Introduction
Antiphospholipid syndrome (APS) is an autoimmune
disorder caused by the production of antiphospholipid antibodies
(aPLs) which contribute to thrombosis (1). In addition to anionic phospholipids,
aPLs also recognize phospholipid binding proteins, including
β2-glycoprotein I (β2GPI) and prothrombin
(2). Among these,
β2GPI has emerged as the major antigenic target for
aPLs. Anti-β2GPI antibodies are found abundantly in the
plasma of patients with APS, suggesting its important role in the
pathophysiology of APS (3).
The anti-β2GPI/β2GPI complex
activates endothelial cells and monocytes upon binding to the
surface membrane of endothelial cells and monocytes, promoting
tissue factor (TF) activity, thereby increasing the risk of
thrombosis, and enhancing the expression and secretion of
pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α)
and interleukin (IL)-1β, which are beneficial to thrombus formation
in APS (4–6). The stimulation of endothelial cells
or monocytes by the anti-β2GPI/β2GPI complex
has been shown to be mediated by intracellular pathways dependent
on certain receptors, such as Toll-like receptor 4 (TLR4), which
help ligand recognition and binding. As a pathogen recognition
protein, the activation of TLR4 by its natural ligand,
lipopolysaccharide (LPS), or by other ligands, plays an important
role in activating the innate immune system (7), in recognizing microbes and in
initiating inflammatory responses. In recent studies, we revealed
that TLR4 and its signal transduction pathway contribute to
anti-β2GPI/β2GPI complex-induced TF and TNF-α
expression in THP-1 cells and monocytes (8), and that the
anti-β2GPI/β2GPI complex upregulates TF
expression in THP-1 cells or monocytes following the activation of
nuclear transcription factors, including nuclear factor-κB (NF-κB)
and activator protein-1 (AP-1) (9). As crucial downstream molecules of
the TLR4 signaling pathway, mitogen-activated protein kinases
(MAPKs), including p38, extracellular signal-regulated kinase 1/2
(ERK1/2) and c-Jun N-terminal kinase (JNK) are involved in the
development of a number of diseases (10), and are activated by the
anti-β2GPI/β2GPI complex through myeloid
differentiation protein 2 (MD-2) and myeloid differentiation factor
88 (MyD88) in THP-1 cells (11).
These results led us to hypothesize that the intracellular signal
transduction pathway of TLRs-MAPKs-NF-κB in the
anti-β2GPI/β2GPI complex-induced activation
of cells may be a potential therapeutic target for APS.
Polyphenols of green tea, which comprise 30% of the
dry weight of green tea leaves, include epigallocatechin-3-gallate
(EGCG), epigallocatechin (EGC), epicatechin-3-gallate (ECG) and
epicatechin (EC). Among these, EGCG is the most abundant catechin
and has a variety of biological and pharmacological properties,
preventing cancer, allergies, oxidation, microbes, thrombosis,
inflammation and cardiovascular diseases. Previous studies have
demonstrated that EGCG exerts a number of beneficial effects by
affecting a wide array of signal transduction pathways, including
Notch (12), Wnt (13), JAK/STAT (14) and MAPK (15).
It is known that EGCG has beneficial effects;
however, whether it affects the anti-β2GPI/β2GPI complex-stimulated
activation of THP-1 cells remains to be determined. In the present
study, we investigated the ability of EGCG to block the effects of
the anti-β2GPI/β2GPI complex on THP-1 cells
and the possible mechanisms involved in this process.
Materials and methods
Cell lines and cell culture
The human acute monocytic leukemia cell line, THP-1,
was obtained from Shanghai Institutes Biological Sciences
(Shanghai, China). The cells were cultured in RPMI-1640 medium
(Gibco-BRL, Grand Island, NY, USA) supplemented with 1% glutamine,
1% penicillin/streptomycin and 10% fetal bovine serum (FBS)
(Gibco-BRL). The cells were cultured at 37°C in a humidified
incubator supplemented with 5% CO2 near confluence and
deprived of serum for 16 h prior to being used in the experiments.
All experimental data were obtained from cells at passages
3–10.
Quantitative reverse transcription PCR
(qRT-PCR)
THP-1 cells were seeded at 2×106
cells/well into 6-well plates and serum-starved for 16 h prior to
stimulation with monoclonal anti-β2GPI (10 μg/ml;
Chemicon, Temecula, CA, USA)/β2GPI (100 μg/ml; US
Biological, Swampscott, MC, USA) complex, anti-β2GPI (10
μg/ml)/bovine serum albumin (BSA) (100 μg/ml; Sigma, St. Louis, MO,
USA), control rabbit immunoglobulin G isotype (R-IgG) (10 μg/ml;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA)/β2GPI (100 μg/ml) or 500 ng/ml of LPS
(Escherichia coli, strain 0128:B12; Sigma) for 2 h [the
concentrations of the above reagents are based on those described
in our previous studies (16–18)]. Cells in some wells were
pre-treated with various concentrations of EGCG (0–50 μg/ml; Sigma)
for 1 h, and EGCG was not removed. Subsequently, total RNA was
extracted from the cells using TRIzol reagent (Invitrogen,
Carlsbad, CA, USA) according to the manufacturer’s instructions.
Oligo(dT)-primers were used for reverse transcription with 2 μg of
total RNA in a 25 μl reaction volume using a 2720 Thermal Cycler
(Toyobo Biotechnology, Osaka, Japan). The expression levels of the
target mRNA in the cells were analyzed by qRT-PCR using SYBR-Green
I dye (Takara Bio, Kyoto, Japan). The primers used for PCR were as
follows: TF forward, 5′-TCAGGTG ATCCACCCACCTT-3′ and reverse,
5′-GCACCCAATTT CCTTCCATTT-3′; TNF-α forward, 5′-CCCAGGCAGTCA
GATCATCTTCT-3′ and reverse, 5′-ATGAGGTACAGGCCC TCTGAT-3′; TLR4
forward, 5′-CCTGTGCAATTTGACC ATTG-3′ and reverse,
5′-AAGCATTCCCACCTTTGTTG-3′. Primers for the control housekeeping
gene β-actin were forward, 5′-CACGAAACTACCTTCAACTCC-3′ and reverse,
5′-CATACTCCTGCTTGCTGATC-3′. Each pair of primers was shown to yield
only one product. The amplifications were performed in triplicate
on a Mx3000P qPCR System (Agilent Technologies, Santa Rosa, CA,
USA) for 35 cycles of denaturation for 30 sec at 95°C, annealing
for 30 sec at 60°C for TF and TLR4, 58°C for TNF-α and 56°C for
β-actin, and extension for 30 sec at 72°C. The relative mRNA levels
of target genes to the control β-actin were calculated using a
standard curve.
Detection of TNF-α secretion
THP-1 cells were seeded at 2×106
cells/well into 6-well plates and serum-starved for 16 h prior to
stimulation with anti-β2GPI (10 μg/ml)/β2GPI
(100 μg/ml) complex, anti-β2GPI (10 μg/ml)/ BSA (100
μg/ml), R-IgG (10 μg/ml)/β2GPI (100 μg/ml) or 500 ng/ml
of LPS for 24 h. The cells in some wells were pre-treated with
various concentrations of EGCG (0–50 μg/ml) for 1 h, and EGCG was
not removed. TNF-α protein, secreted into the cell culture medium,
was measured using the TNF-α ELISA kit (Neobioscience, Shenzhen,
China), following the manufacturer’s instructions. The TNF-α
protein concentration in the cell culture medium was expressed as
pg/ml.
Measurement of TF activity
THP-1 cells 2×106 cells/well were treated
as described above for the indicated periods of time. Cell lysates
were collected and assayed using TF activity kits (Assaypro,
Greenwich, CT, USA) according to the manufacturer’s instructions.
The TF activity in the cells was determined as factor X activation
by the TF/VIIa complex as described in our previous studies
(16–18). The color development in the assay
was monitored by the absorbance at 405 nm using a kinetic
microplate reader (Gene Co., Ltd., Hong Kong, China). The
concentration of generated factor Xa was calculated as Vmax
(mOD/min) using a standard curve.
Western blot analysis
The THP-1 cells were seeded at 2×106
cells/well into 6-well plates and serum-starved for 16 h prior to
stimulation with the complex of monoclonal anti-β2GPI
(10 μg/ml)/β2GPI (100 μg/ml), LPS (500 ng/ml) for 6 h.
The cells in some wells were pre-treated with various
concentrations of EGCG (0–50 μg/ml) for 1 h, and EGCG was not
removed. For the determination of total cellular protein, the cells
were collected and lysed with lysis buffer containing 20 mM
Tris-HCl (pH 7.5), 1% Triton X-100, 150 mM NaCl, 2.5 mM EDTA and 1
mM PMSF. The lysates were centrifuged at 10,000 rpm for 30 min
using the Compact High Speed Refrigerated Centrifuge 6930. (Kubota,
Tokyo, Japan) to remove unbroken cells, nuclei and other
organelles. The supernatant containing plasma membrane was
recovered and stored at −70°C for analysis. Equal amounts of
protein (5 μg) were electrophoresed in 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis gels (SDS-PAGE) and
transferred onto a polyvinylidene difluoride (PVDF) membranes
(Bio-Rad, Hercules, CA, USA). The membranes were blocked in fresh
5% dry non-fat milk in Tris-buffered saline/0.05% Tween-20 (TBST)
for 1 h at room temperature, washed with TBST 3 times, and then
incubated with the primary antibodies against p38 MAPK, phospho-p38
MAPK (p-p38), ERK1/2, phospho-ERK1/2 (p-ERK1/2), JNK, phospho-JNK
(p-JNK), NF-κB (p65), phospho-NF-κB (p-p65), IκB-α (1:1,000; Cell
Signaling Technology, Beverly, MA, USA), TLR4 (1:500; eBioscience,
San Diego, CA, USA) and β-actin (1:2,500; Proteintech Group, Inc.,
Chicago, IL, USA) overnight at 4°C. Following 3 washes with TBST,
the membranes were incubated with horseradish peroxidase
(HRP)-conjugated goat anti-mouse or goat anti-rabbit secondary
antibodies (1:2,000; Santa Cruz Biotechnology, Inc.) for 1 h at
room temperature. Finally, the immunoblot signals were developed
using ECL detection reagents (Millipore, Billerica, MA, USA),
imaged and quantified using a Bio-Rad Fluor-S MultiImager (Typhoon
9400; Amersham, Uppsala, Sweden).
Statistical analysis
Data are expressed as the means ± SEM. The
statistically significant differences were calculated by applying
analysis of variance (ANOVA) using SPSS software (version 16.0).
Values of P<0.05 were considered to indicate statistically
significant differences.
Results
Anti-β2GPI/β2GPI
complex induces TF and TNF-α expression in THP-1 cells
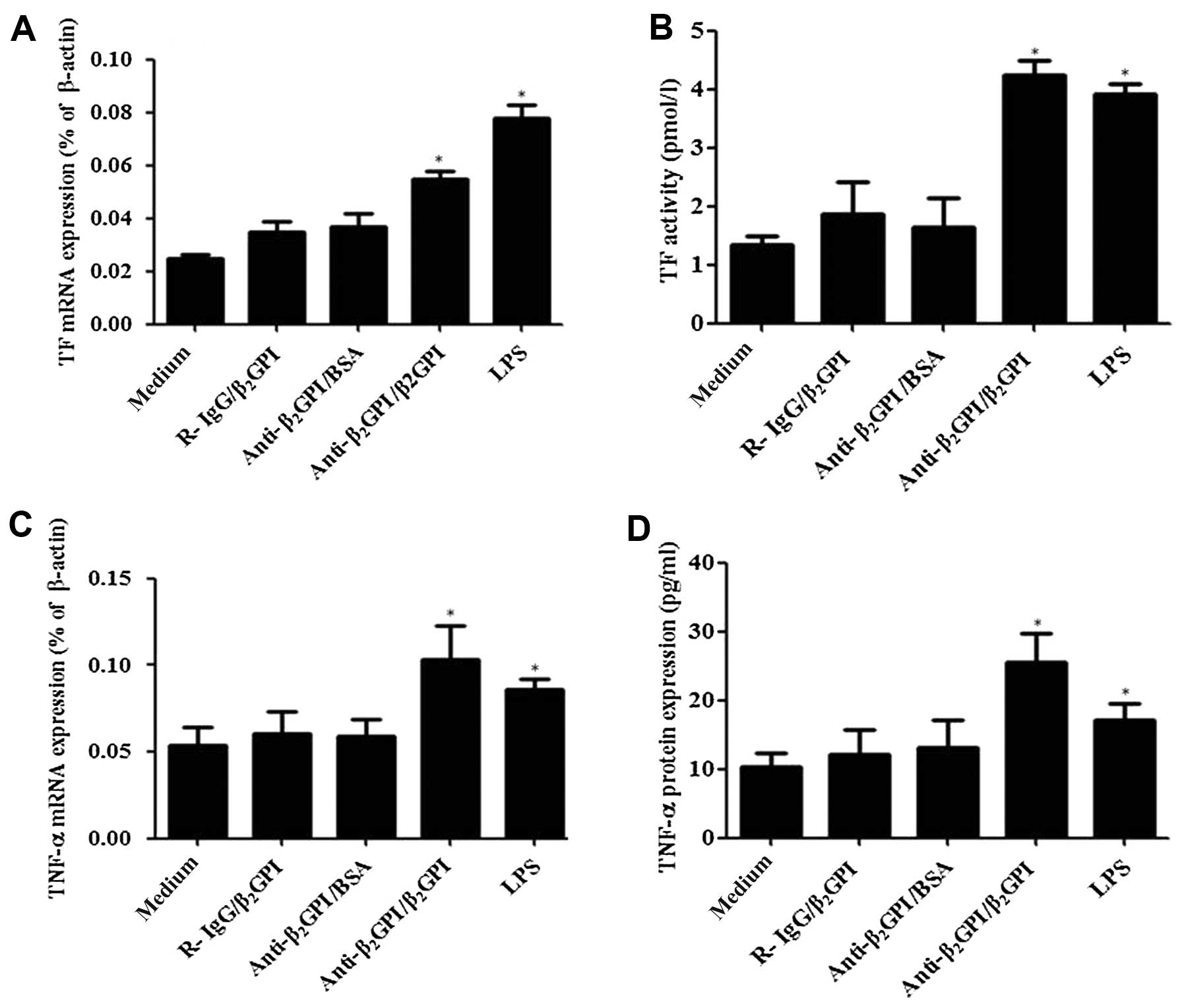
Treatment of the THP-1 cells with the
anti-β2GPI (10 μg/ml)/β2GPI (100 μg/ml)
complex significantly enhanced the TF mRNA levels (Fig. 1A), and its activity (Fig. 1B) compared to the untreated cells
(P<0.05). TNF-α mRNA (Fig. 1C)
and protein levels (Fig. 1D) were
also increased (P<0.05 vs. medium only), and these effects were
comparable to those induced by LPS (500 ng/ml). However, TF and
TNF-α expression did not increase in the cells treated with
anti-β2GPI (10 μg/ml)/BSA (100 μg/ml) or
R-IgG/β2GPI at the same concentration as
anti-β2GPI/β2GPI. These results were similar
to those obtained in our previous studies using THP-1 cells
(16–18).
EGCG inhibits TF expression induced by
anti-β2GPI/β2GPI complex in THP-1 cells
In this study, we first investigated whether EGCG
decreases the effects of anti-β2GPI/β2GPI
complex-induced TF expression in THP-1 cells. The cells were
treated with various concentrations of EGCG (0–50 μg/ml) and then
stimulated with anti-β2GPI (10 μg/ml)/β2GPI
(100 μg/ml) complex for the indicated periods of time.
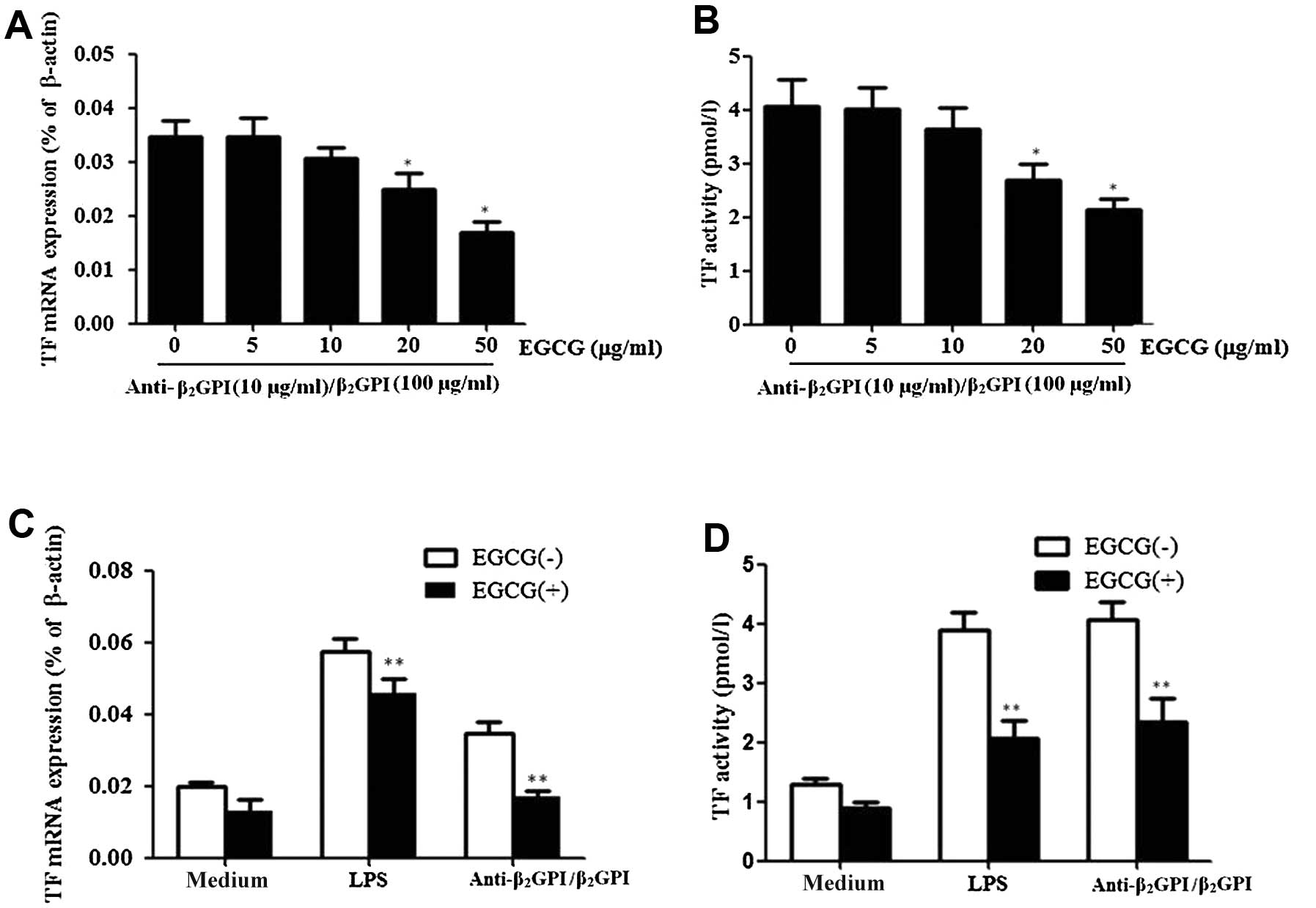
Pre-treatment with EGCG (0–50 μg/ml) inhibited
anti-β2GPI/β2GPI complex-induced TF
expression and activation in a dose-dependent manner, showing
statistical significance at 20–50 μg/ml [P<0.05 vs. control
(medium only)] (Fig. 2A and B).
The maximal inhibition rate of EGCG (50 μg/ml) on TF mRNA
expression and activity was approximately 48 and 50%,
respectively.
We then explored the specific effects of EGCG on
anti-β2GPI/β2GPI complex-enhanced TF
expression in THP-1 cells. Pre-treatment with 50 μg/ml EGCG
significantly reduced the anti-β2GPI/β2GPI
complex- or LPS-enhanced TF mRNA levels in THP-1 cells (P<0.05
vs. anti-β2GPI/β2GPI complex or LPS
stimulation alone) (Fig. 2C). In
addition, the enhanced TF activity level by the
anti-β2GPI/β2GPI complex or LPS was also
reduced by the presence of EGCG (50 μg/ml) (Fig. 2D) (P>0.05). EGCG alone had no
significant inhibitory effect on the TF level.
EGCG inhibits TNF-α expression induced by
the anti-β2GPI/β2GPI complex in THP-1
cells
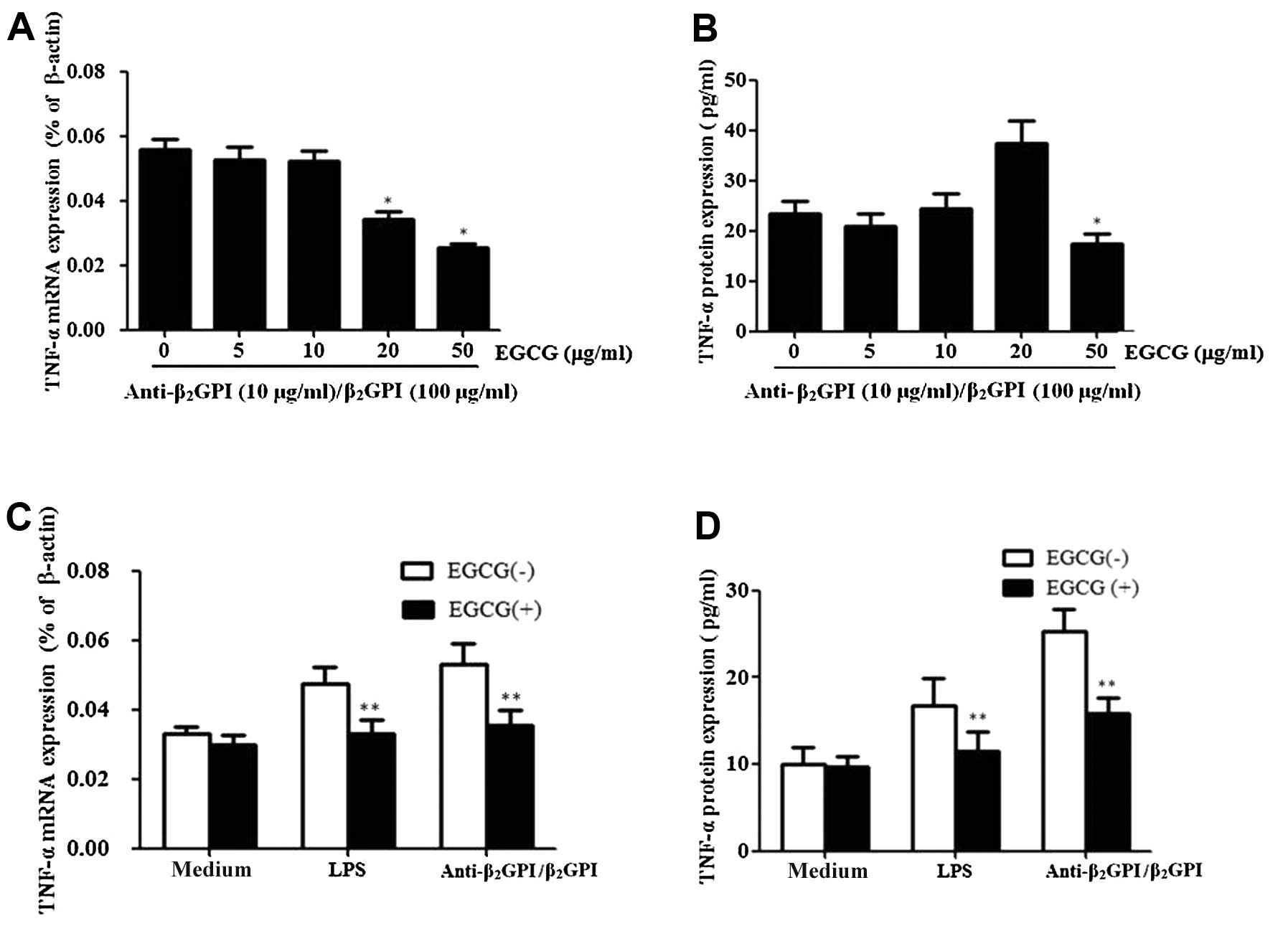
In order to investigate the effects of EGCG on the
expression of TNF-α induced by the
anti-β2GPI/β2GPI complex, the cells were
pre-treated with EGCG (0–50 μg/ml) prior to stimulation with the
anti-β2GPI/β2GPI complex for the indicated
periods of time. Pre-treatment with EGCG (5–50 μg/ml) inhibited the
mRNA expression levels of TNF-α in response to the
anti-β2GPI/β2GPI complex in a dose-dependent
manner, presenting statistical significance at 20–50 μg/ml
(P<0.05 vs. control) (Fig.
3A). EGCG also inhibited the protein expression of TNF-α
induced by the anti-β2GPI/β2GPI complex
(Fig. 3B). The maximal inhibition
rate of TNF-α mRNA and protein by EGCG (50 μg/ml) was approximately
51 and 30%, respectively.
We further determined the specific effects of EGCG
on TNF-α expression in THP-1 cells. Pre-treatment with 50 μg/ml
EGCG significantly reduced the
anti-β2GPI/β2GPI complex-enhanced TNF-α mRNA
and protein expression levels (Fig.
3C and D) (P<0.05 vs. anti-β2GPI/β2GPI
complex stimulation alone). Similarly, pre-treatment with 50 μg/ml
EGCG significantly decreased the LPS induced TNF-α mRNA and protein
expression levels (P>0.05). By contrast, EGCG alone had no
significant effects on the TNF-α level (medium only).
Effect of EGCG on the expression of TLR4
in THP-1 cells
TLR4, a family of integral membrane proteins, has
been reported to mediate aPL-induced endothelial cell or monocytic
cell activation (19). We have
previously demonstrated that TLR4 and its signal transduction
pathway contribute to anti-β2GPI/β2GPI
complex-induced TF and TNF-α expression in THP-1 cells (8). In this study, to examine whether
EGCG blocks the effects of the
anti-β2GPI/β2GPI complex on TLR4 expression
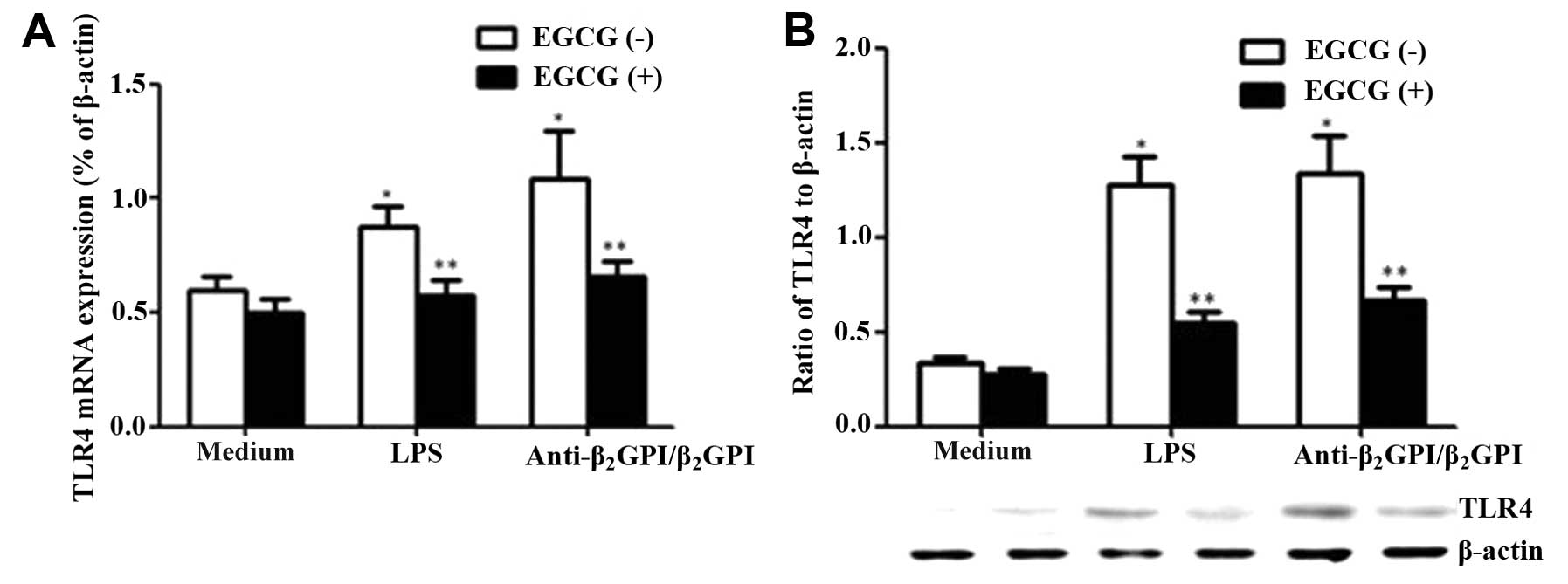
in THP-1 cells, TLR4 mRNA and protein levels in these cells were
evaluated under different conditions. We found that both the
anti-β2GPI (10 μg/ml)/β2GPI (100 μg/ml)
complex and LPS (500 ng/ml) increased the mRNA levels of TLR4
(Fig. 4A, white column)
(P<0.05 vs. medium only). However, pre-incubation of the THP-1
cells with EGCG (50 μg/ml) significantly decreased TLR4 mRNA
expression (P<0.05), even though the cells were treated with
similar concentrations of the anti-β2GPI (10
μg/ml)/β2GPI (100 μg/ml) complex or LPS (Fig. 4A, black column). Similarly, the
TLR4 protein expression levels decreased following treatment with
EGCG prior to stimulation with the
anti-β2GPI/β2GPI complex or LPS (Fig. 4B) (P<0.05 vs.
anti-β2GPI/β2GPI complex or LPS stimulation
alone). Compared with the corresponding controls
(anti-β2GPI/β2GPI complex alone), EGCG (50
μg/ml) decreased the TLR4 mRNA and protein expression levels by
approximately 40 and 50%, respectively (P>0.05). EGCG alone had
no significant effect on TLR4 expression compared to treatment with
medium alone.
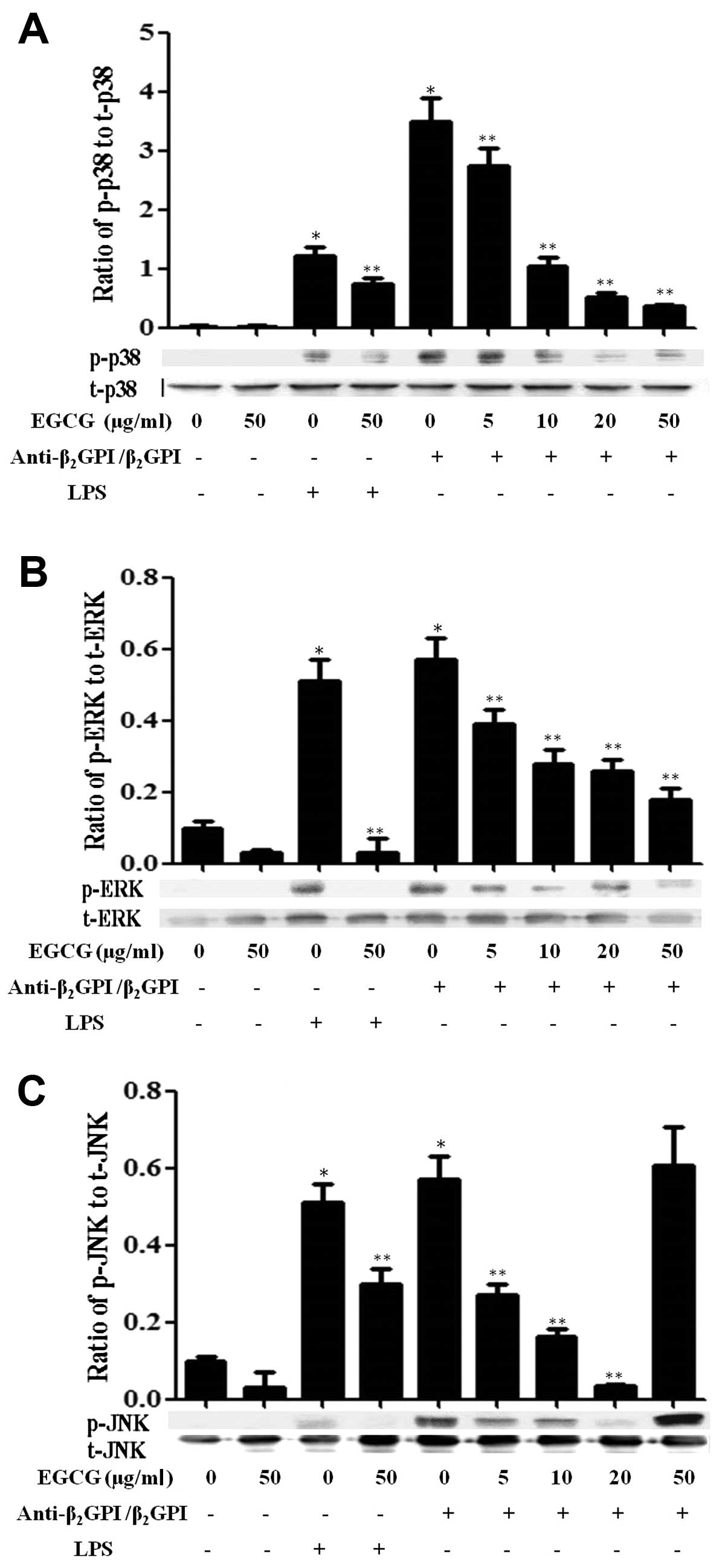
Effect of EGCG on MAPK signaling
pathways
Previously, we found that the
anti-β2GPI/β2GPI complex or LPS stimulated
the activation of MAPK pathways in THP-1 cells within 30 min of
treatment (11). In this study,
we further investigated whether EGCG suppresses the activation of
MAPKs induced by the anti-β2GPI/β2GPI complex
or LPS in THP-1 cells. The anti-β2GPI/β2GPI
and LPS significantly increased the phosphorylation of p38 MAPK
(p-p38), ERK1/2 (p-ERK) and JNK1/2 (p-JNK) in the cells (P<0.05
vs. medium only) (Fig. 5). As the
concentration of EGCG increased (0–50 μg/ml), the expression levels
of total p38 MAPK (t-p38), ERK1/2 (t-ERK) and JNK1/2 (t-JNK) were
not altered, but the levels of p-p38 (Fig. 5A) and p-ERK1/2 (Fig. 5B) in the THP-1 cells stimulated
with the anti-β2GPI/β2GPI complex gradually
decreased, indicating a dose-dependent inhibitory effect of EGCG
(P<0.05 vs. anti-β2GPI/β2GPI complex
alone). On the other hand, EGCG (5–20 μg/ml) inhibited the
phosphorylation of JNK induced by the
anti-β2GPI/β2GPI complex gradually; however,
a high concentration of EGCG (50 μg/ml) did not reduce the
phosphorylation of JNK in the
anti-β2GPI/β2GPI complex-stimulated THP-1
cells (Fig. 5C). In addition, the
stimulatory effects of LPS (500 ng/ml) on MAPKs, including p-p38,
p-ERK1/2 and p-JNK were also blocked by EGCG (50 μg/ml) (P<0.05
vs LPS stimulation alone).
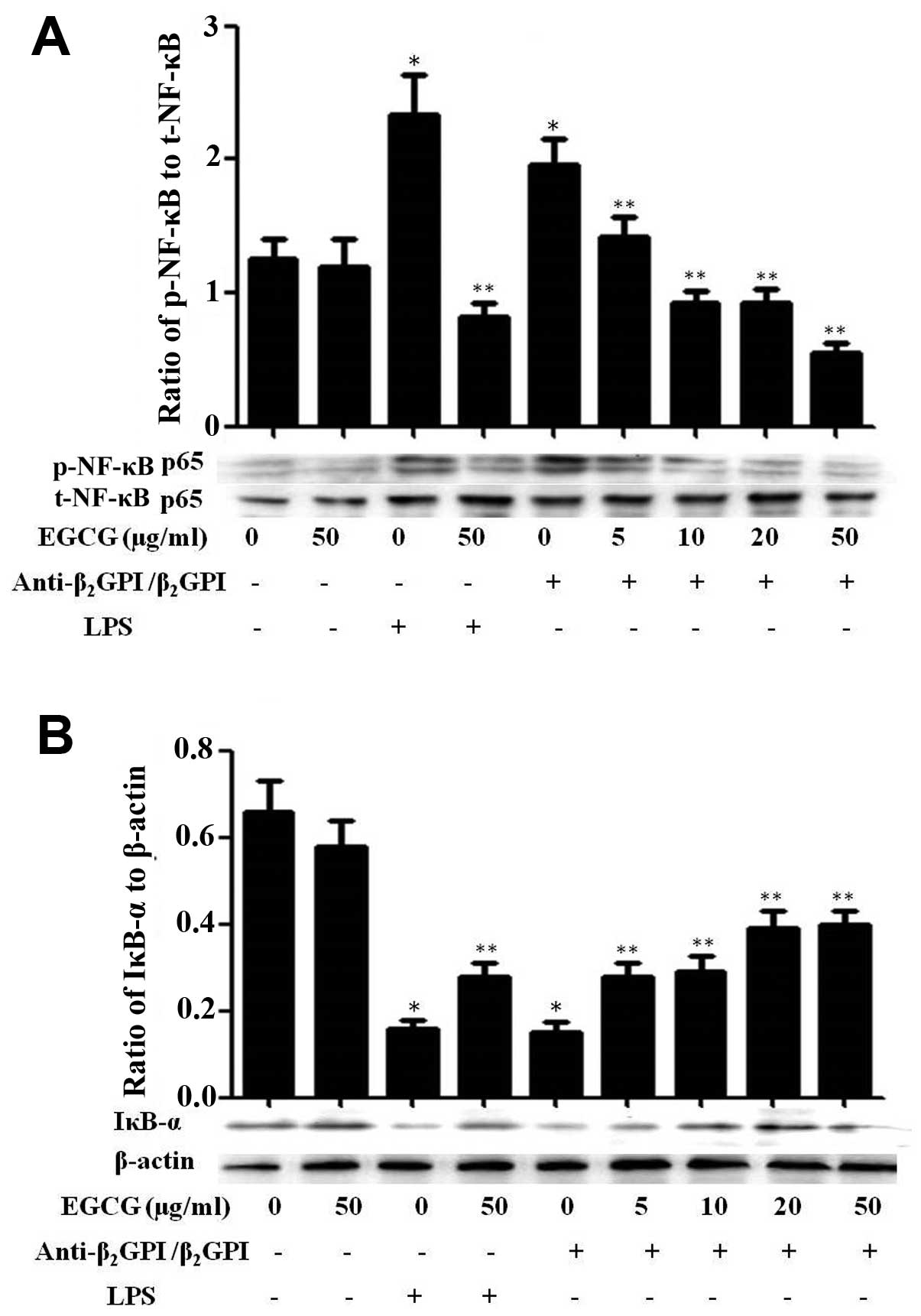
Effect of EGCG on NF-κB activation in
THP-1 cells
NF-κB, originally emerged as a major regulator of
innate and adaptive immunity and inflammatory responses, and has
been shown to be involved in the signal transduction of TLR4/LPS
(20). In a previous study, we
indicated that NF-κB can be activated and plays important roles in
the process of anti-β2GPI/β2GPI-induced TF
expression in THP-1 cells, thereby contributing to the pathological
processes of APS (9). In this
study, we further investigated the effects of EGCG on the
activation of the NF-κB pathway in THP-1 cells. Treatment with
anti-β2GPI (10 μg/ml)/β2GPI (100 μg/ml)
complex or LPS (500 ng/ml) considerably increased the
phosphorylation of NF-κB, which was inhibited by pre-treatment with
EGCG (5–50 μg/ml) in a dose-dependent manner (Fig. 6A). On the other hand, treatment
with the anti-β2GPI (10 μg/ml)/β2GPI (100
μg/ml) complex or LPS (500 ng/ml) effectively inhibited the
expression of IκB-α (inhibitor of NF-κB) in THP-1 cells and this
inhibitory effect was reversed by EGCG (5–50 μg/ml) pre-treatment
in a dose-dependent manner (Fig.
6B). Similarly, the stimulatory effects of LPS (100 ng/ml) on
NF-κB were also blocked by EGCG (50 μg/ml). EGCG alone had no
significant effect on the NF-κB pathway compared to treatment with
medium alone. These results suggest that EGCG blocks the
transduction of the NF-κB pathway induced by the
anti-β2GPI/β2GPI complex.
Discussion
APS is defined by one or more episodes of thrombosis
or unexplained pregnancy loss in association with persisting
positive aPLs, either anticardiolipin (aCL), anti-β2GPI,
and/or lupus anticoagulant (LAC) (21). APS that is frequently associated
with underlying autoimmune disorders, most commonly systemic lupus
erythematosus (SLE), is known as secondary APS, otherwise it is
identified as primary APS. In its most severe and life-threatening
form, APS is termed as catastrophic APS. Since the identification
of APS by Hughes in 1985, the mechanisms or targets of injury
underlying thrombotic events have been vigorously debated. aPL
antibodies, which bind to a range of cellular targets, such as
platelets, monocytes or/and endothelial cells, can upregulate TF.
TF as the main initiation factor of the blood coagulation cascade,
has been suggested to be a main potential mechanism of APS-related
thrombosis (22). On the other
hand, the enhanced expression and secretion of cytokines,
particularly TNF-α, may contribute to the thrombotic activity in
patients with APS (23). In the
present study, we first demonstrate that the
anti-β2GPI/β2GPI complex or LPS activate the
monocytic THP-1 cell line, by increasing TF and TNF-α expression
(Fig. 1). The current finding
suggests that the anti-β2GPI/β2GPI complex
can induce pro-inflammatory and procoagulant phenotypes
characterized by the release of TNF-α and TF, respectively.
Recent improvements in the understanding of the
pathogenic mechanisms of APS, including the aPL-induced activation
of platelets, endothelial cells, monocytes, complement and
coagulation cascade, has led to the identification of potential
targets and future therapies for APS. In general, treatment
regimens for APS must be individualized according to the current
clinical status of the patient and the history of thrombotic
events. Low-dose aspirin is used widely in the treatment of
patients with APS. However, the effectiveness of low-dose aspirin
as the primary prevention therapy for APS remains unproven.
Clopidogrel has anecdotally been reported to be helpful in
individuals with APS and may be useful in patients allergic to
aspirin (24). In patients with
SLE, hydroxychloroquine is also considered to be helpful as it has
intrinsic antithrombotic properties (25). If thrombotic events reoccur in
patients with APS, a combination of warfarin and aspirin may be
used (26). Treatment for
significant thrombotic events in patients with APS is generally
lifelong. Current management strategies for patients with APS are
restricted mainly to anticoagulation therapy, which is not
effective in all patients (27).
Despite antithrombotic therapy, up to 30% of patients with APS have
recurrent thrombotic events (28). Furthermore, patients (2–3%)may
experience bleeding complications with conventional anticoagulation
therapy (29). The optimal
treatment strategy for patients with APS resistant or intolerant to
long-term anticoagulation remains to be discovered.
Based on recent advances in the pathophysiology of
APS, many new therapeutic modalities for treating and/or preventing
thrombosis in patients with APS have been reported, such as B-cell
targeted therapies (30),
eaulizumab (a monoclonal antibody directed against complement C5)
(31), defibrotide (an adenosine
receptor against that blocks monocyte TF expression) (31), statins (32), antiplatelet agents (33) and intracellular pathway inhibitors
[SB203580, a specific inhibitor of p38 MAPK (34), MG132, a specific inhibitor of
NF-κB (35)]. However, available
data on humans are limited to support these innovative
approaches.
Green tea, produced from the tea plant Camellia
sinensis, has been consumed as a popular beverage worldwide for
thousands of years. The most significant phytochemical in green tea
is a polyphenol termed EGCG. EGCG has been the subject of interest
in a number of studies investigating its potential use as a
therapeutic agent for a broad range of disorders over the past few
decades. For example, EGCG (1–30 μM) has been shown to inhibit
TNF-α and histamine induced endothelial TF expression and activity
in a concentration dependent manner, resulting in a 87% reduction
in TF expression (36). EGCG has
been shown to have an enhanced inhibitory effect on the release of
TNF-α from BALB/3T3 cells treated with okadaic acid (37). EGCG has also been shown to inhibit
the production of inflammatory mediators, such as TNF-α, IL-6 and
IL-8, through the inhibition of intracellular Ca2+
levels (38). These results are
generally in accordance with those in our current study. However,
to our knowledge, EGCG has never been investigated as a therapeutic
strategy for APS. As demonstrated in this study, the
anti-β2GPI/β2GPI complex-enhanced TF and
TNF-α mRNA expression, as well as TF activity were inhibited in a
dose-dependent manner by EGCG. The maximal inhibition rates of EGCG
(50 μg/ml) on TF (mRNA level and activity) and TNF-α expression
(mRNA and protein) (induced by
anti-β2GPI/β2GPI complex) were approximately
48, 50, 51 or 30%, respectively and were comparable to those of
EGCG on LPS-induced expression (Figs.
2 and 3). However, it is
puzzling that EGCG (0–20 μg/ml) had irregular effects on the TNF-α
levels of cells stimulated with the
anti-β2GPI/β2GPI complex. Our study
demonstrated that EGCG may be effective in preventing thrombosis in
patients with APS. However, the biological mechanisms of action of
EGCG and its use as a primary prevention treatment for APS remain
unproven.
In our previous studies, we revealed that TLR4 acts
as a co-factor for Annexin A2 on the THP-1 cell surface, and
contributes to anti-β2GPI/β2GPI
complex-enhanced TF expression in THP-1 cells (6,8,16,17). In addition, MD-2 and MyD88 are
also upregulated and cooperate with TLR4, thus leading to the
activation of MAPKs in this process (11). We further demonstrated that the
anti-β2GPI/β2GPI complex upregulated TF
expression in THP-1 cells, following the activation of nuclear
transcription factors, including NF-κB and AP-1 (9). In addition, the intracellular signal
transduction pathway of TLRs-MAPKs-NF-κB/AP-1 axis in
anti-β2GPI/β2GPI complex-induced TF and TNF-α
expression may contribute to the pathological mechanisms
responsible for causing thrombosis in patients with APS.
In a previous study, EGCG was reported to affect an
array of signal pathways through which it exerts its
pharmacological activities. EGCG was shown to inhibit the
degradation of IRAK induced by IL-1β in A549 cells (39). This polyphenol has also been shown
to inhibit the LPS-induced activation of MAPK pathways, including
ERK1/2, p38 and JNK (40). It has
been described that EGCG suppresses the LPS-induced activation of
NF-κB by blocking the degradation of IκB-α following IκB-α
phosphorylation (41). Moreover,
EGCG inhibits MyD88-dependent signaling pathways and TIR
domain-containing adaptor inducing IFN-β (TRIF)-dependent signaling
pathways of TLRs in RAW264.7 cells, which suppresses inflammatory
responses (42).
In the present study, we further demonstrate that
EGCG (50 μg/ml) significantly suppresses TLR4 mRNA and protein
expression in cells stimulated with the
anti-β2GPI/β2GPI complex or LPS (Fig. 4). Our results suggest that EGCG is
capable of blocking the expression of TLR4 in THP-1 cells and
therefore, can contribute to the suppression of THP-1 cell
activation by inhibiting TF and TNF-α expression. As shown by our
results, the anti-β2GPI/β2GPI complex or LPS
activated MAPK pathways, as indicated by the increased
phosphorylation of p38, ERK1/2 and JNK, as well as by the NF-κB
(p65/RelA) pathway in the nuclear fraction (Figs. 5 and 6). Furthermore, EGCG (5–50 μg/ml)
inhibited both MAPK (including p38, ERK1/2 as well as JNK) and
NF-κB activation induced by the
anti-β2GPI/β2GPI complex in a dose-dependent
manner. Similarly, EGCG at a concentration of 50 μg/ml markedly
inhibited LPS-induced MAPK and NF-κB activation. These results
indicate that the blockade of MAPK and NF-κB activation is the
major mechanism responsible for the inhibitory effects of EGCG on
the anti-β2GPI/β2GPI complex-mediated
expression of TF and TNF-α. However, it is worth mentioning that
EGCG (5–10 μg/ml) only slightly inhibited TF and TNF-α expression,
but significantly blocked the activation of MAPKs and NF-κB. This
may explain the fact that the
anti-β2GPI/β2GPI complex stimulated cells not
only throught he MAPKs/NF-κB pathway, but also through other
receptors. Zhang et al (43) recently demonstrated that WNT
signaling activation stimulates the production of the
pro-inflammatory cytokines, IL-18 and TNF-α, in the spinal cord,
suggesting that it may contribute to the uptake of the
anti-β2GPI/β2GPI complex.
In conclusion, data from our present study, as well
as from our previous studies, strongly indicate that EGCG inhibits
the anti-β2GPI/β2GPI-induced activation of
THP-1 cells by decreasing TF and TNF-α expression levels via
blocking the intracellular signal transduction pathway of
TLRs-MAPKs-NF-κB axis, and may serve as a preventive and
therapeutic agent for APS.
Acknowledgements
This study was supported by grants from the
National Natural Science Foundation of China (no. 81370614) to H.Z.
and the Student’s Scientific Research of Jiangsu University (no.
CX08 B_16x) to T.W.
References
|
1
|
de Groot PG and Derksen RH:
Antiphospholipid antibodies: update on detection pathophysiology,
and treatment. Curr Opin Hematol. 11:165–169. 2004.PubMed/NCBI
|
|
2
|
Miyakis S, Lockshin MD, Atsumi T, et al:
International consensus statement on an update of the
classification criteria for definite antiphospholipid syndrome
(APS). J Thromb Haemost. 4:295–306. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bas de Laat H, Derksen RH and de Groot PG:
beta2-glycoprotein I, the playmaker of the antiphospholipid
syndrome. Clin Immunol. 112:161–168. 2004.PubMed/NCBI
|
|
4
|
Mulla MJ, Brosens JJ, Chamley LW, et al:
Antiphospholipid antibodies induce a pro-inflammatory response in
first trimester trophoblast via the TLR4/MyD88 pathway. Am J Reprod
Immunol. 62:96–111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mehdi AA, Uthman I and Khamashta M:
Antiphospholipid syndrome: pathogenesis and a window of treatment
opportunities in the future. Eur J Clin Invest. 40:451–464. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xie H, Zhou H, Wang H, Chen D, Xia L, Wang
T and Yan J: Anti-β(2)GPI/β(2)GPI induced TF and TNF-α expression
in monocytes involving both TLR4/MyD88 and TLR4/TRIF signaling
pathways. Mol Immunol. 53:246–254. 2013.
|
|
7
|
Lu YC, Yeh WC and Ohashi PS: LPS/TLR4
signal transduction pathway. Cytokine. 42:145–151. 2008. View Article : Google Scholar
|
|
8
|
Zhou H, Yan Y, Xu G, et al: Toll-like
receptor (TLR)-4 mediates anti-β(2) GPI/β(2) GPI-induced tissue
factor expression in THP-1 cells. Clin Exp Immunol. 163:189–198.
2011.
|
|
9
|
Xia L, Zhou H, Hu L, et al: Both NF-κB and
c-Jun/AP-1 involved in
anti-β2GPI/β2GPI-induced tissue factor
expression in monocytes. Thromb Haemost. 109:643–651. 2013.
|
|
10
|
Lawrence MC, Jivan A, Shao C, et al: The
roles of MAPKs in disease. Cell Res. 18:436–442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou H, Chen D, Xie H, Xia L, Wang T, Yuan
W and Yan J: Activation of MAPKs in the
anti-β2GPI/β2GPI-induced tissue factor
expression through TLR4/IRAKs pathway in THP-1 cells. Thromb Res.
130:e229–e235. 2012.
|
|
12
|
Lee SH, Nam HJ, Kang HJ, Kwon HW and Lim
YC: Epigallocatechin-3-gallate attenuates head and neck cancer stem
cell traits through suppression of Notch pathway. Eur J Cancer.
49:3210–3218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sarkar FH, Li Y, Wang Z and Kong D: The
role of nutraceuticals in the regulation of Wnt and Hedgehog
signaling in cancer. Cancer Metastasis Rev. 29:383–394. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin HY, Hou SC, Chen SC, et al:
(−)-Epigallocatechin gallate induces Fas/CD95-mediated apoptosis
through inhibiting constitutive and IL-6-induced JAK/STAT3
signaling in head and neck squamous cell carcinoma cells. J Agric
Food Chem. 60:2480–2489. 2012.
|
|
15
|
Kim SJ, Jeong HJ, Lee KM, et al:
Epigallocatechin-3-gallate suppresses NF-kappaB activation and
phosphorylation of p38 MAPK and JNK in human astrocytoma U373MG
cells. J Nutr Biochem. 18:587–596. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou H, Wolberg AS and Roubey RA:
Characterization of monocyte tissue factor activity induced by IgG
antiphospholipid antibodies and inhibition by dilazep. Blood.
104:2353–2358. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou H, Wang H, Li N, Yu Y, Huang H, Yan Y
and Wang T: Annexin A2 mediates
anti-β2GPI/β2GPI-induced tissue factor
expression on monocytes. Int J Mol Med. 24:557–562. 2009.
|
|
18
|
Xu G, Wen H, Zhou H, et al: Involvement of
IRAKs and TRAFs in anti-β2GPI/β2GPI-induced
tissue factor expression in THP-1 cells. Thromb Haemost.
106:1158–1169. 2011.PubMed/NCBI
|
|
19
|
Pierangeli SS, Vega-Ostertag ME, Raschi E,
et al: Toll-like receptor and antiphospholipid mediated thrombosis:
in vivo studies. Ann Rheum Dis. 66:1327–1333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vallabhapurapu S and Karin M: Regulation
and function of NF-kappaB transcription factors in the immune
system. Annu Rev Immunol. 27:693–733. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McIntyre JA, Wagenknecht DR and Faulk WP:
Antiphospholipid antibodies: discovery, definitions, detection and
disease. Prog Lipid Res. 42:176–237. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boles J and Mackman N: Role of tissue
factor in thrombosis in antiphospholipid antibody syndrome. Lupus.
19:370–378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Swadzba J, Iwaniec T and Musial J:
Increased level of tumor necrosis factor-α in patients with
antiphospholipid syndrome: marker not only of inflammation but also
of the prothrombotic state. Rheumatol Int. 31:307–313. 2011.
|
|
24
|
Agaba AE, Charaklias N, Babu-Victor A, et
al: Antiphospholipid syndrome: a series of surgical emergencies and
the current evidence for its managemen. Ann R Coll Surg Engl.
88:370–374. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Petri M: Use of hydroxychloroquine to
prevent thrombosis in systemic lupus erythematosus and in
antiphospholipid antibody-positive patients. Curr Rheumatol Rep.
13:77–80. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Les I, Guillermo RL, Munther A, et al:
Intensity and duration of anticoagulation therapy in
antiphospholipid syndrome. Semin Thromb Hemost. 38:339–347. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pierangeli SS, Colden-Stanfield M, Liu X,
Barker JH, Anderson GL and Harris EN: Antiphospholipid antibodies
from antiphospholipid syndrome patients activate endothelial cells
in vitro and in vivo. Circulation. 99:1997–2002. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gharavi AE, Pierangeli SS,
Colden-Stanfield M, Liu XW, Espinola RG and Harris EN: GDKV-induced
antiphospholipid antibodies enhance thrombosis and activate
endothelial cells in vivo and in vitro. J Immunol. 163:2922–2927.
1999.PubMed/NCBI
|
|
29
|
Pierangeli SS, Liu SW, Anderson G, Barker
JH and Harris EN: Thrombogenic properties of murine
anti-cardiolipin antibodies induced by beta 2 glycoprotein 1 and
human immunoglobulin G antiphospholipid antibodies. Circulation.
94:1746–1751. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Iglesias Jiménez E, Camacho-Lovillo M,
Falcón-Neyra D, Lirola-Cruz J and Neth O: Infant with probable
catastrophic antiphospholipid syndrome successfully managed with
rituximab. Pediatrics. 125:e1523–e1528. 2010.PubMed/NCBI
|
|
31
|
Corbacioglu S, Cesaro S, Faraci M, et al:
Defibrotide for prophylaxis of hepatic veno-occlusive disease in
paediatric haemopoietic stem-cell transplantation: an open-label,
phase 3, randomised controlled trial. Lancet. 379:1301–1309. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Espinosa G, Berman H and Cervera R:
Management of refractory cases of catastrophic antiphospholipid
syndrome. Autoimmun Rev. 10:664–668. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Forastiero RR, Martinuzzo ME and de
Larrañaga GF: Circulating levels of tissue factor and
proinflammatory cytokines in patients with primary antiphospholipid
syndrome or leprosy related antiphospholipid antibodies. Lupus.
14:129–136. 2005. View Article : Google Scholar
|
|
34
|
Yoon KH: Sufficient evidence to consider
hydroxychloroquine as an adjunct therapy in antiphospholipid
antibody (Hughes’) syndome. J Rheumatol. 29:1574–1575.
2002.PubMed/NCBI
|
|
35
|
Montiel-Manzano G, Romay-Penabad Z,
Papalardo de Martínez E, Meillon-García LA, García-Latorre E,
Reyes-Maldonado E and Pierangeli SS: In vivo effects of an
inhibitor of nuclear factor-kappa B on thrombogenic properties of
antiphospholipid antibodies. Ann NY Acad Sci. 1108:540–553. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Holy EW, Stämpfli SF, Akhmedov A, et al:
Laminin receptor activation inhibits endothelial tissue factor
expression. J Mol Cell Cardiol. 48:1138–1145. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Suganuma M, Sueoka E, Sueoka N, et al:
Mechanisms of cancer prevention by tea polyphenols based on
inhibition of TNF-α expression. Biofactors. 13:67–72.
2000.PubMed/NCBI
|
|
38
|
Shin HY, Kim SH, Jeong HJ, et al:
Epigallocatechin-3-gallate inhibits secretion of TNF-alpha, IL-6
and IL-8 through the attenuation of ERK and NF-kappaB in HMC-1
cells. Int Arch Allergy Immunol. 142:335–344. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wheeler DS, Catravas JD, Odoms K,
Denenberg A, Malhotra V and Wong HR: Epigallocatechin-3-gallate, a
green tea-derived polyphenol, inhibits IL-1 beta-dependent
proinflammatory signal transduction in cultured respiratory
epithelial cells. J Nutr. 134:1039–1044. 2004.
|
|
40
|
Lin YL and Lin JK:
(−)-Epigallocatechin-3-gallate blocks the induction of nitric oxide
synthase by down-regulating lipopolysaccharide-induced activity of
transcription factor nuclear factor-kappaB. Mol Pharmacol.
52:465–472. 1997.
|
|
41
|
Ahn SC, Kim GY, Kim JH, et al:
Epigallocatechin-3-gallate, constituent of green tea, suppresses
the LPS-induced phenotypic and functional maturation of murine
dendritic cells through inhibition of mitogen-activated protein
kinases and NF-kappaB. Biochem Biophys Res Commun. 313:148–155.
2004. View Article : Google Scholar
|
|
42
|
Youn HS, Lee JY, Saitoh SI, Miyake K, Kang
KW, Choi YJ and Hwang DH: Suppression of MyD88- and TRIF-dependent
signaling pathways of Toll-like receptor by
(−)-epigallocatechin-3-gallate, a polyphenol component of green
tea. Biochem Pharmacol. 72:850–859. 2006.
|
|
43
|
Zhang YK, Huang ZJ, Liu S, Liu YP, Song AA
and Song XJ: WNT signaling underlies the pathogenesis of
neuropathic pain in rodents. J Clin Invest. 123:2268–2286. 2013.
View Article : Google Scholar : PubMed/NCBI
|