Introduction
Microglia, which are a crucial component of the
innate immune system of the central nervous system (CNS), are known
to play crucial roles in regulating neuroinflammation (1). Activated microglia are known to
perform beneficial functions in defense and tissue repair in the
CNS (1). Findings of previous
studies have shown that activated microglia also propagate
inflammation in the CNS through antigen presentation and production
of proinflammatory cytokines or chemokines, reactive oxygen
species, and nitric oxide (NO) (2–4).
For example, Toll-like receptor 4 (TLR4), a member of the Toll-like
receptor family, is involved as a lipopolysaccharide (LPS) receptor
in the activation of microglia (5). Activation of TLR4 expressed in
microglia leads to neuronal injury through its ligation with LPS
(6). Blockade of microglial
activity suppresses the development of inflammatory lesions in an
experimental autoimmune encephalitis model (7). However, the manner in which
immunoregulatory molecules controlling microglial activity are
involved in the development of neuroinflammation remains to be
determined.
Semaphorins and their receptors exhibit various
functions in axon guidance, organogenesis, angiogenesis,
tumorigenesis, and immune regulation (8–18).
The primary receptors for semaphorins are known to be members of
the plexin family (9,19–21). Plexin-A1, in combination with
ligand-binding neuropilins, transmits repulsive axon guidance
signals for soluble class III semaphorins inside the axonal growth
cone (22). Plexin-A1 has also
been shown to play important roles in the developmental stages of
chick heart by working as a receptor for the transmembrane
semaphorin, Sema6D, in a neuropilin-independent manner (23). Furthermore, Plexin-A1 expressed in
dendritic cells is involved in T-dendritic cell interactions in the
immune system (24).
Sema3A, a ligand of the Neuropilin-1-Plexin-A1
coreceptor complex also plays a role as an inducer of neuronal
apoptosis during the embryonic stage (25). However, recent identification of
Sema3A expression in injured adult brain suggests that semaphorins
affect neural regeneration (26,27). Upregulation of Sema3A mRNA and
Neuropilin-1 and -2 mRNA after middle cerebral artery occlusion
suggested that reciprocal contact between injured neurons and
activated microglia may promote phagocytosis of damaged neurons
(26). The expression of
Plexin-A1 in rat microglia has also been identified, suggesting
that Sema3A produced by injured neurons suppresses
neuroinflammation by inducing apoptosis of activated microglia
through binding to the Neuropilin-1-Plexin-A1 coreceptor complex
(28). As described above,
neuroinflammation may be induced extensively in Plexin-A1-deficient
(Plexin-A1−/−) brain by abnormally activated microglia, since
apoptosis hardly occurs in Plexin-A1−/− microglia. Therefore,
microglial responses to inflammatory stimuli may be more
intensified in Plexin-A1−/− brain than in wild-type (WT) brain. To
explore this possibility, we compared the level of
neuroinflammation induced by intracerebroventricular (ICV)
administration of LPS to WT and to Plexin-A1−/− mice. Contrary to
our prediction, LPS-induced neuroinflammation was significantly
weaker in Plexin-A1−/− mice than in WT mice. Thus, the present
findings indicated a mechanism in which Plexin-A1 expressed in
microglia is integral to the optimal production of
inflammation-related mediators, such as cytokines, following TLR4
stimulation.
Materials and methods
Animals
Plexin-A1−/− mice were generated by gene targeting
with E14.1 embryonic stem (ES) cells (29). Briefly, the gene targeting vector
was designed to replace the genomic region containing the
initiation codon and the Sema domain-coding sequence with a
neomycin-resistance gene, and then transfected into E14.1 ES cells
by electroporation. G418- and ganciclovir-resistant clones were
screened by polymerase chain reaction (PCR) and confirmed by
Southern blotting. Mutant ES cells were introduced into mouse
blastocysts and then transferred into pseudopregnant mice to
generate chimeras. F1 heterozygous knockout mice were generated by
breeding the chimeras with Balb/c mice and were then backcrossed
with Balb/c mice for 10 generations. Pairs of resultant
heterozygous mice were bred to gain homozygous knockout mice and
their WT littermates as controls.
The mice were housed in the Wakayama Medical
University animal facilities and the animal center in the Faculty
of Pharmacy of Meijo University. The care and use of mice as well
as other experimental protocols were conducted in accordance with
the guidelines promulgated by the Physiological Society of Japan
and the guidelines on animal experimentation of both Wakayama
Medical University and Meijo University. The Animal Ethics Review
Committee of both institutions approved the experimental
protocol.
Genotype analysis
Genotyping was performed by PCR with mouse tail DNA
as the template and a Plexin-A1 gene-specific primer set as
previously reported (29).
Isolation of primary microglia and
immunocytochemistry
For optimal dissociation of tissue samples, brain
tissue of WT and Plexin-A1−/− pups from postnatal day 1–3 (P1–3)
was dissociated using a Neural Tissue Dissociation Kit (Sumitomo
Bakelite Co., Ltd., Tokyo, Japan). Microglia were isolated from the
single-cell suspension by MACS Technology using CD11b MicroBeads
(Miltenyi Biotec, Bergisch Gladbach, Germany) according to the
manufacturer’s instructions. The isolated microglia were then
cultured for 24 h in a culture medium (Sumitomo Bakelite). The
cells were then fixed in 4% paraformaldehyde in phosphate-buffered
saline (PBS) and processed for immunocytochemistry. After fixed
cultures were permeabilized with 0.2% Triton X-100 in PBS for 5
min, the microglia were stained with isolectin IB4 conjugated with
Alexa 488 (Life Technologies, Carlsbad, CA, USA) for microglial
identification and visualization with anti-Plexin-A1 antibodies
(Abcam, Cambridge, MA, USA). Alexa 594-conjugated secondary
antibodies were used to visualize primary antibody staining, and
DAPI was added for the final 20 min of incubation for nuclear
identification.
Reverse transcription (RT)-PCR analysis
for Plexin-A1 and Neuropilin-1 gene transcripts
RNA was isolated from primary microglia or mouse
hippocampi by the SV total RNA isolation system (Promega, Madison,
WI, USA). RT of RNA was performed with Super-Script II reverse
transcriptase and random primers (Life Technologies). The samples
were normalized with β-actin amplification for semiquantification.
The primers used for PCR amplification were: Plexin-A1 forward,
5′-GTGTGTGGATAGCCATCA-3′ and reverse, 5′-CCAGCCTCTCGAACACT-3′;
Neuropilin-1 forward, 5′-GGCCTCCTGCGATTCGTTACTGCT-3′ and reverse,
5′-CTTAGCCTTGCGCTTGCTGTCATC-3′; and Sema3A forward,
5′-ATTGAATTCAACTATGCAAACGGAAA GAA-3′ and reverse,
5′-TAAGCGGCCGCGACACTTCTG GGTGCCCGCT-3′. Control primers used were:
β-actin forward, 5′-GGGACGACATGGAGAAGATC-3′ and reverse,
5′-AGGTCTTTACGGATGTCAACG-3′. All the primers were annealed at 63ºC,
and 35 cycles of amplification were performed.
Immunohistochemistry
Mice were anesthetized by intraperitoneal injection
of pentobarbital sodium (0.648 mg/10 g body weight; Kyoritsu
Seiyaku Co., Tokyo, Japan) and perfused intracardially with 4%
paraformaldehyde. The brain excised from each mouse was fixed in 4%
paraformaldehyde, and brain injury was estimated based on the
results of hematoxylin and eosin (H&E) staining and
immunohistochemistry in consecutive frozen sections of 10 μm
prepared from the mouse brain 18 h after ICV injection of the mice.
The sections were immunolabeled with anti-Iba-1 antibody (Wako,
Osaka, Japan). Microglia were detected by Iba-1 immunostaining,
which recognizes both resting and activated microglia.
Concurrently, DAPI was used to identify nuclei in the final
visualization. Sections incubated in the absence of primary
antibody were used as negative controls. To determine the number of
Iba-1-positive microglia in the entire hippocampus, three
representative images were taken at ×20 magnification in the
dentate gyrus and the CA1 and CA3 regions. Using Image J software
(National Institutes of Health, Bethesda, MD, USA), a threshold for
positive staining was determined for each image in order to include
all cell bodies and processes but exclude background staining. The
number of Iba-1-positive microglia within the threshold range was
manually counted and the average number was calculated for all the
representative images. Infiltrated neutrophils were detected by
dichloroacetate esterase staining (Muto Pure Chemicals Co., Ltd.,
Tokyo, Japan). The number of neutrophils that infiltrated into the
cerebral cortex following the administration of saline or LPS to
the left lateral ventricle was counted at the bregma level of the
left hemisphere. Neutrophils found in the meninges were not
considered. H&E staining was performed on consecutive 10 μm
frozen sections. Left lateral ventricles were sized using Image J
software and calculated as a percentage of the total area of the
left hemisphere.
Western blotting
For western blotting analysis, tissue extracts were
prepared by homogenizing mouse hippocampus tissue in T-PER Tissue
Protein Extraction Reagent (Thermo Scientific Inc., Waltham, MA,
USA) containing protease inhibitor, α-complete (Roche Applied
Science, Penzberg, Germany). Fifteen micrograms of each tissue
extract sample were adjusted to give a final solution of 60 mM
Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 0.1% bromophenol blue, and
5% β-mercaptoethanol. The solution was heated at 100ºC for 5 min,
electrophoresed through a 10% SDS-polyacrylamide gel, and
transferred to polyvinylidine difluoride membranes (Amersham
Pharmacia Biotech, Buckinghamshire, UK). Plexin-A1, Neuropilin-1,
COX-2, iNOS, IL-1β, TNF-α and β-actin were detected by their
respective antibodies using an enhanced chemiluminescence western
blot detection system (Amersham Pharmacia Biotech) according to the
manufacturer’s instructions. Anti-Plexin-A1 antibody (Abcam),
anti-Neuropilin-1 antibody (Abcam), anti-COX-2 antibody (Santa Cruz
Biotechnology, Inc., Dallas, Texas, USA), anti-iNOS antibody (Merck
Millipore, Darmstadt, Germany), anti-IL-1β antibody (Santa Cruz
Biotechnology), anti-TNF-α antibody (Santa Cruz Biotechnology), and
anti-β-actin antibody (Cell Signaling Technology, Danvers, MA, USA)
were used.
ICV LPS injection
Ultra-pure LPS (Escherichia coli serotype
055:B5; Sigma, St. Louis, MO, USA) was dissolved in sterile saline
at a concentration of 5 mg/ml. Mice were anesthetized by
intraperitoneal injection of pentobarbital sodium (0.648 mg/10 g
body weight) and placed into a rodent stereotaxic frame (David Kopf
Instruments, Tujunga, CA, USA). The scalp was shaved and a burr
hole was drilled 0.5 mm caudal to the bregma and 1.0 mm lateral to
the midline. LPS (200 μg/kg) was injected via a Hamilton
microsyringe (Hamilton Co., Reno, NV, USA) into the ventricle over
a 5-min period. Sham animals received an isovolumetric ICV
injection of saline.
NO assay and cell viability assay
To investigate the effect of Sema3A on the
microglial production of NO, the nitrite content was measured with
Griess reagent (1% sulfanilamide/0.1%
N-(1-naphthyl)-ethylenediamine dihydrochloride in 5% phosphoric
acid; Sigma) according to the manufacturer’s instructions. Primary
microglia were seeded on a 96-well plate at 2×104
cells/well, and incubated overnight in a CO2 incubator
at 37ºC. Eighteen hours after stimulation of the primary microglia
with LPS and Sema3A or control IgG, 50 μl of the culture
supernatant were mixed with 50 μl of Griess reagent and incubated
for 15 min. Absorbance values were measured at 540 nm in a plate
reader, and fresh Dulbecco’s modified Eagle’s medium served as a
blank in all the experiments. The NO concentration was calculated
with reference to the nitrite standard curve. To analyze cell
viability of the primary microglia subjected to NO assay, 5 μl of
MTT (5 mg/ml, Sigma, Tokyo, Japan) was added to the primary
microglia and incubated for 4 h at 37ºC. Formazan, a product of the
MTT tetrazolium ring that was precipitated by the action of
mitochondrial dehydrogenases, was solubilized with 0.1 N HCl in
anhydrous isopropanol containing 10% Triton X-100 and quantified
spectrophotometrically at 595 nm for the measurement of cell
viability.
Statistical analysis
Data are presented as the means ± standard error of
mean (SEM). Comparisons between WT and Plexin-A1−/− mice were
performed with the Student’s t-test or one-way analysis of variance
followed by post-hoc analysis. Statistical significance was
established at a level of p<0.05.
Results
Plexin-A1 is expressed in mouse
microglia
Primary microglia were isolated and purified from
postnatal mouse brain tissue. For all microglial cultures, purity
as determined by lectin cytochemistry analysis was >95%. RNA was
prepared from hippocampi and primary microglia and analyzed by
RT-PCR. As a result, gene transcripts for Plexin-A1 and
Neuropilin-1 were detected in the hippocampus and in primary
microglia (Fig. 1A). RT-PCR also
detected the expression of Sema3A mRNA in the mouse hippocampus
(Fig. 1A). To confirm the
expression of Plexin-A1 protein in mouse microglia, western
blotting was performed using protein extracts from WT and
PlexinA1−/− primary microglia (Fig.
1B). The analysis detected Plexin-A1 protein in WT, but not in
Plexin-A1−/− microglia (Fig. 1B).
Western blotting also detected Sema3A protein, which in mouse
hippocampus is a ligand of Plexin-A1 (Fig. 1B). Expression of Plexin-A1 was
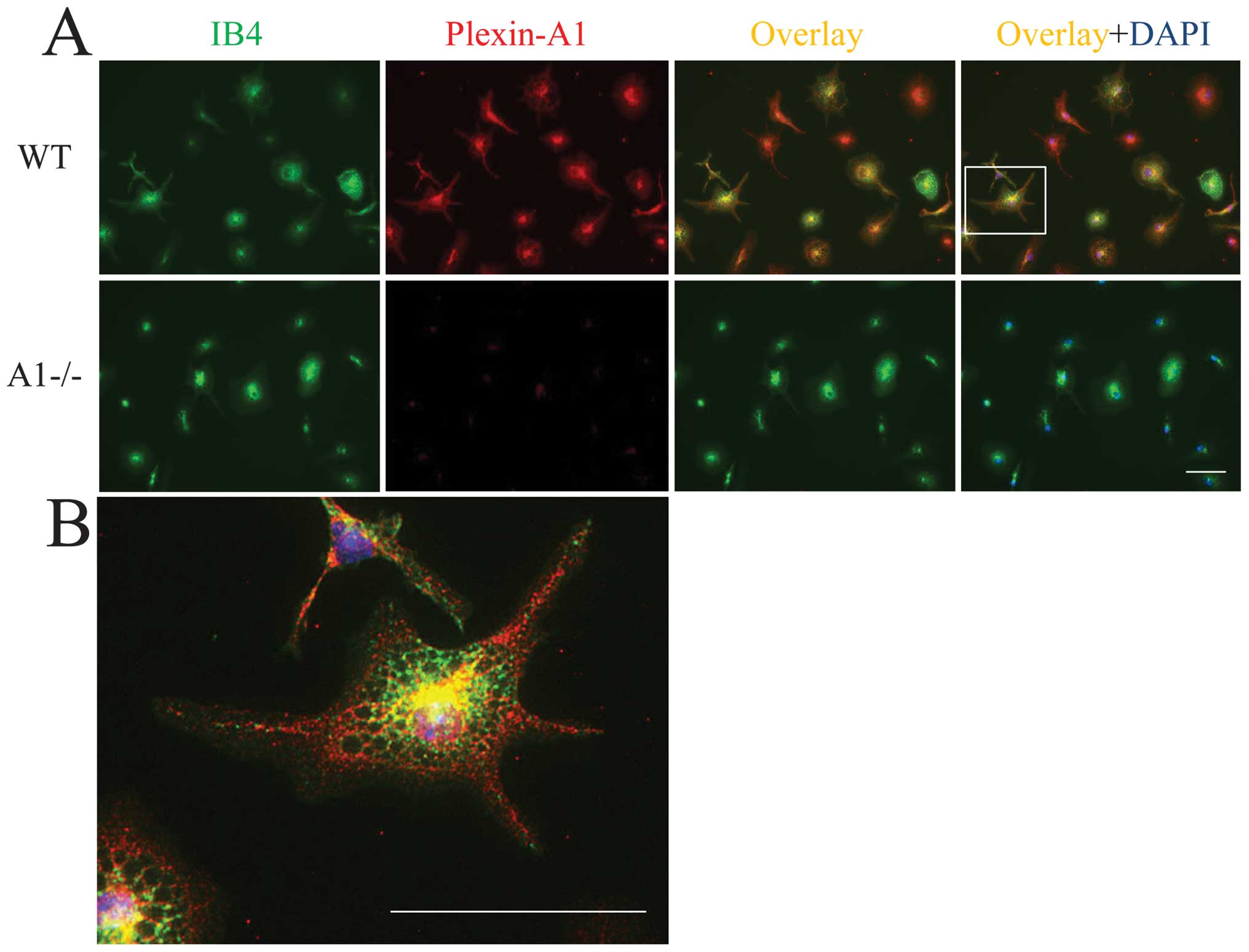
detected in primary microglia by double labeling with lectin
staining for microglia identification and by Plexin-A1
immunocytochemistry (Fig. 2A and
B). In total, 98.6±2.3% (mean ± SEM) of the primary microglia
exhibited positive staining for Plexin-A1. To examine the
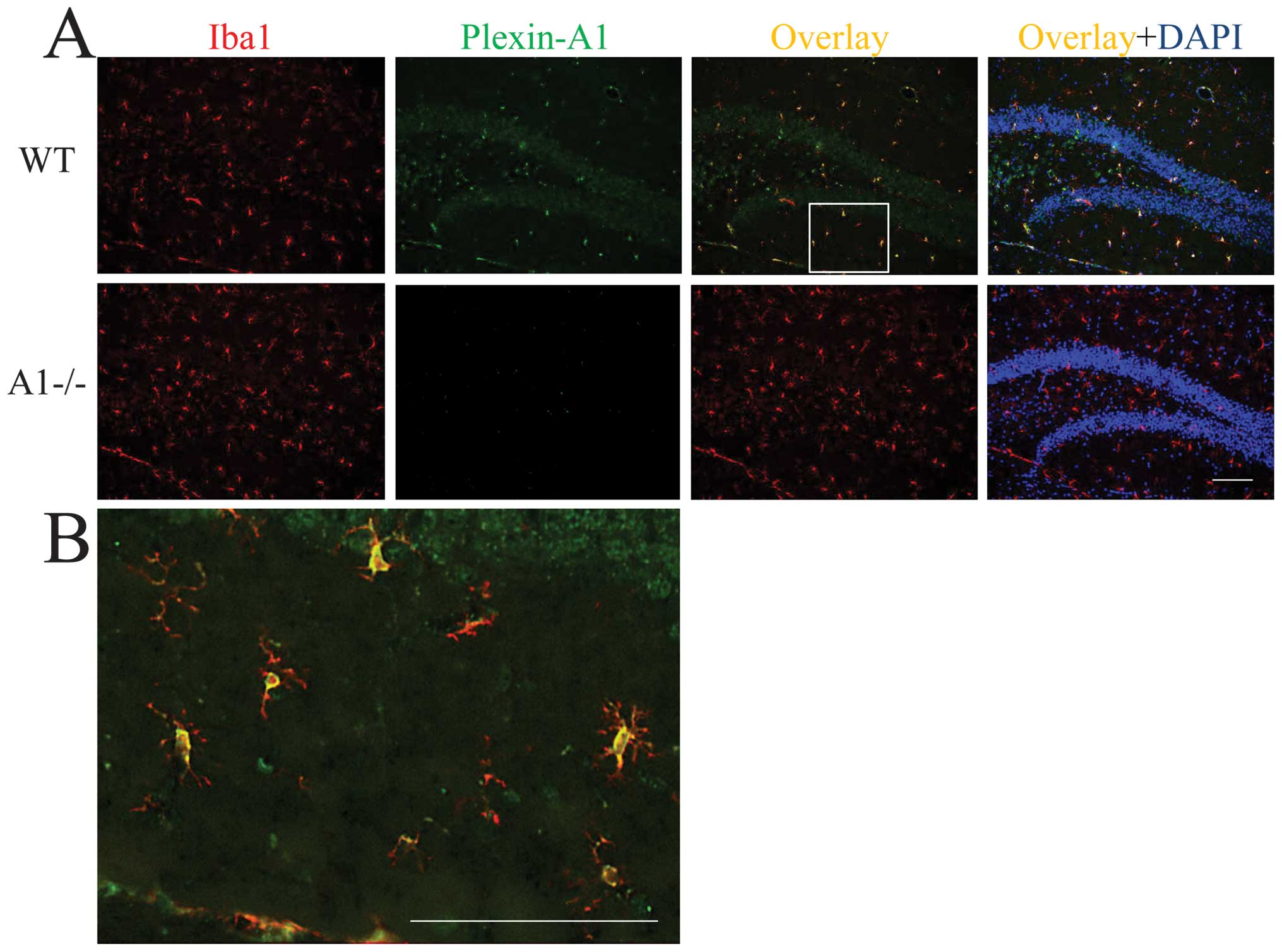
localization of Plexin-A1 in mouse brain, immunohistochemical
analyses were performed on hippocampi of WT and Plexin-A1−/− mice.
The antibodies against Plexin-A1 detected Plexin-A1 in the
microglia in WT (Fig. 3A and B),
but not Plexin-A1−/− mice (Fig.
3A).
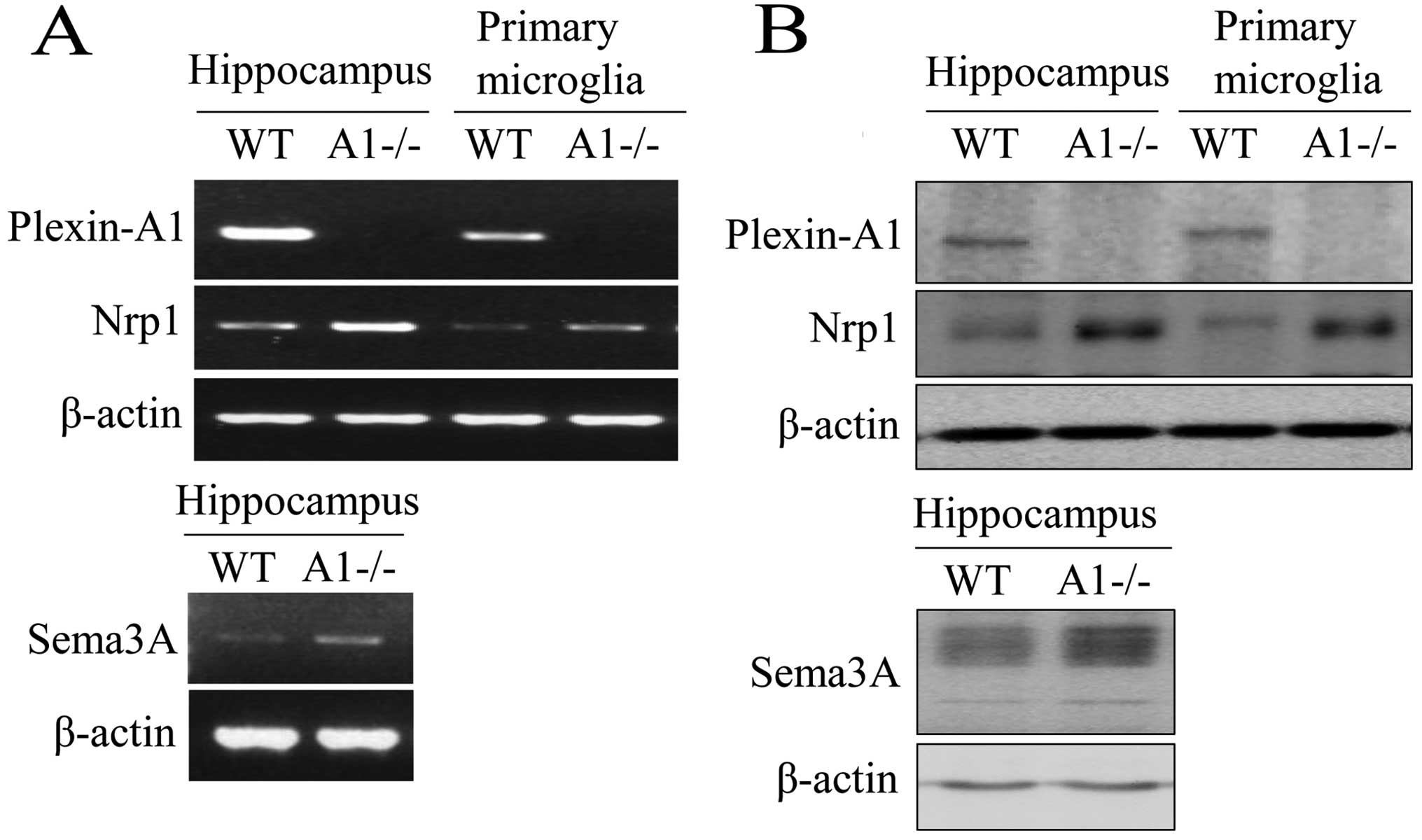 | Figure 1Mouse microglia express both
Plexin-A1 and Neuropilin-1. (A) RT-PCR detects mRNAs of Plexin-A1,
Neuropilin-1, and Sema3A in mouse hippocampus, and Plexin-A1 and
Neuropilin-1 in primary microglia. The method detects no Plexin-A1
mRNA in Plexin-A1-deficient (−/−) hippocampus or Plexin-A1−/−
primary microglia. (B) Western blotting detects Plexin-A1,
Neuropilin-1, and Sema3A protein in mouse hippocampus, and
Plexin-A1 and Neuropilin-1 protein in primary microglia.
Immunoblotting detects no Plexin-A1 protein in the Plexin-A1−/−
hippocampus or Plexin-A1−/− primary microglia. WT, wild-type;
A1−/−, Plexin-A1−/− hippocampus or Plexin-A1−/− primary microglia;
Nrp1, Neuropilin-1; RT-PCR, reverse transcriptase polymerase chain
reaction. |
Deletion of Plexin-A1 modulates
microglial activation status in the LPS response and reduces
neutrophil infiltration into the brain
LPS activates microglia through its binding to the
LPS receptor, TLR4, on the microglial cell surface (5,6).
Plexin-A1−/− microglia may be overactivated in the LPS response due
to their inability to use Sema3A to induce apoptosis of activated
microglia (28). To test this
hypothesis, we compared the microglial responses of WT and
Plexin-A1−/− mice following acute administration of LPS to the
lateral ventricles. Direct LPS ICV injection led to robust
neuroinflammatory responses in the brain, not through activation of
peripheral inflammatory cells such as macrophages, but through
direct microglial activation (30,31). Immunoblotting studies confirmed
the expression of Plexin-A1, Neuropilin-1, Sema3A, and TLR4 in the
hippocampus 18 h after ICV injection of saline or LPS (Fig. 4). To elucidate whether the
deletion of Plexin-A1 would affect classical microglial activation
and the associated inflammatory response, we examined the number of
microglia and the expression level of inflammation-related
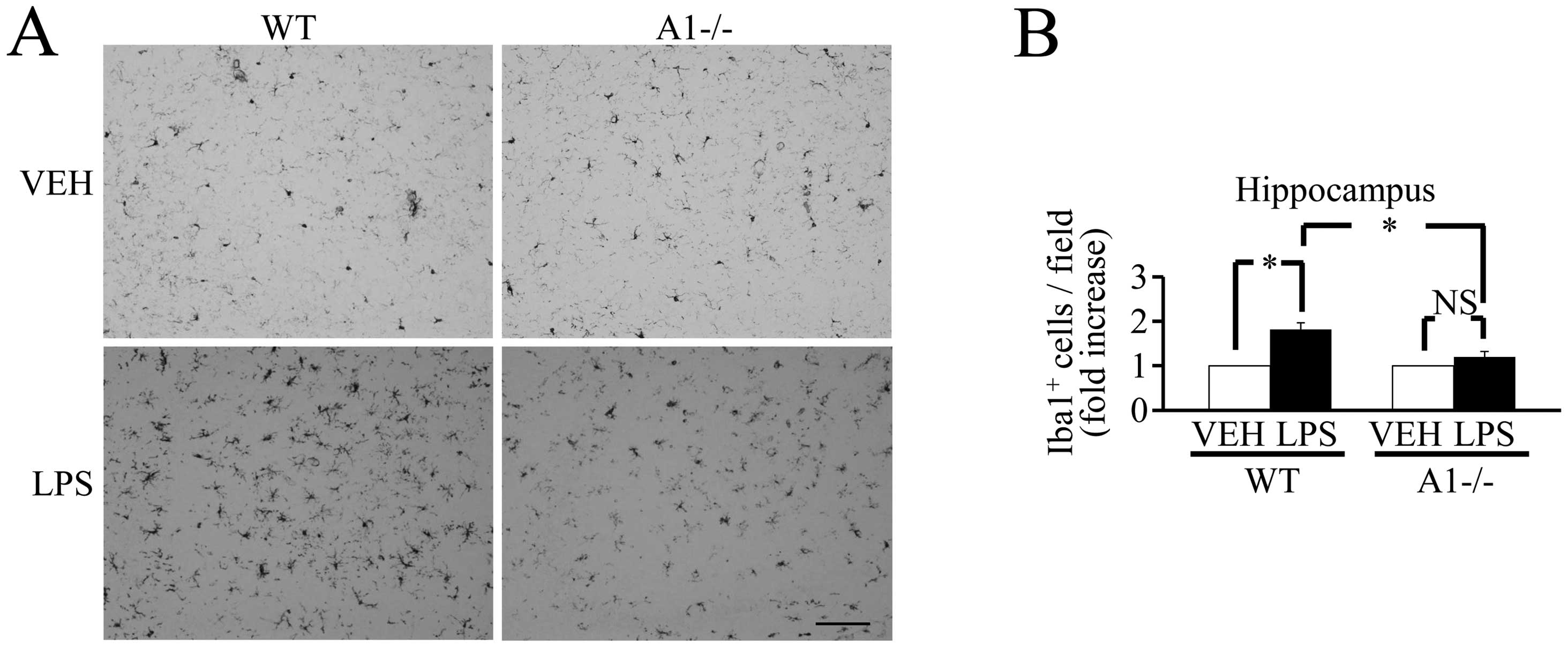
mediators. Of note, 18 h after the ICV injection of LPS, the number
of Iba-1-positive cells did not increase significantly in
Plexin-A1−/− mice compared with WT mice (Fig. 5), and there was no significant
increase in inflammation-related mediators such as COX-2, IL-1β,
TNF-α and iNOS (Fig. 6). Thus,
levels of brain inflammation after ICV injection of LPS were
significantly reduced in Plexin-A1−/− mice as compared to WT mice.
During neuroinflammation, changes in vascular permeability lead to
the development of brain edema, which in turn results in the
enlargement of the lateral ventricles of the brain (32). H&E-stained coronal brain
sections revealed that the ratio of the lateral ventricular area to
the cerebral hemisphere ipsilateral to the injection site was
significantly larger in LPS-injected WT mice as compared to
saline-treated mice (Fig. 7A).
However, significant enlargement of the lateral ventricle following
LPS administration was not observed in Plexin-A1−/− mice (Fig. 7B). Neutrophil leukocytes migrate
into the CNS towards various inflammatory stimuli that exacerbate
brain tissue damage through the release of inflammation-related
mediators and through an increase in vascular permeability
(33,34). To examine whether the absence of
Plexin-A1 affected neutrophil recruitment, we quantified their
infiltration using an esterase stain 18 h after ICV administration
of LPS to specifically mark neutrophils. Following administration,
the number of esterase-positive cells was significantly fewer in
the Plexin-A1−/− cortex as compared to the WT controls (Fig. 7B). Mice administered saline did
not show any significant difference based on genotype (Fig. 7B).
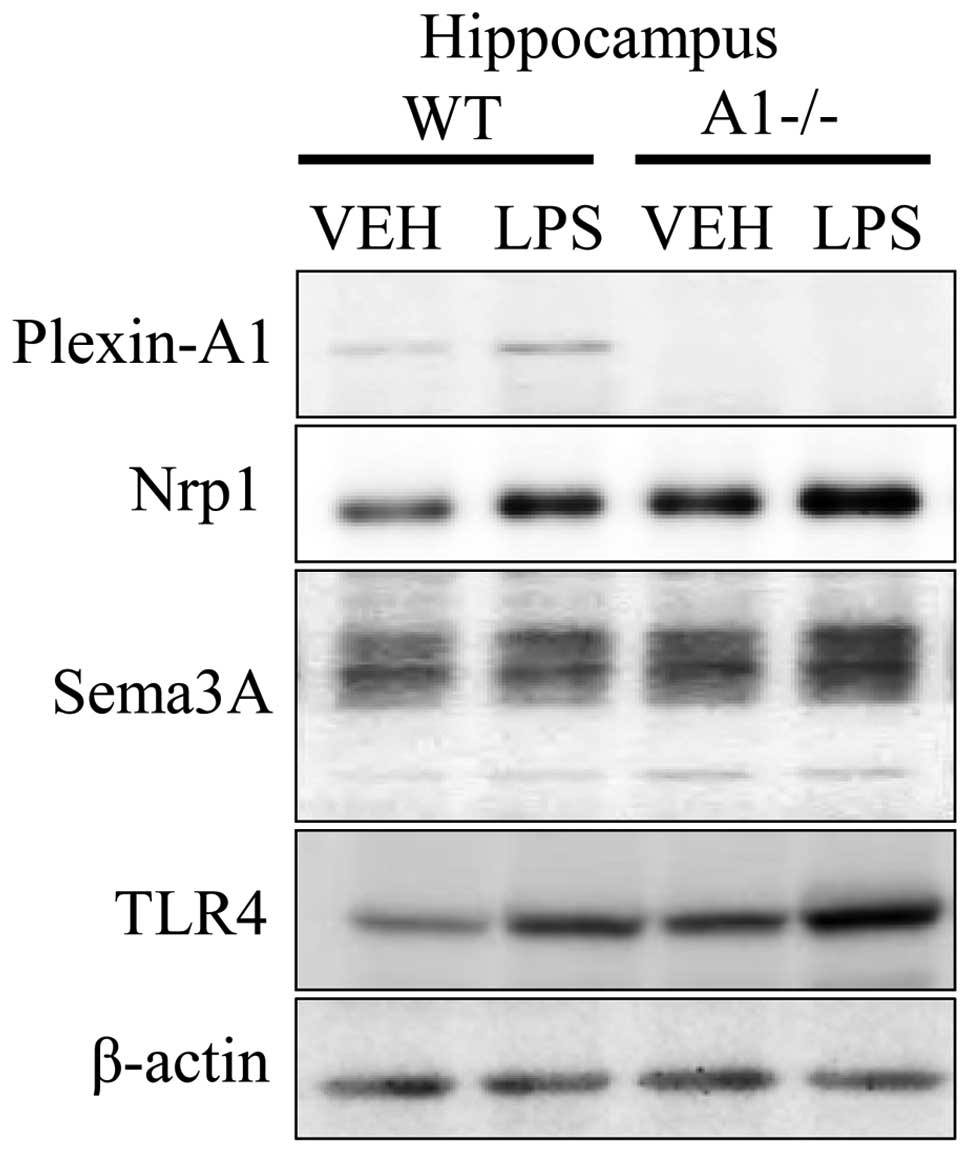 | Figure 4Expression of Plexin-A1,
Neuropilin-1, Sema3A, and TLR4 in hippocampus of WT and
Plexin-A1−/− mice administered lipopolysaccharide. WT or
Plexin-A1−/− mice were subjected to lateral ventricular injection
with saline or lipopolysaccharide (LPS; 200 μg/kg). Western
blotting detected Plexin-A1, Neuropilin-1, Sema3A, and TLR4 in the
mouse hippocampus in all experimental conditions except for
Plexin-A1 in the Plexin-A1−/− hippocampus. Nrp1, Neuropilin-1;
TLR4, Toll-like receptor 4; VEH, vehicle (saline); LPS,
lipopolysaccharide; WT, wild-type; A1−/−, Plexin-A1−/−. |
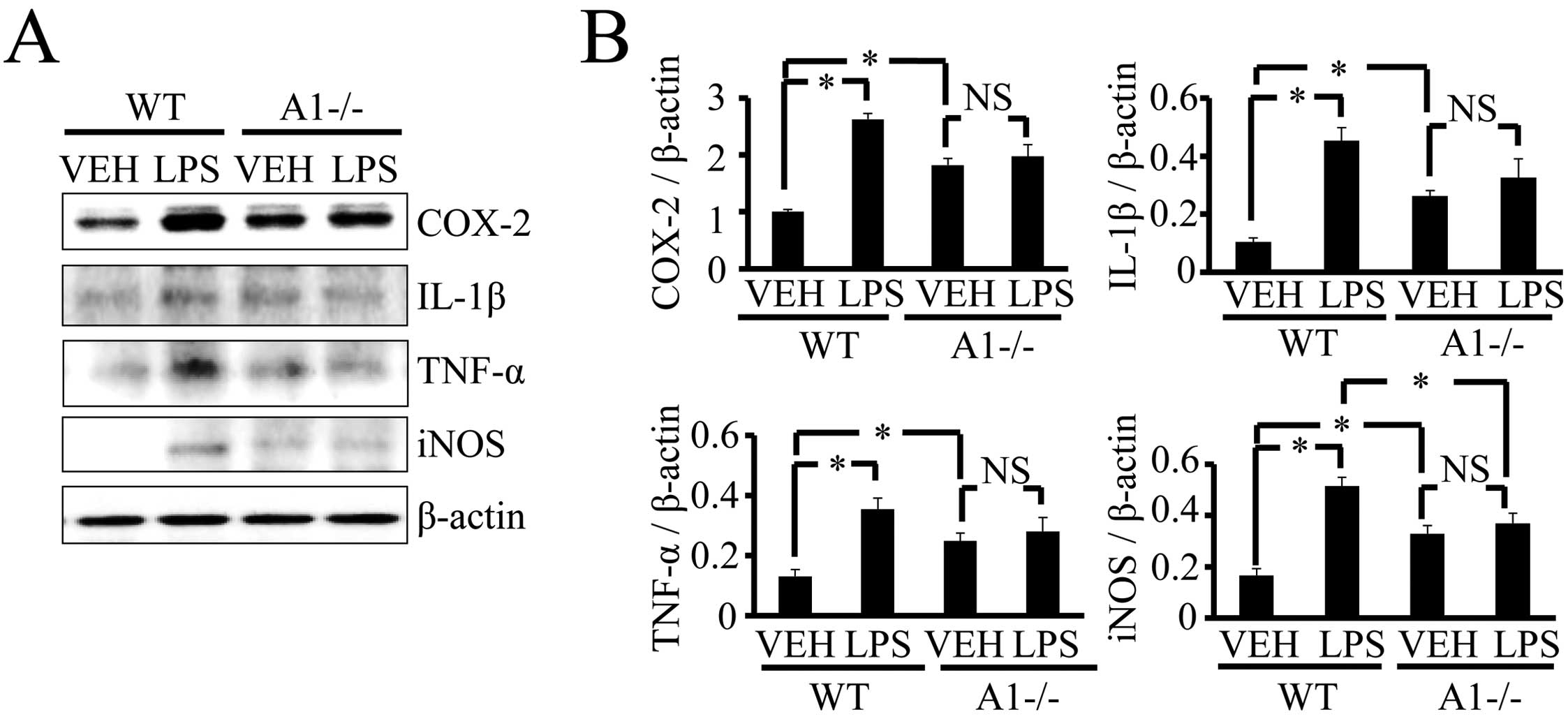 | Figure 6Plexin-A1−/− mice do not show an
increase of inflammation-related mediators in the hippocampus after
LPS injection. (A) Western blotting shows the increase of
inflammation-related mediators in WT hippocampus after LPS
stimulation, but not in Plexin-A1−/− hippocampus after LPS
administration. (B) Quantification of the immunoblot reveals that
the LPS-treated WT group had significantly increased levels of
COX-2, IL-1β, TNF-α and iNOS in the hippocampus as compared with
the saline-treated WT group. By contrast, the LPS-treated
Plexin-A1−/− group does not show any significant increase in levels
of COX-2, IL-1β, TNF-α or iNOS in the hippocampus as compared with
the saline-treated Plexin-A1−/− group. Results are shown as means ±
SEM, *p<0.05. VEH, vehicle (saline); LPS,
lipopolysaccharide; WT, wild-type; A1−/−, Plexin-A1−/−. |
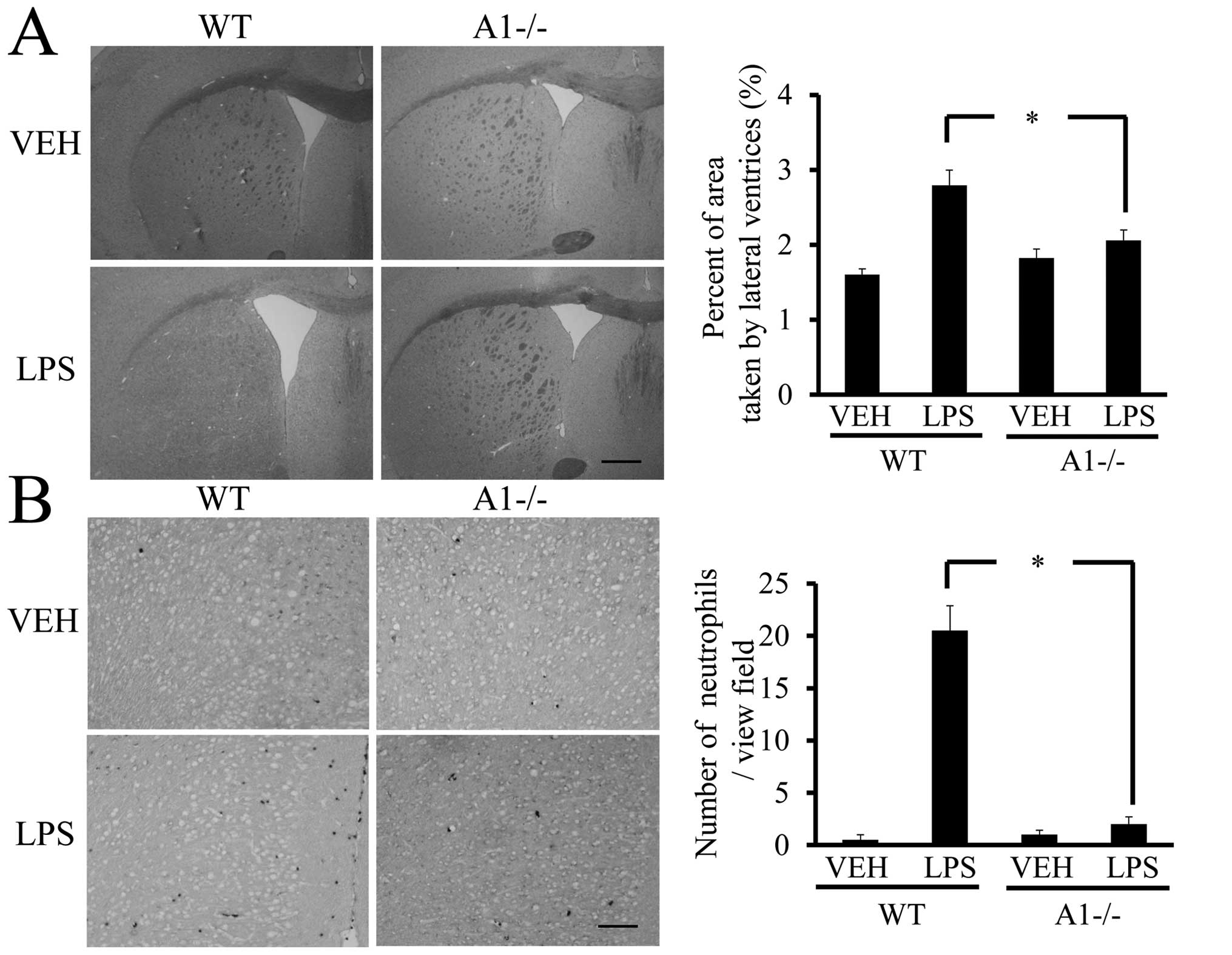 | Figure 7Plexin-A1−/− mice show reduced
ventricular enlargement and neutrophil infiltration after LPS
injection. (A) LPS injection leads to the enlargement of lateral
ventricle in WT mice, but not in Plexin-A1−/− mice. Scale bar is
1,000 μm. Quantification of the area of the lateral ventricle
revealed a significant increase in WT mice treated with LPS as
compared with saline-treated mice. By contrast, there was no
significant increase in lateral ventricular area in the LPS-treated
Plexin-A1−/− mice as compared with saline-treated Plexin-A1−/−
mice. (B) Esterase staining to detect neutrophil was performed with
brain sections from mice injected with saline or LPS. Saline
administration to WT mice did not induce any infiltration of
neutrophil detected by esterase staining in the cerebral cortex,
while WT mice injected with LPS show many infiltrating neutrophils
in the cortical area. Esterase staining hardly detects neutrophil
in the cerebral cortex even after administration of LPS to
Plexin-A1−/− mice. Quantification of neutrophil number shows a
significant increase in neutrophil in the cerebral cortex in
LPS-treated WT mice as compared with saline-treated WT mice. By
contrast, there was no significant increase of neutrophil
infiltration into the cerebral cortex in LPS-treated Plexin-A1−/−
mice compared with saline-treated control mice. Results are shown
as means ± SEM, *p<0.05. VEH, vehicle (saline); LPS,
lipopolysaccharide; WT, wild-type; A1−/−, Plexin-A1−/−. |
Sema3A increases NO production through
Plexin-A1 expression on the microglial cell surface in the LPS
response
To directly demonstrate the role of microglial
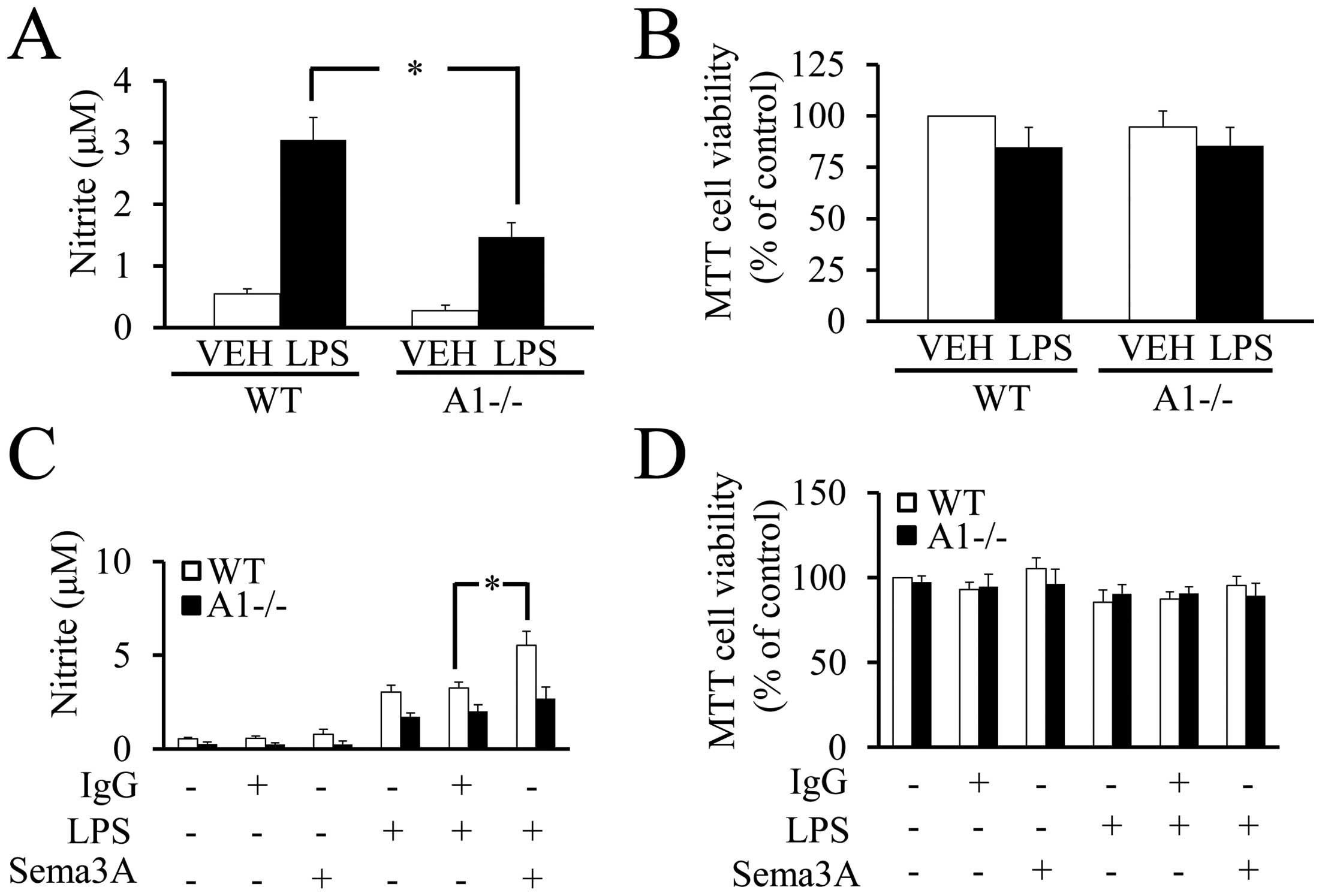
Plexin-A1 in the response to LPS, NO production was measured by the
Griess reaction in cultures of WT or Plexin-A1−/− primary microglia
stimulated with LPS. The Griess reaction revealed a significant
reduction of NO production in Plexin-A1−/− microglia stimulated
with LPS as compared with LPS-treated WT microglia (Fig. 8A). There were no significant
differences in cell viability among WT and Plexin-A1−/− primary
microglia with or without LPS stimulation (Fig. 8B). To examine whether Sema3A
enhances the cell response to LPS, NO production was quantified in
WT and Plexin-A1−/− primary microglia stimulated with LPS and
Sema3A (Fig. 8C). Under
stimulation with LPS, the addition of Sema3A to WT microglia
exhibited a significant increase of NO production as compared with
the addition of control IgG. By contrast, Plexin-A1−/− microglia
stimulated with LPS and Sema3A did not exhibit any significant
increase in NO production as compared with Plexin-A1−/− microglia
stimulated by LPS and control IgG (Fig. 8C). There were no significant
differences in cell viability as determined by MTT assay among any
of the experimental groups of WT and Plexin-A1−/− microglia
(Fig. 8D). Thus, these data
suggest a synergistic action of Plexin-A1 activation and
TLR4-mediated signaling in microglia.
Discussion
Results of the present study demonstrated a novel
finding regarding the crucial role of Plexin-A1 expressed in mouse
microglia, i.e., its activity in enhancing microglial TLR4-mediated
signaling in the development of LPS-induced encephalopathy in mice.
Although crosstalk between the TLR4 pathway and Plexin-A4-mediated
signal was found to play a role in the macrophage response to LPS
(35), it remained unclear which
plexin signal interacts with the TLR4 pathway to fully activate
microglia. Accordingly, to the best of our knowledge, the present
study is the first to indicate the importance of synergistic
crosstalk between TLR4-mediated signaling and Plexin-A1 activation
in the LPS response of mouse microglia.
The present study results demonstrated that the LPS
receptor TLR4 and the semaphorin receptor Plexin-A1 acted
synergistically in the microglial signal transduction pathway,
enhanced microglial activation resulting in neuroinflammation, and
contributed to the development of LPS-induced encephalopathy.
Microglia activated by LPS are induced to proliferate and produce
inflammation-related mediators, suggesting involvement in neuronal
injury (6,36). Since microglia activated by
neuronal injury in Plexin-A1−/− mice are considered resistant to
the induction of apoptosis by the Sema3A secreted from injured
neurons (28), Plexin-A1−/−
microglia were predicted to react excessively with LPS and
facilitate neuroinflammation. Contrary to this prediction,
Plexin-A1−/− mice ICV administered LPS, as compared with
Plexin-A1−/− mice injected with saline, did not show a significant
increase in microglial cell number or in the expression level of
inflammation-related mediators (Figs.
5 and 6). These
inflammation-related mediators excessively produced by
over-activated microglia may act on themselves, thereby inducing
microglial cell death (37–40). Since microglia in Plexin-A1−/−
mice were significantly decreased compared with the WT in the
response to LPS (Fig. 5B), it is
possible that overactivation of microglia in Plexin-A1−/− mouse
brain induces microglial cell death. However, the TUNEL-positive
cell number significantly decreased after LPS administration in the
brain of LPS-treated Plexin-A1−/− mice as compared with WT mice
(unpublished data). The data indicate a reduced possibility that
the decrease of microglial cell number is due to the induction of
their cell death by overactivation of microglia. Accordingly, the
significant decrease of microglia in Plexin-A1−/− mice after
administration of LPS may be derived, not from the overactivation
of microglia, but from the inability of Plexin-A1−/− microglia to
fully respond to LPS. Activation of intracerebral microglia by LPS
administration causes the ventricles to enlarge due to the
neuroinflammation-induced brain edema and the increase of leukocyte
infiltration migrating towards the TNF-α secreted by activated
microglia in the brain (30,41). A significant decrease in the
lateral ventricle area and neutrophil invasion in Plexin-A1−/−
brain after LPS administration suggests that LPS-dependent
activation of Plexin-A1−/− microglia is attenuated (Fig. 7). Furthermore, a significantly
lower production of NO after LPS stimulation in Plexin-A1−/−
microglia compared with WT microglia demonstrated the essential
role of Plexin-A1 for the full enhancement of LPS-induced
microglial activation (Fig. 8A).
The data suggest that a crosstalk between Plexin-A1 and the TLR4
pathway synergistically enhances the activation of microglia, and
thus Plexin-A1-mediated signaling in microglia has an essential
role in the development of LPS-induced encephalopathy.
Sema3A may have dual roles in inducing either
apoptosis or microglial activation through the Plexin-A1 receptor,
depending on the cellular context. A previous study regarding the
apoptosis-inducing activity of Sema3A towards activated microglia
detected apoptotic microglia with morphological identification of
condensed nuclei without using TUNEL or activated caspase-3
staining (28). The expression of
activated caspase-3 has been demonstrated to be essential for
LPS-dependent microglial activation (42). Therefore, the inhibition of
Sema3A-induced cell death by the activated caspase-3 inhibitor
reported in a previous study (28) may have an alternative
interpretation in which the inhibitor instead suppressed
overactivation of microglia. Accordingly, Sema3A may be involved in
the crucial autoregulatory mechanism of microglia and in the
strengthening of microglial activation through synergistic
activation of Plexin-A1-mediated signaling and the TLR4 pathway,
but it may also induce apoptosis of excessively activated microglia
in order not to kill neurons in close proximity to the
over-activated microglia. Our in vitro model, however,
suggests that Sema3A-induced Plexin-A1 signaling is required for
LPS-induced microglial activation, but not for the apoptotic
induction of activated microglia (Fig. 8C and D). In the LPS response
outside the brain, the signaling of Sema3A through Plexin-A4
(another member of the Plexin-A family) has crosstalk with
TLR4-mediated signaling in macrophages, and plays a crucial role in
exacerbating cytokine storms (35). Furthermore, Plexin-B1 on microglia
bound with the Sema4D ligand has been demonstrated to be necessary
for the inflammatory mediator-dependent activation of microglia
(43). Our findings further
develop the study of the regulatory mechanisms of the Plexin family
in the inflammatory response. Therefore, results of the present
study suggest that the regulatory mechanism of the
semaphorin-Plexin signaling system may be applicable to the
treatment of LPS-induced encephalopathy and other psychiatric
diseases associated with neuroinflammation.
Acknowledgements
We acknowledge the members of the Department of
Physiology of Meijo University for helpful discussion and technical
assistance. The study was primarily supported by a Grant-in-Aid for
Scientific Research from the Ministry of Education, Science, Sports
and Culture, Japan (No. 22590195).
References
|
1
|
Ransohoff RM and Perry VH: Microglial
physiology: unique stimuli, specialized responses. Annu Rev
Immunol. 27:119–145. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heppner FL, Greter M, Marino D, et al:
Experimental autoimmune encephalomyelitis repressed by microglial
paralysis. Nat Med. 11:146–152. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jack C, Ruffini F, Bar-Or A and Antel JP:
Microglia and multiple sclerosis. J Neurosci Res. 81:363–373. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ponomarev ED, Shriver LP and Dittel BN:
CD40 expression by microglial cells is required for their
completion of a two-step activation process during central nervous
system autoimmune inflammation. J Immunol. 176:1402–1410. 2006.
View Article : Google Scholar
|
|
5
|
Lehnardt S, Lachance C, Patrizi S, et al:
The toll-like receptor TLR4 is necessary for
lipopolysaccharide-induced oligodendrocyte injury in the CNS. J
Neurosci. 22:2478–2486. 2002.PubMed/NCBI
|
|
6
|
Lehnardt S, Massillon L, Follett P, et al:
Activation of innate immunity in the CNS triggers neurodegeneration
through a Toll-like receptor 4-dependent pathway. Proc Natl Acad
Sci USA. 100:8514–8519. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heppner FL, Greter M, Marino D, et al:
Experimental autoimmune encephalomyelitis repressed by microglial
paralysis. Nat Med. 11:146–152. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tessier-Lavigne M and Goodman SC: The
molecular biology of axon guidance. Science. 274:1123–1133. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pasterkamp RJ and Kolodkin AL: Semaphorin
junction: making tracks toward neural connectivity. Curr Opin
Neurobiol. 13:79–89. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sekido Y, Bader S, Latif F, et al: Human
semaphorins A(V) and IV reside in the 3p21.3 small cell lung cancer
deletion region and demonstrate distinct expression patterns. Proc
Natl Acad Sci USA. 93:4120–4125. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gu C, Rodriguez ER, Reimert DV, et al:
Neuropilin-1 conveys semaphorin and VEGF signaling during neural
and cardiovascular development. Dev Cell. 5:45–57. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Toyofuku T, Zhang H, Kumanogoh A, et al:
Guidance of myocardial patterning in cardiac development by Sema6D
reverse signalling. Nat Cell Biol. 6:1204–1211. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kumanogoh A, Watanabe C, Lee I, et al:
Identification of CD72 as a lymphocyte receptor for the class IV
semaphorin CD100: a novel mechanism for regulating B cell
signaling. Immunity. 13:621–631. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi W, Kumanogoh A, Watanabe C, et al: The
class IV semaphorin CD100 plays nonredundant roles in the immune
system: defective B and T cell activation in CD100-deficient mice.
Immunity. 13:633–642. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumanogoh A, Marukawa S, Suzuki K, et al:
Class IV semaphorin Sema4A enhances T-cell activation and interacts
with Tim-2. Nature. 419:629–633. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kumanogoh A, Shikina T, Suzuki K, et al:
Nonredundant roles of Sema4A in the immune system: defective T cell
priming and Th1/Th2 regulation in Sema4A-deficient mice. Immunity.
22:305–316. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kikutani H and Kumanogoh A: Semaphorins in
interactions between T cells and antigen-presenting cells. Nat Rev
Immunol. 3:159–167. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Elhabazi A, Marie-Cardine A, Chabbert-de
Ponnat I, Bensussan A and Boumsell L: Structure and function of the
immune semaphorin CD100/SEMA4D. Crit Rev Immunol. 23:65–81. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tamagnone L and Comoglio PM: Signalling by
semaphorin receptors: cell guidance and beyond. Trends Cell Biol.
10:377–383. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Granziero L, Circosta P, Scielzo C, et al:
CD100/Plexin-B1 interactions sustain proliferation and survival of
normal and leukemic CD5+ B lymphocytes. Blood. 101:1962–1969.
2003.PubMed/NCBI
|
|
21
|
Walzer T, Galibert L, Comeau MR and De
Smedt T: Plexin C1 engagement on mouse dendritic cells by viral
semaphorin A39R induces actin cytoskeleton rearrangement and
inhibits integrin-mediated adhesion and chemokine-induced
migration. J Immunol. 174:51–59. 2005. View Article : Google Scholar
|
|
22
|
Takahashi T, Fournier A, Nakamura F, et
al: Plexin-neuropilin-1 complexes form functional semaphorin-3A
receptors. Cell. 99:59–69. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Toyofuku T, Zhang H, Kumanogoh A, et al:
Dual roles of Sema6D in cardiac morphogenesis through
region-specific association of its receptor, Plexin-A1, with
off-track and vascular endothelial growth factor receptor type 2.
Genes Dev. 18:435–447. 2004. View Article : Google Scholar
|
|
24
|
Wong AW, Brickey WJ, Taxman DJ, et al:
CIITA-regulated plexin-A1 affects T-cell-dendritic cell
interactions. Nat Immunol. 4:891–898. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shirvan A, Ziv I, Fleminger G, et al:
Semaphorins as mediators of neuronal apoptosis. J Neurochem.
73:961–971. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fujita H, Zhang B, Sato K, Tanaka J and
Sakanaka M: Expressions of neuropilin-1, neuropilin-2 and
semaphorin 3A mRNA in the rat brain after middle cerebral artery
occlusion. Brain Res. 914:1–14. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pasterkamp RJ and Verhaagen J: Emerging
roles for semaphorins in neural regeneration. Brain Res Brain Res
Rev. 35:36–54. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Majed HH, Chandran S, Niclou SP, et al: A
novel role for Sema3A in neuroprotection from injury mediated by
activated microglia. J Neurosci. 26:1730–1738. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takegahara N, Takamatsu H, Toyofuku T, et
al: Plexin-A1 and its interaction with DAP12 in immune responses
and bone homeostasis. Nat Cell Biol. 8:615–622. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou H, Lapointe BM, Clark SR, Zbytnuik L
and Kubes P: A requirement for microglial TLR4 in leukocyte
recruitment into brain in response to lipopolysaccharide. J
Immunol. 177:8103–8110. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Buttni M, Limonta S and Boddeke HW:
Peripheral administration of lipopolysaccharide induces activation
of microglial cells in rat brain. Neurochem Int. 29:25–35.
1996.PubMed/NCBI
|
|
32
|
Grin’kina NM, Karnabi EE, Damania D,
Wadgaonkar S, Muslimov IA and Wadgaonkar R: Sphingosine kinase 1
deficiency exacerbates LPS-induced neuroinflammation. PLoS One.
7:e364752012.PubMed/NCBI
|
|
33
|
Banks WA and Erickson MA: The blood-brain
barrier and immune function and dysfunction. Neurobiol Dis.
37:26–32. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shaftel SS, Griffin WS and O’Banion MK:
The role of interleukin-1 in neuroinflammation and Alzheimer
disease: an evolving perspective. J Neuroinflammation. 5:72008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wen H, Lei Y, Eun SY and Ting JP:
Plexin-A4-semaphorin 3A signaling is required for Toll-like
receptor- and sepsis-induced cytokine storm. J Exp Med.
207:2943–2957. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Durrenberger PF, Facer P, Gray RA, et al:
Cyclooxygenase-2 (Cox-2) in injured human nerve and a rat model of
nerve injury. J Peripher Nerv Syst. 9:15–25. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lehnardt S: Innate immunity and
neuroinflammation in the CNS: the role of microglia in Toll-like
receptor-mediated neuronal injury. Glia. 58:253–263.
2010.PubMed/NCBI
|
|
38
|
Shin WH, Lee DY, Park KW, et al: Microglia
expressing interleukin-13 undergo cell death and contribute to
neuronal survival in vivo. Glia. 46:142–152. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee J, Hur J, Lee P, et al: Dual role of
inflammatory stimuli in activation-induced cell death of mouse
microglial cells. Initiation of two separate apoptotic pathways via
induction of interferon regulatory factor-1 and caspase-11. J Biol
Chem. 276:32956–32965. 2001. View Article : Google Scholar
|
|
40
|
Yang MS, Ji KA, Jeon SB, et al:
Interleukin-13 enhances cyclooxygenase-2 expression in activated
rat brain microglia: implications for death of activated microglia.
J Immunol. 177:1323–1329. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Choi SH, Aid S, Choi U and Bosetti F:
Cyclooxygenases-1 and -2 differentially modulate leukocyte
recruitment into the inflamed brain. Pharmacogenomics J.
10:448–457. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Burguillos MA, Deierborg T, Kavanagh E, et
al: Caspase signalling controls microglia activation and
neurotoxicity. Nature. 472:319–324. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Okuno T, Nakatsuji Y, Moriya M, et al:
Roles of Sema4D-plexin-B1 interactions in the central nervous
system for pathogenesis of experimental autoimmune
encephalomyelitis. J Immunol. 184:1499–1506. 2010. View Article : Google Scholar : PubMed/NCBI
|