Introduction
Hereditary retinal dystrophy is a broad group of
clinically and genetically heterogeneous diseases that usually
cause irreversible blindness. Retinitis pigmentosa (RP) is the most
common form, with Leber congenital amaurosis (LCA) being the most
severe form. The clinical manifestations as well as the causative
genes frequently overlap in the different forms of these diseases
(1). Mutations in almost 200
genes have been described in association with hereditary retinal
diseases (RetNet: http://www.sph.uth.tmc.edu/Retnet/sum-dis.htm).
However, mutations in each of these genes usually contribute to
only a small fraction of the diseases (2–4).
Thus, detection of disease-causing mutations among these genes is a
great challenge in individual patients, and involves great effort
and expense. Identification of a gene-specific genotype-phenotype
correlation would facilitate mutation detection, similar to CYP4V2
mutations and Bietti crystalline corneoretinal dystrophy (5,6).
Mutations in crumbs homolog (CRB)1 are among the
common causes of severe early onset retinal dystrophy, including
LCA and early onset RP (4,7–31).
Although a firm genotype-phenotype correlation between CRB1
mutations and specific phenotypes has yet to be determined, some
characteristic clinical findings have been suggested to associate
with CRB1 mutations.
The aim of the present study was to investigate
whether characteristic phenotype-directed mutational screening
facilitated the detection of CRB1 mutations. Probands with some of
the CRB1-associated phenotypes were selected for this study. The
Sanger sequencing of CRB1 coding and adjacent intronic regions
revealed homozygous and compound heterozygous mutations in CRB1 in
seven of 22 Chinese probands with at least one of the
CRB1-associated phenotypes.
Materials and methods
Clinical characteristics of the
probands
Probands from 22 unrelated families with at least
one of the CRB1-associated phenotypes participated in this study
(Table I). Clinical phenotypes in
this study were mainly based on the fundus photos and the age of
onset (1–10 years of age). CRB1-associated characteristic clinical
findings included: i) severe early onset retinal dystrophy
(including LCA and early onset RP) (4,7–31);
ii) nummular pigmentary changes at the posterior fundus; iii)
preservation of the para-arteriolar retinal pigment epithelium
(PPRPE) (14); iv) Coats-like
vasculopathy (CLV) (12); and v)
pigmented paravenous chorioretinal atrophy (PPCRA) (20). The LCA, PPRPE and CLV phenotypes
were not studied since LCA has been previously analyzed, whereas
PPRPE and CLV were not identified in our patients. Macular dot
pigmentation in seven probands was also selected to examine the
specificity of macular nummular pigmentation.
 | Table IClassification of the probands in this
study. |
Table I
Classification of the probands in this
study.
| No. of cases |
|---|
|
|
|---|
| Clinical
characteristics | Screened | CRB1 mutations |
|---|
| A. Macular nummular
pigmentation | 4 | 3 |
| B. Pigmented
paravenous chorioretinal atrophy | 4 | 0 |
| C. Early onset
retinitis pigmentosa with macular involvement (<10 years) | 7 | 4 |
| D. Other retinal
dystrophy with macular dot pigmentation | 7 | 0 |
| Total | 22 | 7 |
Informed consent was obtained from each
participating individual or their guardians prior to the collection
of clinical data and venous blood. This study was approved by the
Institutional Review Board of the Zhongshan Ophthalmic Center and
followed the tenets of the Declaration of Helsinki.
Genomic DNA
Genomic DNA was prepared from venous leukocytes
using a method described previously (32). The procedure for Sanger sequencing
of CRB1 was the same as that described in our previous study and
the same sets of primers were used (4). The informatics analysis for the
detected variants was also as described previously (4). Only homozygous or compound
heterozygous variants that were predicted to be pathogenic were
considered to be disease-causing because: i) CRB1 mutations were
only reported to associate with recessive retinal dystrophy
(8); and ii) single heterozygous
null mutations for recessive genes were common in the general
population (33). Controls were
healthy university students with normal visual acuity without any
known hereditary retinopathy and family history of such diseases as
those controls used in the genetic study of myopia (34).
Results
Homozygous or compound heterozygous
mutations in CRB1
Complete sequencing analysis of CRB1 revealed
homozygous and compound heterozygous variants in seven of the 22
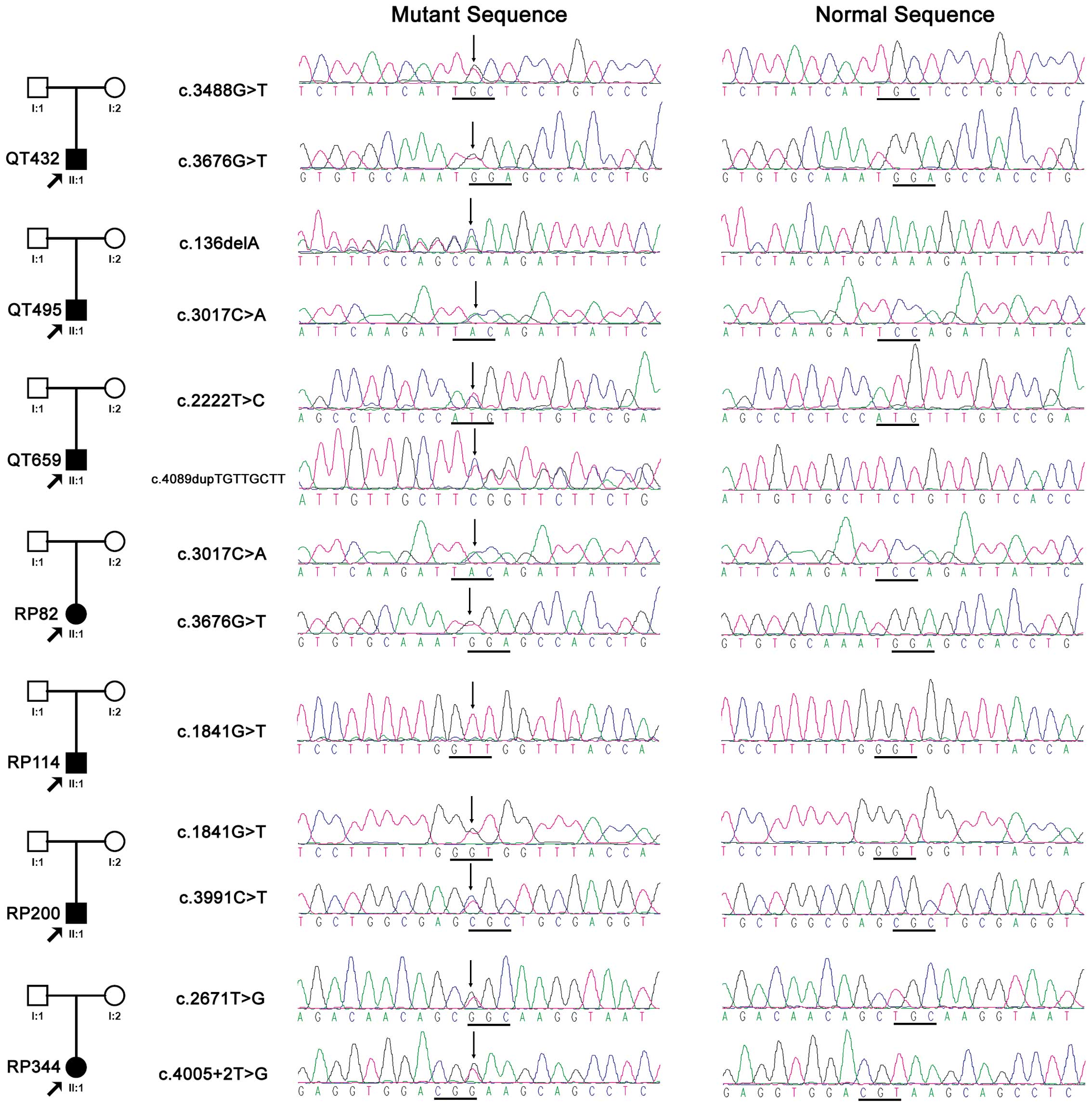
probands (Fig. 1), involving six
novel and four known variations (Table II). Of the 10 variants, four were
predicted to be truncated and the remaining six variants were
missense. The residues with the six missense changes were
relatively conserved in different species (Table II and Fig. 2) and involved in the functional
domain of the CRB1 protein (Table
II). Five of the six missense variants were predicted to affect
the encoded protein by PolyPhen-2, while two variants were
predicted to be damaging by sorting intolerant from tolerant (SIFT)
(Table II). Only the missense
change p.M741T, was predicted to be benign or tolerated by
PolyPhen-2 or SIFT, but it was reported to be causative in a
previous study (31). Therefore,
all 10 variants detected in this study were considered to be
pathogenic.
| Table IIPotential pathogenic mutations
detected in 7 of the 22 probands. |
Table II
Potential pathogenic mutations
detected in 7 of the 22 probands.
| Nucleotide
change | Effects | Protein domain | Bioinformatics
analysis | Status in
probands | Novel or
reported |
|---|
|
|
|---|
| Conservation | PolyPhen or splice
site | SIFT | Homozygous | Heterozygous |
|---|
| c.136delA | Frameshift | - | - | - | - | | QT495 | Novel |
| c.1841G>T | p.G614V | LamG | Yes | Missense, PBD
(1.00) | Tolerated (0.18) | RP114 | RP200 | Novel |
| c.2222T>C | p.M741T | LamG | Yes | Missense, benign
(0.395) | Tolerated (0.41) | | QT659 | Hanein et al
(31) |
| c.2671T>G | p.C891G | EGF | Yes | Missense, PD
(0.475) | Damaging (0) | | RP344 | Bernal et al
(28) |
| c.3017C>A | p.S1006Y | LamG | Yes | Missense, PBD
(0.964) | Tolerated (0.14) | | QT495, RP82 | Novel |
| c.3488G>T | p.C1163F | EGF | Yes | Missense, PBD
(0.999) | Damaging (0) | | QT432 | Novel |
| c.3676G>T | p.G1226X | - | - | - | - | | QT432, RP82 | Li et al
(4) |
| c.3991C>T | p.R1331C | EGF | Yes | Missense, PBD
(0.941) | Tolerated
(0.18) | | RP200 | Novel |
| c.4005+2T>G | Splicing site | - | - | Splicing site
eliminated | - | | RP344 | Li et al
(4) |
|
c.4089dupTGTTGCTT | Frameshift | - | - | - | - | | QT659 | Novel |
Clinical data of the seven probands with
CRB1 mutations
Each proband had either poor vision or nystagmus at
<8 years of age. Visual acuity ranged from finger counting to
0.6 (Table III). Homozygous or
compound heterozygous mutations were identified in three of four
probands with macular nummular pigmentation and in four of seven
probands with early onset RP with macular involvement. However, no
mutations were identified in the 11 probands with either pigmented
paravenous chorioretinal atrophy or other retinal dystrophy with
macular dot pigmentation (Tables
I and III). All seven
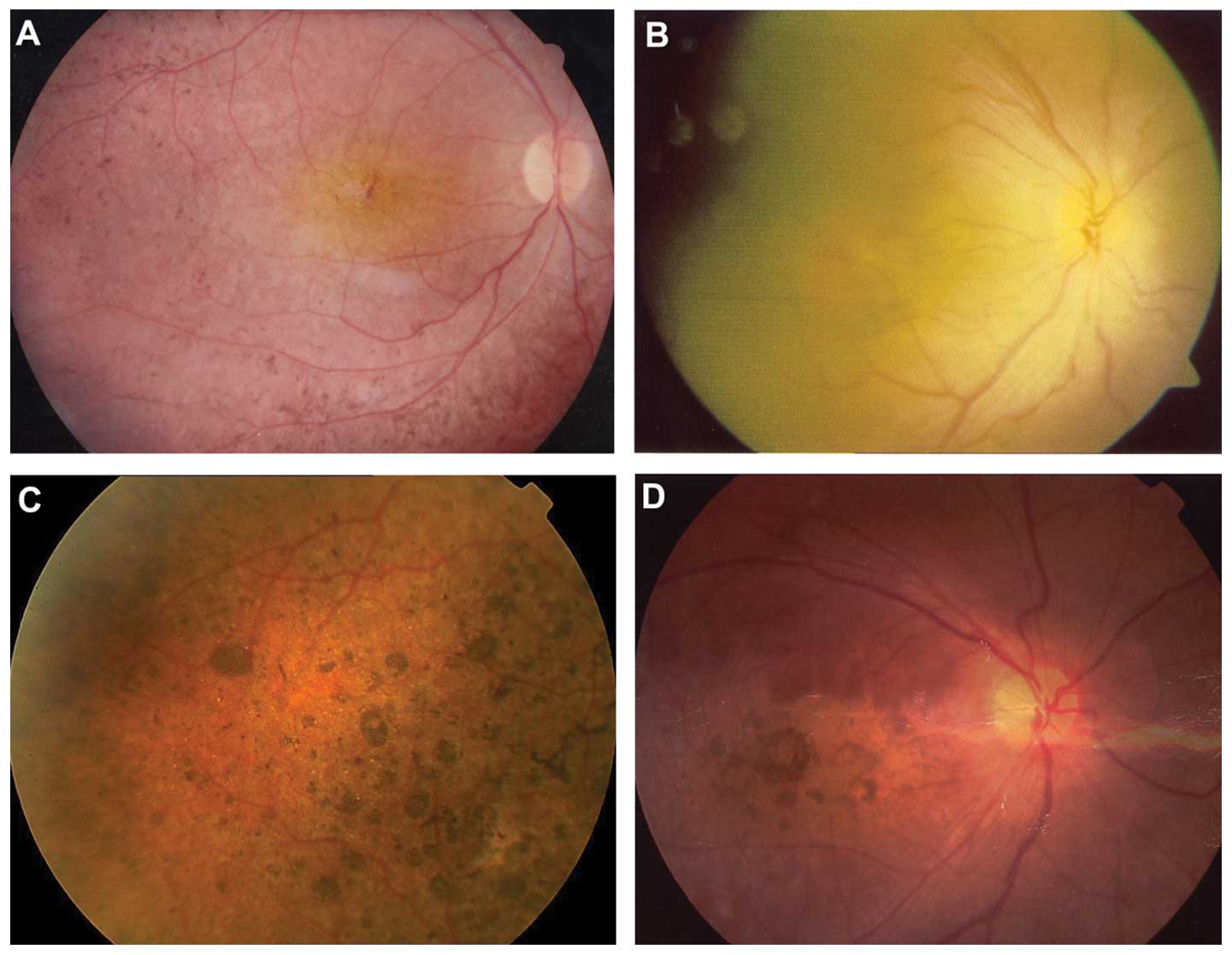
probands with CRB1 mutations were isolated cases (Fig. 1). Fig. 3 shows the representative fundus
changes of three probands with CRB1 mutations (Fig. 3A–C) and one proband without CRB1
mutation (Fig. 3D) but with
nummular pigmentation.
| Table IIIClinical data of the probands with
CRB1 homozygous or compound heterozygous mutations. |
Table III
Clinical data of the probands with
CRB1 homozygous or compound heterozygous mutations.
| Proband ID | CRB1 mutations | Gender | Age (years) at | First symptom | Visual acuity
(right; left) | Fundus changes | ERG responses | Classification in
Table I |
|---|
|
|
|---|
| Exam | Onset | Rod | Cone |
|---|
| QT432 |
c.[3488G>T];[3676G>T] | M | 4.5 | 1 | NYS | FC | GRD, MNP, YCLS,
BSP | Not detectable | Not detectable | A |
| QT495 |
c.[136delA];[3017C>A] | M | 22 | EC | PV; NYS | 0.02; 0.04 | GRD, BSP | NA | NA | C |
| QT659 |
c.[2222T>C];[4089dupTGTTGCTT] | M | 4.5 | EC | NYS; HH | NA | GRD | Not detectable | Not detectable | C |
| RP82 |
c.[3017C>A];[3676G>T] | F | 5 | 3 | PV | 0.6; 0.6 | GRD, BSP | Not detectable | Reduced | C |
| RP114 |
c.[1841G>T];[1841G>T] | M | 6 | 2 | PV; NYS | 0.3; 0.3 | GRD, MNP, YCLS,
BSP | Not detectable | Not detectable | A |
| RP200 |
c.[1841G>T];[3991C>T] | M | 20 | 8 | PV | 0.1; FC | GRD | Not detectable | Not detectable | C |
| RP344 |
c.[2671T>G];[4005+2T>G] | F | 7 | 6 | PV | 0.2;0.2 | GRD, MNP, BSP | Not detectable | Reduced | A |
Discussion
A recent review on CRB1 mutations suggested an
average prevalence of homozygous or compound heterozygous CRB1
mutations in patients with different forms of retinal dystrophy as
follows: 6.6% (109/1,645) for LCA or early onset retinal dystrophy,
1.2% (4/335) for RP, 66.7% (18/27) for RP with PPRPE and 26.7%
(8/30) for RP with CLV (8). Those
findings suggested that the sequencing analysis of patients with
CRB1-specific phenotypes may facilitate mutation detection of
CRB1-associated retinopathy. The frequency of CRB1 mutations in
patients with macular nummular pigmentation was not available due
to the unavailability of the frequency of this fundus change in
patients analyzed in previous studies. However, macular nummular
pigmentation is rarely observed in patients with mutations in other
genes, although it has been described in different studies in a
number of patients with CRB1 mutations (9,16,24,25,35–37). Therefore, it may be considered a
characteristic indication for CRB1-associated retinal
dystrophy.
In the present study, the entire coding region and
adjacent intronic sequences of CRB1 were analyzed in 22 probands
with one of the possible CRB1-associated phenotypes, using Sanger
sequencing. Homozygous and compound heterozygous mutations in the
CRB1 gene, involving six novel and four known variants, were
identified in 31.8% (7/22) of the probands, including 75% (3/4)
with macular nummular pigmentation and 57.2% (4/7) with early onset
RP with macular involvement. No novel mutations were detected in
192 alleles of 96 healthy individuals. The frequent association of
CRB1 mutations with early onset of RP and LCA suggests that early
onset RP is a spectrum from LCA to RP with early onset RP being
detected within this spectrum. Thus, genes associated with early
onset RP are potentially good candidates for LCA and vice
versa.
Results of the bioinformatics analysis suggest that
these mutations are likely to exert pathogenic effects on the
encoded proteins (Fig. 2). Our
study provides additional evidence that macular nummular
pigmentation may be considered a gene-specific indication for
CRB1-associated retinal dystrophy. The analysis also suggests that
CRB1 mutations are common causes for early onset RP, as observed by
their contribution to LCA. Findings of a recent study also showed
that CRB1 mutations are a relatively common cause of autosomal
recessive early onset retinal degeneration (38), suggesting that certain
CRB1-associated specific phenotypes, including PPRPE and CLV, may
not be common in the studied populations. This may also be due to
the relatively small sample size of this study.
Identification of gene-specific phenotypes may
facilitate mutation identification and subsequent genetic
counseling as well as provide useful evidence to determine
causative variants underlying the extremely heterogeneous
hereditary retinal diseases. This is particularly true for isolated
patients with mutations in multiple genes, which are likely to
become increasingly common in the era of next-generation
sequencing. Systematic and careful records of clinical data,
particularly of fundus photos of different regions and at different
time-points, may provide additional gene-specific phenotypes for
CRB1 and for other genes associated with retinal dystrophy. This
record keeping should be encouraged in future investigations of
mutations related to retinal dystrophy.
Acknowledgements
The authors would like to thank the patients for
their participation and the anonymous reviewers for their
constructive advice. This study was supported by the NSFC81170881
(to Q.Z.).
References
|
1
|
Berger W, Kloeckener-Gruissem B and
Neidhardt J: The molecular basis of human retinal and vitreoretinal
diseases. Prog Retin Eye Res. 29:335–375. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
den Hollander AI, Black A, Bennett J and
Cremers FP: Lighting a candle in the dark: advances in genetics and
gene therapy of recessive retinal dystrophies. J Clin Invest.
120:3042–3053. 2010.PubMed/NCBI
|
|
3
|
Hartong DT, Berson EL and Dryja TP:
Retinitis pigmentosa. Lancet. 368:1795–1809. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li L, Xiao X, Li S, et al: Detection of
variants in 15 genes in 87 unrelated Chinese patients with Leber
congenital amaurosis. PLoS One. 6:e194582011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li A, Jiao X, Munier FL, et al: Bietti
crystalline corneoretinal dystrophy is caused by mutations in the
novel gene CYP4V2. Am J Hum Genet. 74:817–826. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiao X, Mai G, Li S, et al: Identification
of CYP4V2 mutation in 21 families and overview of mutation spectrum
in Bietti crystalline corneoretinal dystrophy. Biochem Biophys Res
Commun. 409:181–186. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Corton M, Tatu SD, Avila-Fernandez A, et
al: High frequency of CRB1 mutations as cause of early-onset
retinal dystrophies in the Spanish population. Orphanet J Rare Dis.
8:202013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bujakowska K, Audo I, Mohand-Saïd S, et
al: CRB1 mutations in inherited retinal dystrophies. Hum Mutat.
33:306–315. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abouzeid H, Li Y, Maumenee IH, et al: A
G1103R mutation in CRB1 is co-inherited with high hyperopia and
Leber congenital amaurosis. Ophthalmic Genet. 27:15–20. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Booij JC, Florijn RJ, ten Brink JB, et al:
Identification of mutations in the AIPL1, CRB1, GUCY2D, RPE65, and
RPGRIP1 genes in patients with juvenile retinitis pigmentosa. J Med
Genet. 42:e672005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
den Hollander AI, Davis J, van der
Velde-Visser SD, et al: CRB1 mutation spectrum in inherited retinal
dystrophies. Hum Mutat. 24:355–369. 2004.PubMed/NCBI
|
|
12
|
den Hollander AI, Heckenlively JR, van den
Born LI, et al: Leber congenital amaurosis and retinitis pigmentosa
with coats-like exudative vasculopathy are associated with
mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet.
69:198–203. 2001.PubMed/NCBI
|
|
13
|
den Hollander AI, Lopez I, Yzer S, et al:
Identification of novel mutations in patients with Leber congenital
amaurosis and juvenile RP by genome-wide homozygosity mapping with
SNP microarrays. Invest Ophthalmol Vis Sci. 48:5690–5698.
2007.PubMed/NCBI
|
|
14
|
den Hollander AI, ten Brink JB, de Kok YJ,
et al: Mutations in a human homologue of Drosophila crumbs
cause retinitis pigmentosa (RP12). Nat Genet. 23:217–221.
1999.PubMed/NCBI
|
|
15
|
Gerber S, Perrault I, Hanein S, et al: A
novel mutation disrupting the cytoplasmic domain of CRB1 in a large
consanguineous family of Palestinian origin affected with Leber
congenital amaurosis. Ophthalmic Genet. 23:225–235. 2002.
View Article : Google Scholar
|
|
16
|
Henderson RH, Mackay DS, Li Z, et al:
Phenotypic variability in patients with retinal dystrophies due to
mutations in CRB1. Br J Ophthalmol. 95:811–817. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jalkh N, Guissart C, Chouery E, et al:
Report of a novel mutation in CRB1 in a Lebanese family presenting
retinal dystrophy. Ophthalmic Genet. Jan 30–2013.(Epub ahead of
print).
|
|
18
|
Lotery AJ, Jacobson SG, Fishman GA, et al:
Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch
Ophthalmol. 119:415–420. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lotery AJ, Malik A, Shami SA, et al: CRB1
mutations may result in retinitis pigmentosa without
para-arteriolar RPE preservation. Ophthalmic Genet. 22:163–169.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McKay GJ, Clarke S, Davis JA, et al:
Pigmented paravenous chorioretinal atrophy is associated with a
mutation within the crumbs homolog 1 (CRB1) gene. Invest Ophthalmol
Vis Sci. 46:322–328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McMahon TT, Kim LS, Fishman GA, et al:
CRB1 gene mutations are associated with keratoconus in patients
with leber congenital amaurosis. Invest Ophthalmol Vis Sci.
50:3185–3187. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Paun CC, Pijl BJ, Siemiatkowska AM, et al:
A novel crumbs homolog 1 mutation in a family with retinitis
pigmentosa, nanophthalmos, and optic disc drusen. Mol Vis.
18:2447–2453. 2012.PubMed/NCBI
|
|
23
|
Riveiro-Alvarez R, Vallespin E, Wilke R,
et al: Molecular analysis of ABCA4 and CRB1 genes in a Spanish
family segregating both Stargardt disease and autosomal recessive
retinitis pigmentosa. Mol Vis. 14:262–267. 2008.PubMed/NCBI
|
|
24
|
Yzer S, Fishman GA, Racine J, et al: CRB1
heterozygotes with regional retinal dysfunction: implications for
genetic testing of leber congenital amaurosis. Invest Ophthalmol
Vis Sci. 47:3736–3744. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zenteno JC, Buentello-Volante B,
Ayala-Ramirez R and Villanueva-Mendoza C: Homozygosity mapping
identifies the Crumbs homologue 1 (Crb1) gene as responsible for a
recessive syndrome of retinitis pigmentosa and nanophthalmos. Am J
Med Genet A. 155A:1001–1006. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Benayoun L, Spiegel R, Auslender N, et al:
Genetic heterogeneity in two consanguineous families segregating
early onset retinal degeneration: the pitfalls of homozygosity
mapping. Am J Med Genet A. 149A:650–656. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bustamante-Aragones A, Vallespin E,
Rodriguez de Alba M, et al: Early noninvasive prenatal detection of
a fetal CRB1 mutation causing Leber congenital amaurosis. Mol Vis.
14:1388–1394. 2008.PubMed/NCBI
|
|
28
|
Bernal S, Calaf M, Garcia-Hoyos M, et al:
Study of the involvement of the RGR, CRPB1, and CRB1 genes in the
pathogenesis of autosomal recessive retinitis pigmentosa. J Med
Genet. 40:e892003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Henderson RH, Waseem N, Searle R, et al:
An assessment of the apex microarray technology in genotyping
patients with Leber congenital amaurosis and early-onset severe
retinal dystrophy. Invest Ophthalmol Vis Sci. 48:5684–5689. 2007.
View Article : Google Scholar
|
|
30
|
Simpson DA, Clark GR, Alexander S, et al:
Molecular diagnosis for heterogeneous genetic diseases with
targeted high-throughput DNA sequencing applied to retinitis
pigmentosa. J Med Genet. 48:145–151. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hanein S, Perrault I, Gerber S, et al:
Leber congenital amaurosis: comprehensive survey of the genetic
heterogeneity, refinement of the clinical definition, and
genotype-phenotype correlations as a strategy for molecular
diagnosis. Hum Mutat. 23:306–317. 2004. View Article : Google Scholar
|
|
32
|
Wang Q, Wang P, Li S, et al: Mitochondrial
DNA haplogroup distribution in Chaoshanese with and without myopia.
Mol Vis. 16:303–309. 2010.PubMed/NCBI
|
|
33
|
Nishiguchi KM and Rivolta C: Genes
associated with retinitis pigmentosa and allied diseases are
frequently mutated in the general population. PLoS One.
7:e419022012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiao X, Wang P, Li S, et al: Association
of markers at chromosome 15q14 in Chinese patients with moderate to
high myopia. Mol Vis. 18:2633–2646. 2012.PubMed/NCBI
|
|
35
|
Tosi J, Tsui I, Lima LH, et al: Case
report: autofluorescence imaging and phenotypic variance in a
sibling pair with early-onset retinal dystrophy due to defective
CRB1 function. Curr Eye Res. 34:395–400. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li L, Xiao X, Li S, et al: Lack of
phenotypic effect of triallelic variation in SPATA7 in a family
with Leber congenital amaurosis resulting from CRB1 mutations. Mol
Vis. 17:3326–3332. 2011.PubMed/NCBI
|
|
37
|
Chen Y, Zhang Q, Shen T, et al:
Comprehensive mutation analysis by whole-exome sequencing in 41
Chinese families with Leber congenital amaurosis. Invest Ophthalmol
Vis Sci. 54:4351–4357. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Beryozkin A, Zelinger L, Bandah-Rozenfeld
D, et al: Mutations in CRB1 are a relatively common cause of
autosomal recessive early-onset retinal degeneration in the Israeli
and Palestinian populations. Invest Ophthalmol Vis Sci.
54:2068–2075. 2013. View Article : Google Scholar : PubMed/NCBI
|