount (0.4 μg) of wild-type and mutant
GATA4 activated the ANF promoter by ~10- and 3-fold,
respectively (Fig. 5). When the
same amount of wild-type GATA4 (0.2 μg) was co-transfected
with mutant GATA4 (0.2 μg), the induced activation of the
ANF promoter was ~6-fold. These results suggest that the
GATA4 mutant has a significantly reduced activation activity
compared with the wild-type counterpart.
Discussion
In the present study, a novel heterozygous GATA4
mutation of p.V291L was identified in a family with idiopathic DCM.
The missense mutation co-segregated with DCM in the family and was
absent in the 400 reference chromosomes from an ethnically matched
control population. A cross-species alignment of multiple GATA4
protein sequences showed that the altered amino acid was completely
conserved evolutionarily. Functional analysis revealed that the
mutant was associated with significantly reduced transcriptional
activity. Therefore, it is likely that GATA4 loss-of-function
mutation predisposes these mutation carriers to DCM.
It has been verified that GATA4 is a transcriptional
activator of several genes expressed during cardiac development,
including the ANF gene (7). Hence, the functional effect of the
GATA4 mutation may be investigated by assaying the
transcriptional activity of the ANF promoter in cells
expressing GATA4. In the present study, the functional role
of the novel p.V291L mutation of GATA4 identified in the familial
DCM patients was characterized by transcriptional activity
analysis. The data showed significantly diminished transcriptional
activity on a target gene. These results support that the
haploinsufficiency or dominant-negative effect resulting from the
GATA4 mutation is potentially an alternative pathological mechanism
of DCM.
The findings that GATA4 loss-of-function mutation
enhances the susceptibility to DCM may be partially ascribed to the
developmental and regenerative defects of the myocardium as well as
abnormal heart remodeling (17).
During embryonic genesis, GATA4 is required for activation and
maintenance of the core cardiac regulatory network, and GATA4-null
mutations suppress myocardial specification and maturation, giving
rise to death (35). GATA4 also
plays a central role in the postnatal maturation and homeostasis of
cardiomyocytes and adult heart function as well as adaptation
(21). Mice with
cardiomyocyte-restricted deletion of GATA4 were viable and
survived into adulthood, but they underwent a progressive
deterioration in cardiac function and enlargement of the heart in
adulthood. Furthermore, in this mouse model, pressure overload or
exercise stimulation failed to elicit cardiac hypertrophy, but
induced rapid decompensation, precipitous heart failure and
increased apoptosis (21). In
contrast, overexpression of GATA4 in the heart was sufficient for
inducing cardiac hypertrophy (36). In humans, mutations in GATA4 and
its transcriptionally cooperative partners, including NKX2-2 and
TBX20, have been associated with familial DCM (17,33,37). Taken together, these findings
provide evidence that the GATA4 mutation contributes to DCM.
In addition, transcriptions of a number of important
cardiac genes are activated by GATA4, and mutations in multiple
target molecules have been found to be responsible for DCM,
including α-actin, α-myosin heavy chain, troponin C and troponin I
(3,7). Therefore, functionally compromised
GATA4 increases the vulnerability to DCM probably by reducing
expression of target genes.
Similar with previous studies (8–16,38–40), in the present study, congenital
heart disease and paroxysmal atrial fibrillation were observed in 2
patients harboring the GATA4 mutation. In addition, the other 2
members of the GATA family, GATA5 and GATA6 have similar expression
profile and functional characteristics with GATA4 (5), and mutations in GATA5 and GATA6 are
also involved in congenital heart disease and atrial fibrillation
(41–56). These observational results
underscore the critical role of GATA transcription factors in the
development, remodeling and function of the heart.
In conclusion, the present study expands the
mutational spectrum of GATA4 linked to DCM and provides novel
insight into the molecular pathogenesis of DCM, suggesting
potential implications in prenatal prophylaxis and personalized
treatment of DCM.
Acknowledgements
We are thankful to the participants for their
dedication to the study. The present study was supported by grants
from the National Natural Science Fund of China (81070153, 81270161
and 81270314), and the National Basic Research Program of China
(2010CB912604).
References
|
1
|
Garcia-Pavia P, Cobo-Marcos M,
Guzzo-Merello G, Gomez-Bueno M, Bornstein B, Lara-Pezzi E, Segovia
J and Alonso-Pulpon L: Genetics in dilated cardiomyopathy. Biomark
Med. 7:517–533. 2013. View Article : Google Scholar
|
|
2
|
Lakdawala NK, Winterfield JR and Funke BH:
Dilated cardiomyopathy. Circ Arrhythm Electrophysiol. 6:228–237.
2013. View Article : Google Scholar
|
|
3
|
McNally EM, Golbus JR and Puckelwartz MJ:
Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin
Invest. 123:19–26. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Flack E and Kannankeril PJ: The genetics
of dilated cardiomyopathy. Heart Rhythm. 9:397–398. 2012.
View Article : Google Scholar
|
|
5
|
Pikkarainen S, Tokola H, Kerkelä R and
Ruskoaho H: GATA transcription factors in the developing and adult
heart. Cardiovasc Res. 63:196–207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Perrino C and Rockman HA: GATA4 and the
two sides of gene expression reprogramming. Circ Res. 98:715–716.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brody MJ, Cho E, Mysliwiec MR, Kim TG,
Carlson CD, Lee KH and Lee Y: Lrrc10 is a novel cardiac-specific
target gene of Nkx2-5 and GATA4. J Mol Cell Cardiol. 62:237–246.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garg V, Kathiriya IS, Barnes R,
Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS,
Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC and Srivastava D:
GATA4 mutations cause human congenital heart defects and reveal an
interaction with TBX5. Nature. 424:443–447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rajagopal SK, Ma Q, Obler D, Shen J,
Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V,
Srivastava D, Goldmuntz E, Broman KW, Benson DW, Smoot LB and Pu
WT: Spectrum of heart disease associated with murine and human
GATA4 mutation. J Mol Cell Cardiol. 43:677–685. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang J, Fang M, Liu XY, Xin YF, Liu ZM,
Chen XZ, Wang XZ, Fang WY, Liu X and Yang YQ: A novel GATA4
mutation responsible for congenital ventricular septal defects. Int
J Mol Med. 28:557–564. 2011.
|
|
11
|
Liu XY, Wang J, Zheng JH, Bai K, Liu ZM,
Wang XZ, Liu X, Fang WY and Yang YQ: Involvement of a novel GATA4
mutation in atrial septal defects. Int J Mol Med. 28:17–23.
2011.PubMed/NCBI
|
|
12
|
McCulley DJ and Black BL: Transcription
factor pathways and congenital heart disease. Curr Top Dev Biol.
100:253–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang YQ, Li L, Wang J, Liu XY, Chen XZ,
Zhang W, Wang XZ, Jiang JQ, Liu X and Fang WY: A novel GATA4
loss-of-function mutation associated with congenital ventricular
septal defect. Pediatr Cardiol. 33:539–546. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang YQ, Wang J, Liu XY, Chen XZ, Zhang W,
Wang XZ, Liu X and Fang WY: Novel GATA4 mutations in patients with
congenital ventricular septal defects. Med Sci Monit.
18:CR344–CR350. 2012.PubMed/NCBI
|
|
15
|
Wang E, Sun S, Qiao B, Duan W, Huang G, An
Y, Xu S, Zheng Y, Su Z, Gu X, Jin L and Wang H: Identification of
functional mutations in GATA4 in patients with congenital heart
disease. PLoS One. 8:e621382013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang YQ, Gharibeh L, Li RG, Xin YF, Wang
J, Liu ZM, Qiu XB, Xu YJ, Xu L, Qu XK, Liu X, Fang WY, Huang RT,
Xue S and Nemer G: GATA4 loss-of-function mutations underlie
familial tetralogy of Fallot. Hum Mutat. 34:1662–1671. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li RG, Li L, Qiu XB, Yuan F, Xu L, Li X,
Xu YJ, Jiang WF, Jiang JQ, Liu X, Fang WY, Zhang M, Peng LY, Qu XK
and Yang YQ: GATA4 loss-of-function mutation underlies familial
dilated cardiomyopathy. Biochem Biophys Res Commun. 439:591–596.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Molkentin JD, Lin Q, Duncan SA and Olson
EN: Requirement of the transcription factor GATA4 for heart tube
formation and ventral morphogenesis. Genes Dev. 11:1061–1072. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kuo CT, Morrisey EE, Anandappa R, Sigrist
K, Lu MM, Parmacek MS, Soudais C and Leiden JM: GATA4 transcription
factor is required for ventral morphogenesis and heart tube
formation. Genes Dev. 11:1048–1060. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pu WT, Ishiwata T, Juraszek AL, Ma Q and
Izumo S: GATA4 is a dosage-sensitive regulator of cardiac
morphogenesis. Dev Biol. 275:235–244. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oka T, Maillet M, Watt AJ, Schwartz RJ,
Aronow BJ, Duncan SA and Molkentin JD: Cardiac-specific deletion of
Gata4 reveals its requirement for hypertrophy, compensation, and
myocyte viability. Circ Res. 98:837–845. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Suzuki YJ and Evans TL: Regulation of
cardiac myocyte apoptosis by the GATA-4 transcription factor. Life
Sci. 74:1829–1838. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu X, Zhang L and Liang J: Rosuvastatin
prevents pressure overload-induced myocardial hypertrophy via
inactivation of the Akt, ERK1/2 and GATA4 signaling pathways in
rats. Mol Med Rep. 8:385–392. 2013.PubMed/NCBI
|
|
24
|
Kikuchi K, Holdway JE, Werdich AA,
Anderson RM, Fang Y, Egnaczyk GF, Evans T, Macrae CA, Stainier DY
and Poss KD: Primary contribution to zebrafish heart regeneration
by gata4(+) cardiomyocytes. Nature. 464:601–605. 2010.PubMed/NCBI
|
|
25
|
Qian L, Huang Y, Spencer CI, Foley A,
Vedantham V, Liu L, Conway SJ, Fu JD and Srivastava D: In vivo
reprogramming of murine cardiac fibroblasts into induced
cardiomyocytes. Nature. 485:593–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song K, Nam YJ, Luo X, Qi X, Tan W, Huang
GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, Hill JA,
Bassel-Duby R and Olson EN: Heart repair by reprogramming
non-myocytes with cardiac transcription factors. Nature.
485:599–604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lyons I, Parsons LM, Hartley L, Li R,
Andrews JE, Robb L and Harvey RP: Myogenic and morphogenetic
defects in the heart tubes of murine embryos lacking the homeo box
gene Nkx2-5. Genes Dev. 9:1654–1666. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Prall OW, Menon MK, Solloway MJ, Watanabe
Y, Zaffran S, Bajolle F, Biben C, McBride JJ, Robertson BR, Chaulet
H, Stennard FA, Wise N, Schaft D, Wolstein O, Furtado MB, Shiratori
H, Chien KR, Hamada H, Black BL, Saga Y, Robertson EJ, Buckingham
ME and Harvey RP: An Nkx2-5/Bmp2/Smad1 negative feedback loop
controls heart progenitor specification and proliferation. Cell.
128:947–959. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pashmforoush M, Lu JT, Chen H, Amand TS,
Kondo R, Pradervand S, Evans SM, Clark B, Feramisco JR, Giles W, Ho
SY, Benson DW, Silberbach M, Shou W and Chien KR: Nkx2-5 pathways
and congenital heart disease; loss of ventricular myocyte lineage
specification leads to progressive cardiomyopathy and complete
heart block. Cell. 117:373–386. 2004.
|
|
30
|
Schott JJ, Benson DW, Basson CT, Pease W,
Silberbach GM, Moak JP, Maron BJ, Seidman CE and Seidman JG:
Congenital heart disease caused by mutations in the transcription
factor NKX2-5. Science. 281:108–111. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reamon-Buettner SM and Borlak J: NKX2-5:
an update on this hypermutable homeodomain protein and its role in
human congenital heart disease (CHD). Hum Mutat. 31:1185–1194.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang J, Xin YF, Liu XY, Liu ZM, Wang XZ
and Yang YQ: A novel NKX2-5 mutation in familial ventricular septal
defect. Int J Mol Med. 27:369–375. 2011.PubMed/NCBI
|
|
33
|
Costa MW, Guo G, Wolstein O, Vale M,
Castro ML, Wang L, Otway R, Riek P, Cochrane N, Furtado M,
Semsarian C, Weintraub RG, Yeoh T, Hayward C, Keogh A, Macdonald P,
Feneley M, Graham RM, Seidman JG, Seidman CE, Rosenthal N, Fatkin D
and Harvey RP: Functional characterization of a novel mutation in
NKX2-5 associated with congenital heart disease and adult-onset
cardiomyopathy. Circ Cardiovasc Genet. 6:238–247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Elliott P, O’Mahony C, Syrris P, Evans A,
Rivera Sorensen C, Sheppard MN, Carr-White G, Pantazis A and
McKenna WJ: Prevalence of desmosomal protein gene mutations in
patients with dilated cardiomyopathy. Circ Cardiovasc Genet.
3:314–322. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schlesinger J, Schueler M, Grunert M,
Fischer JJ, Zhang Q, Krueger T, Lange M, Tönjes M, Dunkel I and
Sperling SR: The cardiac transcription network modulated by Gata4,
Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS
Genet. 7:e10013132011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liang Q, De Windt LJ, Witt SA, Kimball TR,
Markham BE and Molkentin JD: The transcription factors GATA4 and
GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J
Biol Chem. 276:30245–30253. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kirk EP, Sunde M, Costa MW, Rankin SA,
Wolstein O, Castro ML, Butler TL, Hyun C, Guo G, Otway R, Mackay
JP, Waddell LB, Cole AD, Hayward C, Keogh A, Macdonald P, Griffiths
L, Fatkin D, Sholler GF, Zorn AM, Feneley MP, Winlaw DS and Harvey
RP: Mutations in cardiac T-box factor gene TBX20 are associated
with diverse cardiac pathologies, including defects of septation
and valvulogenesis and cardiomyopathy. Am J Hum Genet. 81:280–291.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jiang JQ, Shen FF, Fang WY, Liu X and Yang
YQ: Novel GATA4 mutations in lone atrial fibrillation. Int J Mol
Med. 28:1025–1032. 2011.PubMed/NCBI
|
|
39
|
Yang YQ, Wang MY, Zhang XL, Tan HW, Shi
HF, Jiang WF, Wang XH, Fang WY and Liu X: GATA4 loss-of-function
mutations in familial atrial fibrillation. Clin Chim Acta.
412:1825–1830. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang J, Sun YM and Yang YQ: Mutation
spectrum of the GATA4 gene in patients with idiopathic atrial
fibrillation. Mol Biol Rep. 39:8127–8135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jiang JQ, Li RG, Wang J, Liu XY, Xu YJ,
Fang WY, Chen XZ, Zhang W, Wang XZ and Yang YQ: Prevalence and
spectrum of GATA5 mutations associated with congenital heart
disease. Int J Cardiol. 165:570–573. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wei D, Bao H, Liu XY, Zhou N, Wang Q, Li
RG, Xu YJ and Yang YQ: GATA5 loss-of-function mutations underlie
tetralogy of Fallot. Int J Med Sci. 10:34–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wei D, Bao H, Zhou N, Zheng GF, Liu XY and
Yang YQ: GATA5 loss-of-function mutation responsible for the
congenital ventriculoseptal defect. Pediatr Cardiol. 34:504–511.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang YQ, Wang J, Wang XH, Wang Q, Tan HW,
Zhang M, Shen FF, Jiang JQ, Fang WY and Liu X: Mutational spectrum
of the GATA5 gene associated with familial atrial fibrillation. Int
J Cardiol. 157:305–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gu JY, Xu JH, Yu H and Yang YQ: Novel
GATA5 loss-of-function mutations underlie familial atrial
fibrillation. Clinics (Sao Paulo). 67:1393–1399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang XH, Huang CX, Wang Q, Li RG, Xu YJ,
Liu X, Fang WY and Yang YQ: A novel GATA5 loss-of-function mutation
underlies lone atrial fibrillation. Int J Mol Med. 31:43–50.
2013.PubMed/NCBI
|
|
47
|
Kodo K, Nishizawa T, Furutani M, Arai S,
Yamamura E, Joo K, Takahashi T, Matsuoka R and Yamagishi H: GATA6
mutations cause human cardiac outflow tract defects by disrupting
semaphorin-plexin signaling. Proc Natl Acad Sci USA.
106:13933–13938. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Maitra M, Koenig SN, Srivastava D and Garg
V: Identification of GATA6 sequence variants in patients with
congenital heart defects. Pediatr Res. 68:281–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lin X, Huo Z, Liu X, Zhang Y, Li L, Zhao
H, Yan B, Liu Y, Yang Y and Chen YH: A novel GATA6 mutation in
patients with tetralogy of Fallot or atrial septal defect. J Hum
Genet. 55:662–667. 2010. View Article : Google Scholar
|
|
50
|
Wang J, Luo XJ, Xin YF, Liu Y, Liu ZM,
Wang Q, Li RG, Fang WY, Wang XZ and Yang YQ: Novel GATA6 mutations
associated with congenital ventricular septal defect or tetralogy
of Fallot. DNA Cell Biol. 31:1610–1617. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zheng GF, Wei D, Zhao H, Zhou N, Yang YQ
and Liu XY: A novel GATA6 mutation associated with congenital
ventricular septal defect. Int J Mol Med. 29:1065–1071.
2012.PubMed/NCBI
|
|
52
|
Huang RT, Xue S, Xu YJ and Yang YQ:
Somatic mutations in the GATA6 gene underlie sporadic
tetralogy of Fallot. Int J Mol Med. 31:51–58. 2013.
|
|
53
|
Bui PH, Dorrani N, Wong D, Perens G,
Dipple KM and Quintero-Rivera F: First report of a de novo 18q11.2
microdeletion including GATA6 associated with complex congenital
heart disease and renal abnormalities. Am J Med Genet A.
161A:1773–1778. 2013.PubMed/NCBI
|
|
54
|
Yang YQ, Wang XH, Tan HW, Jiang WF, Fang
WY and Liu X: Prevalence and spectrum of GATA6 mutations associated
with familial atrial fibrillation. Int J Cardiol. 155:494–496.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yang YQ, Li L, Wang J, Zhang XL, Li RG, Xu
YJ, Tan HW, Wang XH, Jiang JQ, Fang WY and Liu X: GATA6
loss-of-function mutation in atrial fibrillation. Eur J Med Genet.
55:520–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li J, Liu WD, Yang ZL and Yang YQ: Novel
GATA6 loss-of-function mutation responsible for familial atrial
fibrillation. Int J Mol Med. 30:783–790. 2012.PubMed/NCBI
|