Introduction
Bicuspid aortic valve (BAV) is the most common form
of congenital heart disease in humans, with an estimated prevalence
of 0.5–2% in the general population as well as a pronounced male
predominance of ~3:1 (1–6). While BAV can occur in isolation, it
is frequently associated with other congenital cardiovascular
malformations, such as coarctation of the aorta, interruption of
the aorta, ventricular septal defect, atrial septal defect, patent
ductus arteriosus and hypoplastic left heart syndrome, leading to a
wide spectrum of clinical presentations ranging from severe
disorder detected in utero to asymptomatic condition in old
age (7). Patients with BAV are at
high risk for the development of severe complications, including
aortic valve regurgitation, aortic valvular stenosis, aortic
dilation or even aneurysm, aortic dissection, thrombus formation,
and infective endocarditis (8–11).
BAV accounts for 70–85% of aortic stenosis in pediatric patients
and at least 50% of stenotic aortic valve in adults (12,13). Moreover, individuals with BAV have
an 8-fold increased risk of aortic dissection and 25-year risk of
valve replacement of 53%, aneurysm formation of 26% and aortic
surgery of 25% (14). Therefore,
BAV confers a heavier burden of disease than all other congenital
cardiac lesions combined (3,7).
Despite its high prevalence and significant clinical importance,
the underlying pathogenic basis of BAV remains largely unclear.
Cardiac valve morphogenesis occurring early in fetal
development is a complex and dynamic process that requires the
temporal and spatial cooperation of cardiac cell commitment,
differentiation, proliferation, and migration, and both
environmental and genetic risk factors may interrupt this
biological process, resulting in abnormal valvulogenesis and the
formation of BAV (15,16). Mounting evidence suggests that
genetic defects play an important role in the pathogenesis of BAV
(7,13). Previous studies have established
the substantial familial clustering of BAV, with a prevalence
ranging from 9 to 24% in the first-degree relatives of BAV patients
and a heritability as high as 89%, suggesting a Mendelian pattern
of inheritance (7,13). By genome-wide scan of the
available family members with polymorphic microsatellite markers
and linkage analysis, BAV-susceptibility loci have been mapped on
chromosomes 9q34-35, 18q, 5q15-21 and 13q33-qter, and NOTCH1
has been identified as the first culprit gene accountable for BAV
in the genomic region of 9q34 (17,18). Moreover, candidate gene strategy
has led to the identification of various novel mutations in the
NOTCH1 gene that are associated with BAV (19–21). Additionally, mutations in other
genes, including KCNJ2, TGFBR2 and NKX2-5,
were detected in patients with BAV (22–24). Nevertheless, BAV is of pronounced
genetic heterogeneity and the genetic determinants underpinning BAV
in a large number of patients remain to be determined.
GATA5 was reported to have a crucial role in
cardiovascular development and valvular morphogenesis (25–31), and the targeted deletion of
GATA5 in mice resulted in partially penetrant BAV (31). Furthermore, the endocardial
cell-specific inactivation of GATA5 led to BAV, similar to
that observed in GATA5-null mice (31). GATA5 is a zinc finger-containing
transcription factor that belongs to a subgroup of the GATA family
of DNA binding proteins, which, together with GATA4 and GATA6, is
abundantly expressed in various mesoderm- and endoderm-derived
tissues, predominantly in embryonic heart (32,33). In humans, mutations in
GATA5 have been found to be associated with a wide variety
of congenital cardiovascular anomalies, including ventricular and
atrial septal defect, tetralogy of Fallot, double outlet right
ventricle, aortic stenosis, and BAV (21,34–39). Taken together, these findings
suggested screening GATA5 for mutations in another cohort of
patients with BAV.
Materials and methods
Study population
A cohort of 110 unrelated patients with BAV was
recruited from the Chinese Han population for this study. The
available relatives of the index patients carrying the identified
GATA5 mutations were also enlisted. Patients underwent
clinical evaluation that included individual and familial
histories, medical records, complete physical examination, 12-lead
electrocardiogram, and two-dimensional transthoracic
echocardiography with color flow Doppler. BAV was confirmed by
imaging and/or direct view during aortic valve replacement surgery.
Familial BAV was defined if two or more affected relatives had
proven BAV. The patients with known chromosomal abnormalities or
syndromic cardiovascular defects, such as Marfan syndrome, Turner
syndrome, and Di George syndrome, were excluded from the study.
A total of 200 unrelated, ethnically matched healthy
individuals randomly selected from the subjects undergoing routine
physical examinations were used as controls. In terms of medical
histories and echocardiographic records, the control individuals
had no congenital cardiovascular deformities. The ethnic origin of
a participant was determined by a combination of self-reported
ethnicity and a personal questionnaire regarding birthplace,
language, religion, and ancestry.
Peripheral venous blood samples were obtained from
BAV cases and control individuals. The study protocol conforms to
the principles outlined in the Declaration of Helsinki and was
approved by the local Institutional Ethics Committee. Written
informed consent was obtained from all participants or their
guardians prior to study.
Mutational screening of GATA5
Genomic DNA from each participant was extracted from
blood lymphocytes with a Wizard Genomic DNA Purification kit
(Promega, Madison, WI, USA). The coding regions and flanking
introns of the GATA5 gene were sequenced initially in 110
unrelated patients with BAV, and subsequently in the available
relatives of the index patients carrying identified mutations and
the 200 unrelated, ethnically-matched healthy individuals. The
primer pairs used to amplify the coding exons and exon/intron
boundaries of GATA5 by polymerase chain reaction (PCR) were
described previously (39). PCR
was performed using HotStar Taq DNA Polymerase (Qiagen GmbH,
Hilden, Germany) on a Veriti Thermal Cycler (Applied Biosystems,
Foster, CA, USA), with standard conditions and concentrations of
reagents. The amplified products were analyzed on 1% agarose gels
stained with ethidium bromide and purified with QIAquick Gel
Extraction kit (Qiagen GmbH). Both strands of each PCR product were
sequenced with a BigDye® Terminator v3.1 Cycle
Sequencing kit (Applied Biosystems) under an ABI PRISM 3130 XL DNA
Analyzer (Applied Biosystems). The sequencing primers were the same
as previously designed for specific region amplifications. The DNA
sequences were viewed and analyzed with the DNA Sequencing Analysis
Software v5.1 (Applied Biosystems). The variant was validated by
resequencing an independent PCR-generated amplicon from the subject
and met our quality control thresholds with a call rate of >99%.
Additionally, an identified sequence variation was searched in the
single-nucleotide polymorphism (SNP) database at the National
Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/), the human gene mutation
(HGM) database (http://www.hgmd.org/), and the 1000
genome database (http://www.1000genomes.org/) to confirm its
novelty.
Alignment of multiple GATA5 protein
sequences among species
Multiple GATA5 protein sequences across various
species were aligned using the online program of MUSCLE, version
3.6 (http://www.ncbi.nlm.nih.gov/).
Prediction of the pathogenic potential of
a GATA5 sequence variation
The disease-causing potential of a GATA5
sequence variation was predicted by MutationTaster (an online
program at http://www.mutationtaster.org), which automatically
gave a probability for the variation to be either a pathogenic
mutation or a benign polymorphism. Notably, the P-value used here
is the probability of the correct prediction rather than the
probability of error as used in t-test statistics (i.e., a value
close to 1 indicates a high ‘security’ of the prediction).
Additionally, the online program PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) was used to
predict the possible impact of an amino acid substitution on the
structure and function of GATA5 protein.
Plasmids and site-directed
mutagenesis
The recombinant expression plasmid pcDNA3.1-hGATA5
was constructed as described previously (39). The atrial natriuretic factor
(ANF)-luciferase reporter gene, which contains the 2600-bp
5′-flanking region of the ANF gene, i.e., ANF(−2600)-Luc,
was kindly provided by Dr Ichiro Shiojima, from the Department of
Cardiovascular Science and Medicine, Chiba University Graduate
School of Medicine, Chuo-ku, Chiba, Japan. The identified mutation
was introduced into the wild-type GATA5 using a QuickChange II XL
Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA) with
a complementary pair of primers. The mutant was sequenced to
confirm the appropriate mutation and to exclude any other sequence
variations.
Reporter gene assay
HEK-293 cells were cultured in Dulbecco’s modified
Eagle’s medium supplemented with 10% fetal calf serum and seeded in
12-well plates prior to transfection. The ANF(−2600)-Luc reporter
construct and an internal control reporter plasmid pGL4.75
(hRluc/CMV, Promega) were used in the transient transfection assay
to evaluate the transcriptional activity of the GATA5
mutants. HEK-293 cells were transfected with 0.4 μg of wild-type or
mutant pcDNA3.1-hGATA5 expression vector, 0.4 μg of ANF(−2600)-Luc
reporter construct, and 0.04 μg of pGL4.75 control reporter vector
using a PolyFect Transfection Reagent (Qiagen). For cotransfection
experiments, 0.2 μg of wild-type pcDNA3.1-hGATA5 together with 0.2
μg of mutant pcDNA3.1-hGATA5 or 0.2 μg of empty pcDNA3.1 vector
were used in the presence of 0.4 μg of ANF(−2600)-Luc and 0.04 μg
of pGL4.75. Firefly and Renilla luciferase activities were
measured with the Dual-Glo® luciferase assay system
(Promega) 48 h after transfection. The activity of the ANF promoter
was presented as fold activation of Firefly luciferase relative to
Renilla luciferase. A minimum of three independent
experiments were performed for wild-type and mutant
GATA5.
Statistical analysis
Data are expressed as means ± standard deviation.
Continuous variables were tested for normality of distribution, and
the Student’s unpaired t-test was used to compare the numeric
variables between the two groups. Comparison of the categorical
variables between the two groups was completed by using Pearson’s
χ2 test or Fisher’s exact test when appropriate. A
two-tailed P-value <0.05 was considered to indicate statistical
difference.
Results
Clinical characteristics of the study
subjects
A cohort of 110 unrelated patients with BAV was
enrolled and clinically evaluated as well as 200 unrelated control
individuals. All the participants had no established environmental
risk factors for congenital heart disease, such as maternal illness
and drug use in the first trimester of pregnancy, parental smoking,
and long-term exposure to toxicants and ionizing radiation. The
baseline clinical characteristics of the 110 unrelated BAV cases
are shown in Table I.
 | Table IClinical characteristics of the 110
unrelated patients with bicuspid aortic valve (BAV). |
Table I
Clinical characteristics of the 110
unrelated patients with bicuspid aortic valve (BAV).
| Parameter | Statistic |
|---|
| Age (years) | 45.4±11.8 |
| Male gender
(%) | 72 (65.5) |
| Positive family
history (%) | 36 (32.7) |
| Abnormal valve
functiona (%) | 73 (66.4) |
| Concomitant
aortopathyb (%) | 67 (60.9) |
| Concomitant other
cardiac structural defectsc
(%) | 20 (18.2) |
| Atrial fibrillation
(%) | 5 (4.5) |
| Surgical repair
(%) | 96 (87.3) |
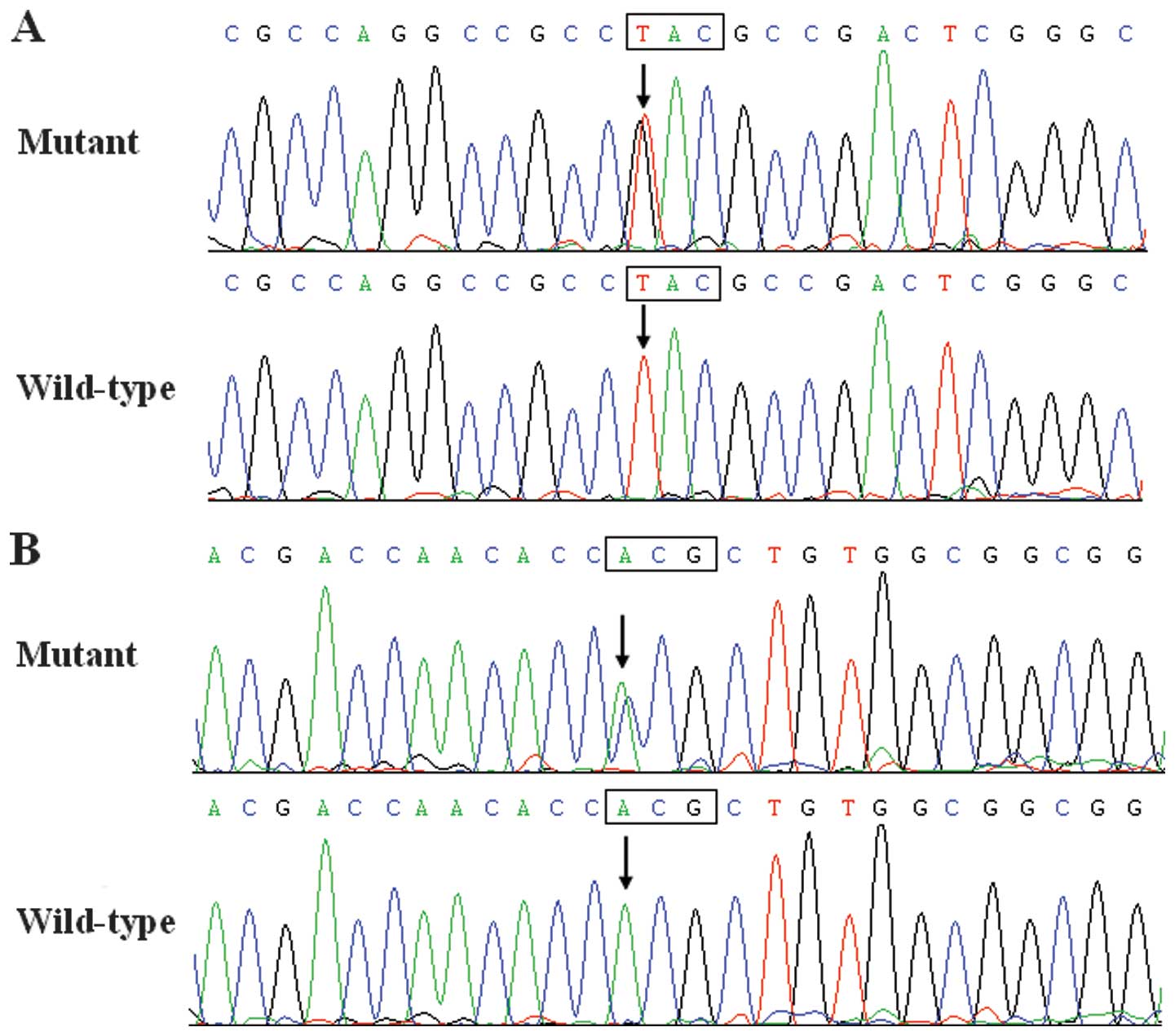
GATA5 sequence variation
Two heterozygous GATA5 sequence variations
were identified in 2 of 110 unrelated BAV patients, respectively,
with a mutational prevalence of ~1.82%. Specifically, a change of
thymine into guanine at the first nucleotide of codon 16 of the
GATA5 gene (c.46T>G), predicting the transition of
tyrosine into aspartic acid at amino acid position 16 (p.Y16D), was
identified in the index patient from family 1. A substitution of
cytosine for adenine in the first nucleotide of codon 252
(c.754A>C), equivalent to the replacement of threonine by
proline at amino acid 252 (p.T252P), was identified in the proband
from family 2. The sequence electropherograms showing the
identified heterozygous GATA5 variations in contrast to
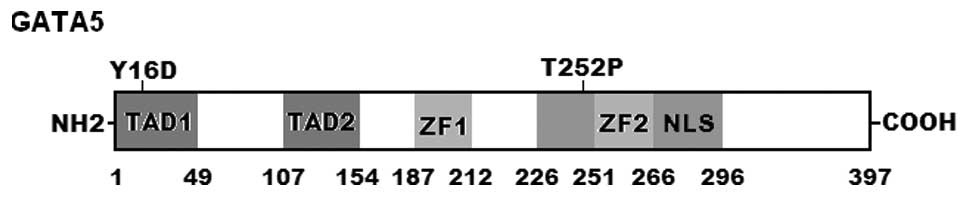
corresponding control sequences are shown in Fig. 1. A schematic diagram of GATA5
protein showing the structural domains and the locations of the
detected mutations is shown in Fig.
2. The variations were not observed in the 400 control
chromosomes or found in the SNP, HGM and 1,000 genome databases,
which were consulted again on November 10, 2013.
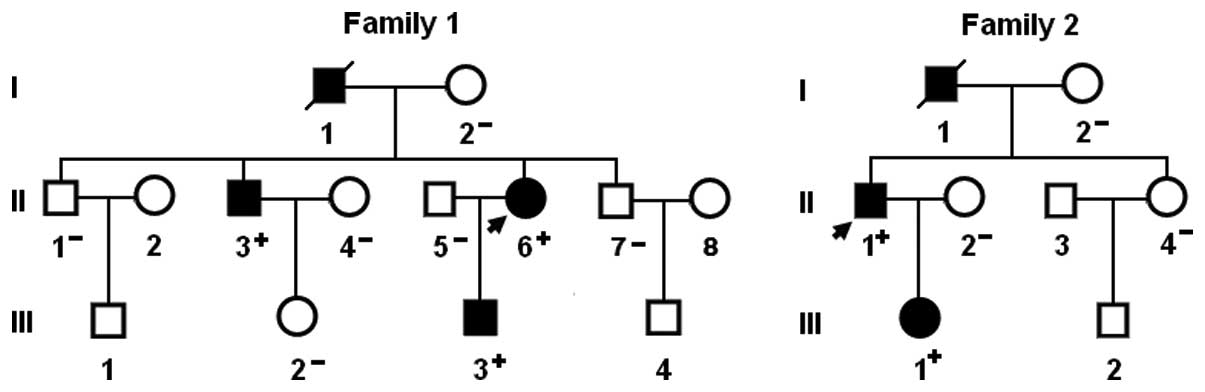
A genetic scan of the family members of the mutation
carriers showed that in each family, the variation was present in
all the affected family members available, but absent in the
unaffected family members examined. Analysis of the pedigrees
revealed that the variations cosegregated with BAV with complete
penetrance. The pedigree structures of the two families are shown
in Fig. 3. In addition, in family
1, the proband’s father (I-1) and brother (II-3) had ventricular
septal defect, aortic stenosis and the electrocardiogram documented
atrial fibrillation. In family 2, the proband’s father (I-1) also
had aortic stenosis. The phenotypic characteristics and results of
genetic screening of the affected pedigree members are listed in
Table II.
| Table IIPhenotypic characteristics and status
of the GATA5 mutations of the affected pedigree members. |
Table II
Phenotypic characteristics and status
of the GATA5 mutations of the affected pedigree members.
| Subject
information | Phenotypes | Genotypes |
|---|
|
|
|
|---|
| Identity | Gender | Age (years) | Congenital cardiac
structural defects | GATA5
mutations |
|---|
| Family 1 | | | | Y16D |
| I-1 | M | 61a | BAV, VSD, AS | NA |
| II-3 | M | 48 | BAV, VSD, AS | +/− |
| II-6 | F | 45 | BAV | +/− |
| III-3 | M | 19 | BAV | +/− |
| Family 2 | | | | T252P |
| I-1 | M | 55a | BAV, AS | NA |
| II-1 | M | 42 | BAV | +/− |
| III-1 | F | 16 | BAV | +/− |
Alignment of multiple GATA5 protein
sequence
As shown in Fig.
4, a cross-species alignment of multiple GATA5 protein
sequences demonstrated that the affected amino acids of p.Y16 and
p.T252 were completely conserved evolutionarily, suggesting that
the amino acids are functionally important.
Causative potential of GATA5 sequence
variations
The GATA5 sequence variations of c.46T>G
and c.754A>C were automatically predicted to be disease-causing,
with P-values of 0.82270 and 1.00000, respectively. No SNPs in the
altered regions were identified in the MutationTaster database. The
two variations were predicted to be probably damaging by the
software PolyPhen-2, with the same score of 1.000 (sensitivity:
0.00; specificity: 1.00)
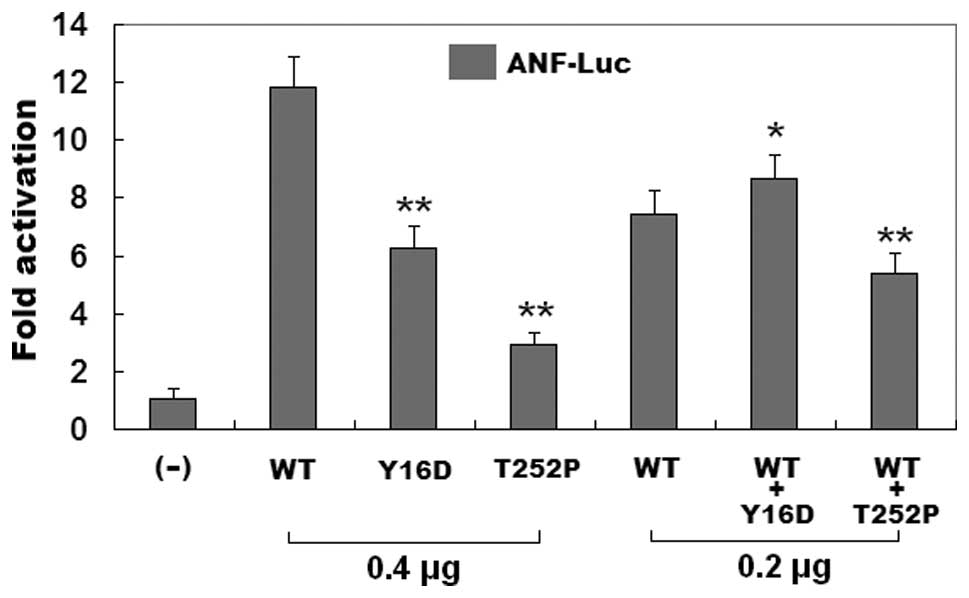
Reduced transcriptional activity of the
GATA5 mutants
As shown in Fig.
5, the wild-type GATA5, the Y16D-mutant, and the T252P-mutant
GATA5 activated the ANF promoter by ~12-, ~6- and ~3-fold,
respectively. When wild-type GATA5 was coexpressed with the same
amount of Y16D- or T252P-mutant GATA5, the induced activation of
the ANF promoter was ~9- or ~5-fold. These results suggest that the
two GATA5 mutants are associated with significantly reduced
transactivational activity compared with their wild-type
counterpart.
Discussion
In the current study, the novel heterozygous
mutations in GATA5, p.Y16D and p.T252P, were identified in two
unrelated families with BAV. In each family, the mutant allele was
present in all the affected family members, but absent in the
unaffected relatives examined and 400 referral chromosomes from an
ethnically-matched control population. A cross-species alignment of
GATA5 amino acid sequences demonstrated that the altered amino
acids were completely conserved evolutionarily. The p.Y16D and
p.T252P variations were predicted to be pathogenic mutations, and
the functional analysis revealed that the GATA5 mutant proteins
were consistently associated with significantly reduced
transcriptional activity. Therefore, it is likely that functionally
impaired GATA5 contributes to BAV in these families. To the best of
our knowledge, this is the first study to associate GATA5
loss-of-function mutations with enhanced susceptibility to BAV in
humans.
At present, six members (GATA1-6) of the GATA
transcription factor family have been identified in vertebrate.
GATA1-3 are important regulators of hematopoietic stem cells and
some ectodermal derivatives, whereas GATA4-6 are associated with
cardiogenesis and the formation of a subset of endoderm-derived
tissues (32,33). The human GATA5 gene was
mapped on chromosome 20q13.33 by fluorescence in situ
hybridization, which encodes for a protein of 397 amino acids
(40). By alignment of GATA5 with
GATA4, the structural domains of GATA5 protein are predicted to
encompass two transcriptional activation domains (TAD1, amino acids
1–49 and TAD2, amino acids 107–154), two adjacent zinc fingers
(ZF1, amino acids 187–212 and ZF2, amino acids 242–266), which
comprise the DNA-binding domain with a
Cys-X2-Cys-X17-Cys-X2-Cys
consensus (where X represents any amino acid), and one nuclear
localization signal (NLS, amino acids 226–396). The two TADs are
required for the normal transcriptional activity of GATA5. The
C-terminal ZF2 is essential for DNA sequence recognition and
binding to the consensus motif (T/A)GATA(A/G), within the promoters
of target genes; while the N-terminal ZF1 is responsible for
sequence specificity and stability of protein-DNA binding, and both
ZFs can also interact directly with other regulatory proteins. The
NLS is crucial to the sub-cellular trafficking and nuclear
distribution of GATA5 (41). The
GATA5 mutations of p.Y16D and p.T252P identified in this study are
located in TAD1 and ZF2, respectively, and may be expected to exert
an effect on the transcriptional activity of GATA5 by direct
inhibition or interfering with the nuclear localization and
specific binding ability of GATA5.
In a previous study, it was substantiated that GATA5
regulates multiple downstream molecules expressed during
embryogenesis and cardiac morphogenesis, including ANF, brain
natriuretic peptide, α- and β-myosin heavy chains, and cardiac
troponin C and I (32). Thus, the
functional characteristics of the GATA5 mutations may be
delineated by analyzing the transcriptional activity of the
ANF promoter in tool cells. In this study, the functional
effect of the novel p.Y16D and p.T252P mutations of GATA5
identified in our familial BAV patients were explored by a
transcriptional activity assay and the results revealed a
significantly reduced transcriptional activation of the ANF
promoter. These data suggest that genetically compromised GATA5 is
potentially an alternative molecular mechanism of BAV.
The relationship between GATA5 variants and
human BAV was previously investigated. Padang et al screened
the coding regions and splice signal sequences of the GATA5
gene in 100 unrelated BAV patients, and found four rare
non-synonymous variations within the GATA5 transcriptional
activation domains, i.e., p.Q3R, p.S19W, p.Y142H and p.G166S, in 4
of 100 unrelated patients, respectively, with a mutational
prevalence of 4% (37). However,
the functional roles of these GATA5 variations remain to be
determined. Foffa and colleagues screened by direct sequencing all
the coding exons including adjacent intronic as well as 5′- and
3′-untranslated of GATA5 in a cohort of 11 index patients
with familial BAV, however, no pathogenetic mutation was identified
in GATA (21). The discrepancy in
the mutational prevalence of these reports including the present
study may be partially explained by different sample size and
ethnicity.
It has been validated in animals that genetically
defective GATA5 predisposes to congenital cardiovascular defects.
In zebrafish, the targeted disruption of GATA5 led to
embryonic lethality due to defects in endocardial and myocardial
differentiation and migration, a phenotype similar to cardia bifida
of GATA4-null zebrafish (43). In mice, GATA5 knockout led
to partially penetrant BAV, with a penetrance of 25%, and
endocardial cell-specific inactivation of GATA5 resulted in
BAV, similar to that observed in GATA5-null mice (31). Furthermore, the mice that were
compound heterozygous for GATA5 and GATA4 or for
GATA5 and GATA6 knockout died embryonically or
perinatally due to severe defects of the outflow tract development
including double outlet right ventricle and ventricular septal
defect (30). These experimental
results emphasize the notable sensitivity of the developing
cardiovascular system to the levels of GATA4, GATA5 and GATA6, and
indicate that these GATA factors may act cooperatively to regulate
some target genes.
Of note, ventricular septal defect and atrial
fibrillation were documented in two BAV patients harboring the
p.Y16D mutation of GATA5. Similar to our findings, GATA5 has been
previously reported to be involved in various congenital heart
diseases as well as atrial fibrillation (38,39,42). Additionally, the GATA family
members GATA4 and GATA6 have an expression profile and functional
characteristics that overlap with those of GATA5, and a long list
of mutations in GATA4 and GATA6 have also been connected with a
large variety of congenital cardiovascular deformations and atrial
fibrillation (44–77). These findings support that the
transcription factors of the GATA family are pivotal for the
cardiovascular morphogenesis.
In conclusion, to the best of our knowledge, this
study provides the first genetic evidence for the association of
functionally compromised GATA5 with increased vulnerability to BAV,
highlighting the role of a GATA signaling pathway in BAV and other
developmental cardiovascular malformations, and suggesting the
potential implications for genetic counseling and clinical care of
the families presenting with BAV.
Acknowledgements
The authors would like to thank the participants for
their dedication to the study. This study was supported in part by
grants from the National Natural Science Fund of China (81070153,
81270161 and 81271927), the Personnel Development Foundation of
Shanghai, China (2010019), the Natural Science Fund of Shanghai,
China (10ZR1428000), and the Key Program of Basic Research of
Shanghai, China (10JC1414002).
References
|
1
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco
S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ,
Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH,
Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK,
Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner
PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D and
Turner MB: American Heart Association Statistics Committee and
Stroke Statistics Subcommittee: Heart disease and stroke
statistics-2013 update: a report from the American Heart
Association. Circulation. 127:e6–e245. 2013. View Article : Google Scholar
|
|
2
|
Roberts WC: The congenitally bicuspid
aortic valve. A study of 85 autopsy cases. Am J Cardiol. 26:72–83.
1970. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ward C: Clinical significance of the
bicuspid aortic valve. Heart. 83:81–85. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Larson EW and Edwards WD: Risk factors for
aortic dissection: a necropsy study of 161 cases. Am J Cardiol.
53:849–855. 1984. View Article : Google Scholar
|
|
5
|
Basso C, Boschello M, Perrone C, Mecenero
A, Cera A, Bicego D, Thiene G and De Dominicis E: An
echocardiographic survey of primary school children for bicuspid
aortic valve. Am J Cardiol. 93:661–663. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tutar E, Ekici F, Atalay S and Nacar N:
The prevalence of bicuspid aortic valve in newborns by
echocardiographic screening. Am Heart J. 150:513–515. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Siu SC and Silversides CK: Bicuspid aortic
valve disease. J Am Coll Cardiol. 55:2789–2800. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fedak PW, Verma S, David TE, Leask RL,
Weisel RD and Butany J: Clinical and pathophysiological
implications of a bicuspid aortic valve. Circulation. 106:900–904.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tadros TM, Klein MD and Shapira OM:
Ascending aortic dilatation associated with bicuspid aortic valve:
pathophysiology, molecular biology, and clinical implications.
Circulation. 119:880–890. 2009. View Article : Google Scholar
|
|
10
|
Alegret JM, Ligero C, Vernis JM,
Beltrán-Debón R, Aragonés G, Duran I, Palazón O and
Hernández-Aparicio A: Factors related to the need for surgery after
the diagnosis of bicuspid aortic valve: one center’s experience
under a conservative approach. Int J Med Sci. 10:176–182.
2013.PubMed/NCBI
|
|
11
|
Baig W: Endocarditis on the bicuspid
aortic valve: what’s the risk? Heart. 96:1689–1690. 2010.
|
|
12
|
Mack G and Silberbach M: Aortic and
pulmonary stenosis. Pediatr Rev. 21:79–85. 2000. View Article : Google Scholar
|
|
13
|
Cripe L, Andelfinger G, Martin LJ, Shooner
K and Benson DW: Bicuspid aortic valve is heritable. J Am Coll
Cardiol. 44:138–143. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Michelena HI, Khanna AD, Mahoney D,
Margaryan E, Topilsky Y, Suri RM, Eidem B, Edwards WD, Sundt TM III
and Enriquez-Sarano M: Incidence of aortic complications in
patients with bicuspid aortic valves. JAMA. 306:1104–1112. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Armstrong EJ and Bischoff J: Heart valve
development: endothelial cell signaling and differentiation. Circ
Res. 95:459–470. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Combs MD and Yutzey KE: Heart valve
development: regulatory networks in development and disease. Circ
Res. 105:408–421. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Garg V, Muth AN, Ransom JF, Schluterman
MK, Barnes R, King IN, Grossfeld PD and Srivastava D: Mutations in
NOTCH1 cause aortic valve disease. Nature. 437:270–274. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martin LJ, Ramachandran V, Cripe LH,
Hinton RB, Andelfinger G, Tabangin M, Shooner K, Keddache M and
Benson DW: Evidence in favor of linkage to human chromosomal
regions 18q, 5q and 13q for bicuspid aortic valve and associated
cardiovascular malformations. Hum Genet. 121:275–284. 2007.
View Article : Google Scholar
|
|
19
|
Mohamed SA, Aherrahrou Z, Liptau H, Erasmi
AW, Hagemann C, Wrobel S, Borzym K, Schunkert H, Sievers HH and
Erdmann J: Novel missense mutations (p. T596M and pP1797H) in
NOTCH1 in patients with bicuspid aortic valve. Biochem Biophys Res
Commun. 345:1460–1465. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McKellar SH, Tester DJ, Yagubyan M,
Majumdar R, Ackerman MJ and Sundt TM III: Novel NOTCH1 mutations in
patients with bicuspid aortic valve disease and thoracic aortic
aneurysms. J Thorac Cardiovasc Surg. 134:290–296. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Foffa I, Ait Alì L, Panesi P, Mariani M,
Festa P, Botto N, Vecoli C and Andreassi MG: Sequencing of NOTCH1,
GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic
valve. BMC Med Genet. 14:442013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Andelfinger G, Tapper AR, Welch RC, Vanoye
CG, George AL Jr and Benson DW: KCNJ2 mutation results in Andersen
syndrome with sex-specific cardiac and skeletal muscle phenotypes.
Am J Hum Genet. 71:663–668. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Girdauskas E, Schulz S, Borger MA, Mierzwa
M and Kuntze T: Transforming growth factor-beta receptor type II
mutation in a patient with bicuspid aortic valve disease and
intraoperative aortic dissection. Ann Thorac Surg. 14:e70–e71.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Beffagna G, Cecchetto A, Dal Bianco L,
Lorenzon A, Angelini A, Padalino M, Vida V, Bhattacharya S, Stellin
G, Rampazzo A and Daliento L: R25C mutation in the NKX2.5 gene in
Italian patients affected with non-syndromic and syndromic
congenital heart disease. J Cardiovasc Med (Hagerstown).
14:582–586. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Reiter JF, Alexander J, Rodaway A, Yelon
D, Patient R, Holder N and Stainier DY: Gata5 is required for the
development of the heart and endoderm in zebrafish. Genes Dev.
13:2983–2995. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nemer G and Nemer M: Cooperative
interaction between GATA5 and NF-ATc regulates
endothelial-endocardial differentiation of cardiogenic cells.
Development. 129:4045–4055. 2002.
|
|
27
|
Stennard FA, Costa MW, Elliott DA, Rankin
S, Haast SJ, Lai D, McDonald LP, Niederreither K, Dolle P, Bruneau
BG, Zorn AM and Harvey RP: Cardiac T-box factor Tbx20 directly
interacts with Nkx2–5, GATA4, and GATA5 in regulation of gene
expression in the developing heart. Dev Biol. 262:206–224.
2003.PubMed/NCBI
|
|
28
|
Haworth KE, Kotecha S, Mohun TJ and
Latinkic BV: GATA4 and GATA5 are essential for heart and liver
development in Xenopus embryos. BMC Dev Biol. 8:742008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Singh MK, Li Y, Li S, Cobb RM, Zhou D, Lu
MM, Epstein JA, Morrisey EE and Gruber PJ: Gata4 and Gata5
cooperatively regulate cardiac myocyte proliferation in mice. J
Biol Chem. 285:1765–72. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laforest B and Nemer M: GATA5 interacts
with GATA4 and GATA6 in outflow tract development. Dev Biol.
358:368–378. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Laforest B, Andelfinger G and Nemer M:
Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin
Invest. 121:2876–2887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pikkarainen S, Tokola H, Kerkelä R and
Ruskoaho H: GATA transcription factors in the developing and adult
heart. Cardiovasc Res. 63:196–207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Peterkin T, Gibson A, Loose M and Patient
R: The roles of GATA-4, -5 and -6 in vertebrate heart development.
Semin Cell Dev Bio. 16:83–94. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wei D, Bao H, Zhou N, Zheng GF, Liu XY and
Yang YQ: GATA5 loss-of-function mutation responsible for the
congenital ventriculoseptal defect. Pediatr Cardiol. 34:504–511.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang JQ, Li RG, Wang J, Liu XY, Xu YJ,
Fang WY, Chen XZ, Zhang W, Wang XZ and Yang YQ: Prevalence and
spectrum of GATA5 mutations associated with congenital heart
disease. Int J Cardiol. 165:570–573. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wei D, Bao H, Liu XY, Zhou N, Wang Q, Li
RG, Xu YJ and Yang YQ: GATA5 loss-of-function mutations underlie
tetralogy of fallot. Int J Med Sci. 10:34–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Padang R, Bagnall RD, Richmond DR, Bannon
PG and Semsarian C: Rare non-synonymous variations in the
transcriptional activation domains of GATA5 in bicuspid aortic
valve disease. J Mol Cell Cardiol. 53:277–281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang YQ, Wang J, Wang XH, Wang Q, Tan HW,
Zhang M, Shen FF, Jiang JQ, Fang WY and Liu X: Mutational spectrum
of the GATA5 gene associated with familial atrial fibrillation. Int
J Cardiol. 157:305–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang XH, Huang CX, Wang Q, Li RG, Xu YJ,
Liu X, Fang WY and Yang YQ: A novel GATA5 loss-of-function mutation
underlies lone atrial fibrillation. Int J Mol Med. 31:43–50.
2013.PubMed/NCBI
|
|
40
|
Nemer G, Qureshi ST, Malo D and Nemer M:
Functional analysis and chromosomal mapping of Gata5, a gene
encoding a zinc finger DNA-binding protein. Mamm Genome.
10:993–999. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Brewer A and Pizzey J: GATA factors in
vertebrate heart development and disease. Expert Rev Mol Med.
15:1–20. 2006.
|
|
42
|
Gu JY, Xu JH, Yu H and Yang YQ: Novel
GATA5 loss-of-function mutations underlie familial atrial
fibrillation. Clinics (Sao Paulo). 67:1393–1399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Heicklen-Klein A, McReynolds LJ and Evans
T: Using the zebrafish model to study GATA transcription factors.
Semin Cell Dev Biol. 16:95–106. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Garg V, Kathiriya IS, Barnes R,
Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS,
Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC and Srivastava D:
GATA4 mutations cause human congenital heart defects and reveal an
interaction with TBX5. Nature. 424:443–447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Okubo A, Miyoshi O, Baba K, Takagi M,
Tsukamoto K, Kinoshita A, Yoshiura K, Kishino T, Ohta T, Niikawa N
and Matsumoto N: A novel GATA4 mutation completely segregated with
atrial septal defect in a large Japanese family. J Med Genet.
41:e972004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sarkozy A, Conti E, Neri C, D’Agostino R,
Digilio MC, Esposito G, Toscano A, Marino B, Pizzuti A and
Dallapiccola B: Spectrum of atrial septal defects associated with
mutations of NKX2.5 and GATA4 transcription factors. J Med Genet.
42:e162005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hirayama-Yamada K, Kamisago M, Akimoto K,
Aotsuka H, Nakamura Y, Tomita H, Furutani M, Imamura S, Takao A,
Nakazawa M and Matsuoka R: Phenotypes with GATA4 or NKX2.5
mutations in familial atrial septal defect. Am J Med Genet A.
135:47–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Reamon-Buettner SM and Borlak J: GATA4
zinc finger mutations as a molecular rationale for septation
defects of the human heart. J Med Genet. 42:e322005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nemer G, Fadlalah F, Usta J, Nemer M,
Dbaibo G, Obeid M and Bitar F: A novel mutation in the GATA4 gene
in patients with Tetralogy of Fallot. Hum Mutat. 27:293–294. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tomita-Mitchell A, Maslen CL, Morris CD,
Garg V and Goldmuntz E: GATA4 sequence variants in patients with
congenital heart disease. J Med Genet. 44:779–783. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rajagopal SK, Ma Q, Obler D, Shen J,
Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V,
Srivastava D, Goldmuntz E, Broman KW, Benson DW, Smoot LB and Pu
WT: Spectrum of heart disease associated with murine and human
GATA4 mutation. J Mol Cell Cardiol. 43:677–685. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang W, Li X, Shen A, Jiao W, Guan X and
Li Z: GATA4 mutations in 486 Chinese patients with congenital heart
disease. Eur J Med Genet. 51:527–535. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hamanoue H, Rahayuningsih SE, Hirahara Y,
Itoh J, Yokoyama U, Mizuguchi T, Saitsu H, Miyake N, Hirahara F and
Matsumoto N: Genetic screening of 104 patients with congenitally
malformed hearts revealed a fresh mutation of GATA4 in those with
atrial septal defects. Cardiol Young. 19:482–485. 2009. View Article : Google Scholar
|
|
54
|
Chen MW, Pang YS, Guo Y, Pan JH, Liu BL,
Shen J and Liu TW: GATA4 mutations in Chinese patients with
congenital cardiac septal defects. Pediatr Cardiol. 31:85–89. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen Y, Mao J, Sun Y, Zhang Q, Cheng HB,
Yan WH, Choy KW and Li H: A novel mutation of GATA4 in a familial
atrial septal defect. Clin Chim Acta. 411:1741–1745. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Butler TL, Esposito G, Blue GM, Cole AD,
Costa MW, Waddell LB, Walizada G, Sholler GF, Kirk EP, Feneley M,
Harvey RP and Winlaw DS: GATA4 mutations in 357 unrelated patients
with congenital heart malformation. Genet Test Mol Biomarkers.
14:797–802. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Salazar M, Consoli F, Villegas V, Caicedo
V, Maddaloni V, Daniele P, Caianiello G, Pachón S, Nuñez F,
Limongelli G, Pacileo G, Marino B, Bernal JE, De Luca A and
Dallapiccola B: Search of somatic GATA4 and NKX2.5 gene mutations
in sporadic septal heart defects. Eur J Med Genet. 54:306–309.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu XY, Wang J, Zheng JH, Bai K, Liu ZM,
Wang XZ, Liu X, Fang WY and Yang YQ: Involvement of a novel
GATA4 mutation in atrial septal defects. Int J Mol Med.
28:17–23. 2011.
|
|
59
|
Wang J, Fang M, Liu XY, Xin YF, Liu ZM,
Chen XZ, Wang XZ, Fang WY, Liu X and Yang YQ: A novel GATA4
mutation responsible for congenital ventricular septal defects. Int
J Mol Med. 28:557–564. 2011.PubMed/NCBI
|
|
60
|
Yang YQ, Li L, Wang J, Liu XY, Chen XZ,
Zhang W, Wang XZ, Jiang JQ, Liu X and Fang WY: A novel GATA4
loss-of-function mutation associated with congenital ventricular
septal defect. Pediatr Cardiol. 33:539–546. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Granados-Riveron JT, Pope M, Bu’lock FA,
Thornborough C, Eason J, Setchfield K, Ketley A, Kirk EP, Fatkin D,
Feneley MP, Harvey RP and Brook JD: Combined mutation screening of
NKX2–5, GATA4, and TBX5 in congenital heart disease: multiple
heterozygosity and novel mutations. Congenit Heart Dis. 7:151–159.
2012.
|
|
62
|
Yang YQ, Wang J, Liu XY, Chen XZ, Zhang W,
Wang XZ, Liu X and Fang WY: Novel GATA4 mutations in patients with
congenital ventricular septal defects. Med Sci Monit.
18:CR344–CR350. 2012.PubMed/NCBI
|
|
63
|
Wang E, Sun S, Qiao B, Duan W, Huang G, An
Y, Xu S, Zheng Y, Su Z, Gu X, Jin L and Wang H: Identification of
functional mutations in GATA4 in patients with congenital heart
disease. PLoS One. 8:e621382013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Li RG, Li L, Qiu XB, Yuan F, Xu L, Li X,
Xu YJ, Jiang WF, Jiang JQ, Liu X, Fang WY, Zhang M, Peng LY, Qu XK
and Yang YQ: GATA4 loss-of-function mutation underlies familial
dilated cardiomyopathy. Biochem Biophys Res Commun. 439:591–596.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yang YQ, Gharibeh L, Li RG, Xin YF, Wang
J, Liu ZM, Qiu XB, Xu YJ, Xu L, Qu XK, Liu X, Fang WY, Huang RT,
Xue S and Nemer G: GATA4 loss-of-function mutations underlie
familial tetralogy of fallot. Hum Mutat. 34:1662–1671. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Jiang JQ, Shen FF, Fang WY, Liu X and Yang
YQ: Novel GATA4 mutations in lone atrial fibrillation. Int J Mol
Med. 28:1025–1032. 2011.PubMed/NCBI
|
|
67
|
Yang YQ, Wang MY, Zhang XL, Tan HW, Shi
HF, Jiang WF, Wang XH, Fang WY and Liu X: GATA4 loss-of-function
mutations in familial atrial fibrillation. Clin Chim Acta.
412:1825–1830. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wang J, Sun YM and Yang YQ: Mutation
spectrum of the GATA4 gene in patients with idiopathic atrial
fibrillation. Mol Biol Rep. 39:8127–8135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kodo K, Nishizawa T, Furutani M, Arai S,
Yamamura E, Joo K, Takahashi T, Matsuoka R and Yamagishi H: GATA6
mutations cause human cardiac outflow tract defects by disrupting
semaphorin-plexin signaling. Proc Natl Acad Sci USA.
106:13933–13938. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Lin X, Huo Z, Liu X, Zhang Y, Li L, Zhao
H, Yan B, Liu Y, Yang Y and Chen YH: A novel GATA6 mutation in
patients with tetralogy of Fallot or atrial septal defect. J Hum
Genet. 55:662–667. 2010. View Article : Google Scholar
|
|
71
|
Maitra M, Koenig SN, Srivastava D and Garg
V: Identification of GATA6 sequence variants in patients with
congenital heart defects. Pediatr Res. 68:281–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zheng GF, Wei D, Zhao H, Zhou N, Yang YQ
and Liu XY: A novel GATA6 mutation associated with congenital
ventricular septal defect. Int J Mol Med. 29:1065–1071.
2012.PubMed/NCBI
|
|
73
|
Wang J, Luo XJ, Xin YF, Liu Y, Liu ZM,
Wang Q, Li RG, Fang WY, Wang XZ and Yang YQ: Novel GATA6
mutations associated with congenital ventricular septal defect or
tetralogy of fallot. DNA Cell Biol. 31:1610–1617. 2012.
|
|
74
|
Huang RT, Xue S, Xu YJ and Yang YQ:
Somatic mutations in the GATA6 gene underlie sporadic
tetralogy of Fallot. Int J Mol Med. 31:51–58. 2013.
|
|
75
|
Yang YQ, Wang XH, Tan HW, Jiang WF, Fang
WY and Liu X: Prevalence and spectrum of GATA6 mutations associated
with familial atrial fibrillation. Int J Cardiol. 155:494–496.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yang YQ, Li L, Wang J, Zhang XL, Li RG, Xu
YJ, Tan HW, Wang XH, Jiang JQ, Fang WY and Liu X: GATA6
loss-of-function mutation in atrial fibrillation. Eur J Med Genet.
55:520–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Li J, Liu WD, Yang ZL and Yang YQ: Novel
GATA6 loss-of-function mutation responsible for familial atrial
fibrillation. Int J Mol Med. 30:783–790. 2012.PubMed/NCBI
|