Introduction
Sporothrix schenckii (S. schenckii), the
causative agent of sporotrichosis, is a dimorphic fungus that
produces lymphocutaneous lesions. The signature characteristic of
S. Schenckii is a temperature-induced phase transition.
S. schenckii grows as a mold in soil at an ambient
temperature and is converted to a yeast form after the spores
infect the skin of a mammalian host by direct inoculation (1). Although the molecular basis of
phenotypic switching in a dimorphic fungus is not well understood,
several possible mechanisms have been considered, including genomic
rearrangements through epigenetic mechanisms (2). Heritable epigenetic changes are well
characterized in Saccharomyces cerevisiae (S. cerevisiae).
Regulatory genes that control colony morphology are located in
chromosomal positions that exist in two alternative states: a
silenced state and an active state. The transition between the two
states results from changes in chromatin structure. Phenotypic
switching in S. cerevisiae occurs when the chromatin state
spontaneously changes, a characteristic of silenced domains
(3). The silent information
regulator (Sir) genes are required to establish the silent state,
and mutations in these genes can affect the efficiency with which
S. cerevisiae cells pass on the silent state to their
daughter cells (4). Phenotypic
switching in Candida albicans (C. albicans) could in
principle be controlled using a similar mechanism (5). In a recent study of ours,
differentially expressed proteins altered by phenotypic switching
in S. schenckii were identified using two-dimensional
electrophoresis. One of these proteins is homologous to the Sir2
protein from Aspergillus fumigatus and its expression
increases in the early yeast form of S. schenckii (6).
In this study, we describe the molecular cloning of
the S. schenckii Sir homologue, designated as SsSir2. We
performed functional analysis of the SsSir2 gene, and detected
differential gene expression in the dimorphic switch of S.
schenckii. The data presented in this study may broaden our
understanding of the function of the SsSir2 gene from S.
Schenckii.
Materials and methods
Fungal strain, media and growth
conditions
The strain of S. schenckii which was used,
ATCC10268, was maintained at the Research Center for Pathogenic
Fungi, Dalian Medical University, Dalian, China. To obtain a
mycelial culture, the ATCC10268 isolate was inoculated on Sabouraud
dextrose agar (SDA) medium (Sigma, St. Louis, MO, USA) and
incubated at 25°C. The mycelial colonies thus obtained were
inoculated in Sabouraud fluid medium (Sigma) and cultured with
shaking at 100 rpm at 25°C for 96 h. To achieve the switch of S.
schenckii from the mycelial phase to the yeast phase, mycelial
colonies were transferred to brain heart infusion (BHI) liquid
medium at 37°C and shaken at 100 rpm for 96 h. The mycelial and
yeast pellets were collected by centrifugation and stored at
−80°C.
Total RNA, genomic DNA isolation and gene
cloning
Approximately 100 mg samples of S. schenckii
in the mycelial and yeast phase were separately pulverized under
liquid nitrogen with a mortar and pestle. Total RNA isolation was
carried out according to the manufacturer’s instructions using
TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA)
and treated with the RNase-free DNase I kit from Takara (Tokyo,
Japan) to eliminate DNA contamination. Genomic DNA was isolated
from the yeast phase colonies following the manufacturer’s
instructions using the InstaGene™ Matrix kit (Bio-Rad Laboratories,
Hercules, CA, USA). cDNA was synthesized from 500 μg of
total RNA of ATCC10268 by murine leukemia virus reverse
transcriptase (MLV-RT) (Takara) primed with oligo(dT) following the
manufacturer’s instructions, and used as a template for PCR.
Degenerate primers, SsSir2-F1 and SsSir2-R1, were designed based on
multiple alignments of the highly conserved Sir2 domains of C.
albicans Sir2 (NW139502), Cryptococcus neoformans Sir2
(NC009183) and Ustilago maydis Sir2 (XM756946) amino acid
sequences. The PCR product of expected size was cloned into the
pMD18 vector (Takara) and sequenced. The degenerate primers yielded
a 248 bp fragment homologous to known Sir2. To obtain the
full-length cDNA sequence of the SsSir2 gene, 5′-RACE and 3′-RACE
experiments were performed using the 5′-Full RACE kit and 3′-Full
RACE Core Set Ver. 2.0 kit (Takara) according to the manufacturer’s
instructions. Nested PCR was then performed. Briefly, five specific
primers, 3′SIR2-1 and 3′SIR2-2 of 3′-RACE and 5′SIR2-1, 5′SIR2-2
and 5′SIR2-3 of 5′-RACE, were synthesized based on the cDNA
sequence obtained by the degenerate primers. PCR products of
5′-RACE and 3′-RACE were both cloned into the pMD18 vector (Takara)
and sequenced. To determine the nucleotide sequence of the genomic
DNA corresponding to SsSir2, PCR was performed using the primers,
SsSir2-G1 and SsSir2-G2, and genomic DNA as a template. The PCR
products were then sequenced. The sequences of all the primers used
in this study are listed in Table
I.
 | Table ISequences of primers used in this
study. |
Table I
Sequences of primers used in this
study.
| Name | Sequence (5′→3′) | Length (bp) |
|---|
| SIR2-F1 |
ACNGCNGCNGGNATHCCYGAYTT | 23 |
| SIR2-R1 |
CARTCDATRTTYTGRGTRAA | 20 |
| 3′SIR2-1 |
GAACCCTCATCCGTTTTACATTCTCGCC | 28 |
| 3′SIR2-2 |
ACGCCTTCTTGTCTCTCCTCGCTGCC | 26 |
| 5′SIR2-1 | GAGAGACAAGAAGGCG | 16 |
| 5′SIR2-2 |
GAGAATGTAAAACGGATGAGGG | 22 |
| 5′SIR2-3 |
AGCATAGGGCAGTTTGAGGCG | 21 |
| G1 |
ATGGGCCAGGAAGTGTCCA | 19 |
| G2 |
GCGGCGGCCGCCAGCTGTGGCCGCTCAATG | 30 |
| 8F |
GGAACTTACAAGACCATCAT | 20 |
| 58R |
GAGTTGGCCACTGGTTT | 17 |
| 24T |
FAM-CAGTGCCTCAAATTCCAA-TAMRA | 19 |
Bioinformatic and phylogenetic analysis
of SsSir2
The nucleotide sequences were analyzed using
Sequence Scanner software (Applied Biosystems/Life Technologies
Corp., Carlsbad, CA, USA) and the BLAST network service of the
National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/blast). The open
reading frame was found by the ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). For the
exact localization of the exon/intron boundaries the
mRNA-to-genomic alignment program, Spidey (http://www.ncbi.nlm.nih.gov/IEB/Research/Ostell/Spidey/index.html),
was used. The deduced amino acid sequence was analyzed with the
Expert Protein Analysis System (http://www.expasy.org/) and the protein domain
characteristics of SsSir2 were determined using the Simple Modular
Architecture Research Tool (http://hits.isb-sib.ch/cgi-bin/PFSCAN). Isoelectric
point and molecular weight prediction were carried out at
(http://cn.expasy.org/tools/pi_tool.html). Multiple
alignments of SsSir2 were performed with the ClustalW Multiple
Alignment Program (http://www.ebi.ac.uk/clustalw/).
Molecular modeling
The SsSir2 sequence was searched for a similar
sequence using the PSI-BLAST against Protein Data Bank (PDB). The
PSI-BLAST results yielded the X-ray structure of the human histone
deacetylase SIRT2 (PDB code: 1J8F) with a 47.7% identity to our
SsSir2 protein. The fold recognition server, pGenTHREADER,
identified SIRT2 as a good template with a good confidence level.
Consequently, the crystal structure of SIRT2 was used as the
template for modeling SsSir2. The amino acid sequence of SsSir2 was
aligned with the sequence of SIRT2 using the align 2D module in
MODELLER. Manual inspection and modification of the alignment was
performed after initial model generation and analysis of the DOPE
score profile of the generated model. Good sequence alignment was
obtained for 280 amino acids of the SsSir2 sequence; however the
first 17 and the last 145 amino acids were not presented in the
alignment. Homology models were built based upon the alignment of
the sequence using the automated homology modeling program,
MODELLER 9v9, with default parameters. Loops were refined using the
loop-model module of MODELLER with a fast molecular dynamics. The
best model was selected from 3,000 candidates based on the
evaluation of PDF total energy, verify score and Ramachandran plot.
Subsequently, to remove bad contacts between side-chains, the best
model was refined by a 3,000 step energy minimization with a
constraint on backbone atoms using the molecular dynamics
simulation program, NAMD. The quality of the final model was
examined by Verify3D, ERRAT and the Ramachandran plot.
Molecular docking
Molecular docking can fit molecules together in a
favorable configuration to form a complex system. The structural
information from the theoretically modeled complex may help us to
clarify the binding mechanism between SsSir2 and NAD. In order to
gain insight into the binding mode of SsSir2 with NAD, the
automated docking program, AutoDock v4.2, was used to perform the
molecular docking. AutoDock uses a Lamarckian genetic algorithm to
explore the binding possibilities of a ligand in a binding pocket.
This program allows the ligand to be flexible, whereas the protein
side-chains remain fixed. For our docking experiment, a grid of
80×70×70 points was generated with a grid SIR2-R1 spacing of 0.375
Å around the large groove which was supposed to be the NAD binding
site. This grid space defines the region of the protein in which
the ligand searched for the most favorable interactions. A total of
200 dockings were carried out with 27,000 cycles per run;
interaction energies were calculated for various docked positions
and ranked in accordance with the interaction energies between the
ligand and the protein. Finally, the docked complex of SsSir2 and
NAD was selected according to the criteria of the hydrogen bond
network combined with the interaction energy.
Differential expression of SsSir2 in two
stages during the dimorphic switch
The expression of the SsSir2 transcript in the
different stages (mycelial and yeast) were measured by real-time
RT-PCR. Primers and a TaqMan probe for target genes were designed
with PrimerSelect in the Lasergene software package (DNAStar Inc.,
Madison, WI, USA) and are listed in Table I (24T, 8F, 58R). Fifty nanograms
of total RNA were assayed from two stages of S. schenckii in
triplicate using the PrimeScript RT-PCR kit (Takara). The
minus-reverse transcriptase control was also performed in
triplicate. The amplification conditions were optimized for the ABI
PRISM 7500 instrument (Applied Biosystems, Carlsbad, CA, USA). The
cycling conditions using TaqMan probe detection were as follows:
95°C for 2 min followed by 40 cycles at 95°C for 10 sec, 61°C for
10 sec, 72°C for 40 sec. 18srDNA was selected as the endogenous
control. The relative quantification of target gene expression was
carried out using the comparative cycle threshold (CT) method as
previously described by Livak and Schmittgen (7). The ΔCT value was determined by
subtracting the target CT value of each sample from its respective
18srDNA CT value. The calculation of ΔΔCT involved using the
mycelial sample ΔCT value as an arbitrary constant to subtract from
yeast sample ΔCT values. Differences in the expression of target
genes were determined by 2−ΔΔCT. Data are expressed as arithmetic
means ± SD unless otherwise indicated. A comparison between
mycelial and yeast samples was performed using the Student’s
t-test. Differences with a P-value of <0.05 were considered to
be statistically significant.
Results
Cloning and sequence analysis of
SsSir2
A full-length SsSir2 cDNA (1753 bp) including an
open reading frame of 1329 bp, encoding 442 amino residues, was
flanked by a 204 bp 5′-untranslated region (5′-UTR) and a 220 bp
3′-UTR (Fig. 1). The SsSir2
genomic DNA is 1472 bp in length. The aligned results revealed that
there were two introns between the sequences of the genomic DNA and
the cDNA. Its 5′ and 3′ ends conformed to the basic consensus,
GT/AG, for the eukaryotic splice donor and acceptor site. Based on
the sequence of the cDNA, the molecular weight of the predicted
amino acid is approximately 48.1 kDa, the theoretical pI is 4.6.
Motif searches and sequence comparison revealed that SsSir2
consists of both a 187-amino acid conserved enzymatic core domain
(residues 43–229) and extended N- and C-terminal sequences usually
present in eukaryotic Sir2 homologues (Fig. 1). Database searches revealed that
there are several short motifs of conserved amino acids present in
the enzymatic core domain of SsSir2; these include GAGISXXXGIPXXR,
PXXXH, TQNID, HG, two sets of CXXXXC that may be a zinc-finger
motif, FGE, GTS and VN (Fig.
1).
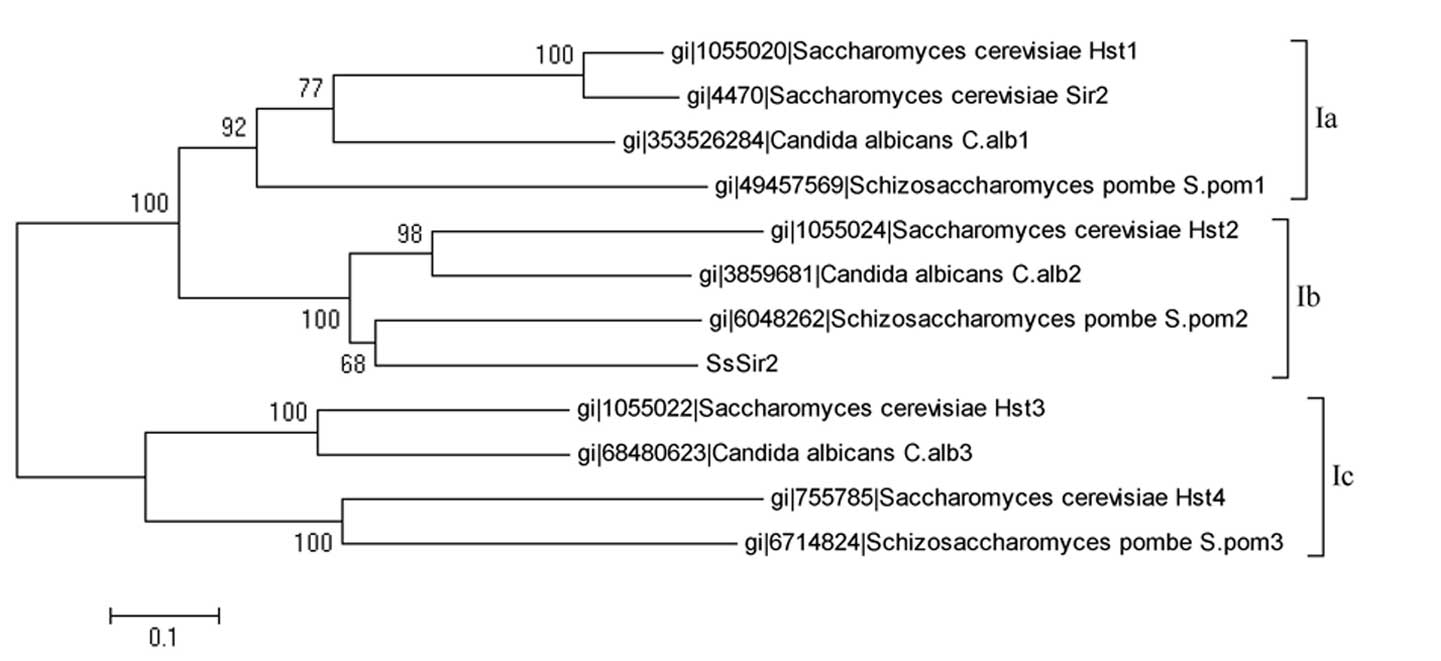
Homology and phylogenetic analysis of
SsSir2
To clarify the association between SsSir2 and other
Sir2-like proteins, we calculated multiple sequence alignments of
these sequences. The derived evolutionary tree is split into two
main branches, one formed by some class Ia and class Ic sirtuins,
the other one by class Ib sirtuins, including the SsSir2 sequence
(Fig. 2). Further alignment
clearly revealed that the SsSir2 kinase core domain is highly
homologous to that of other class Ib sirtuins, such as Hst2
(U39063), C.alb2 (CAA22018 ) and S.pom2 (AL121807) (Fig. 3). These proteins share a similar
structure and have 78, 80 and 75% amino acid similarities in the
core domain, respectively. These proteins have a highly conserved
catalytic core domain in common, but display a variation in their
extended N- and C-terminal domains, which may have a function in
target specificity of Sir2, as mutations outside the enzymatic core
domain of the yeast Sir2 selectively affect distinct forms of
silencing.
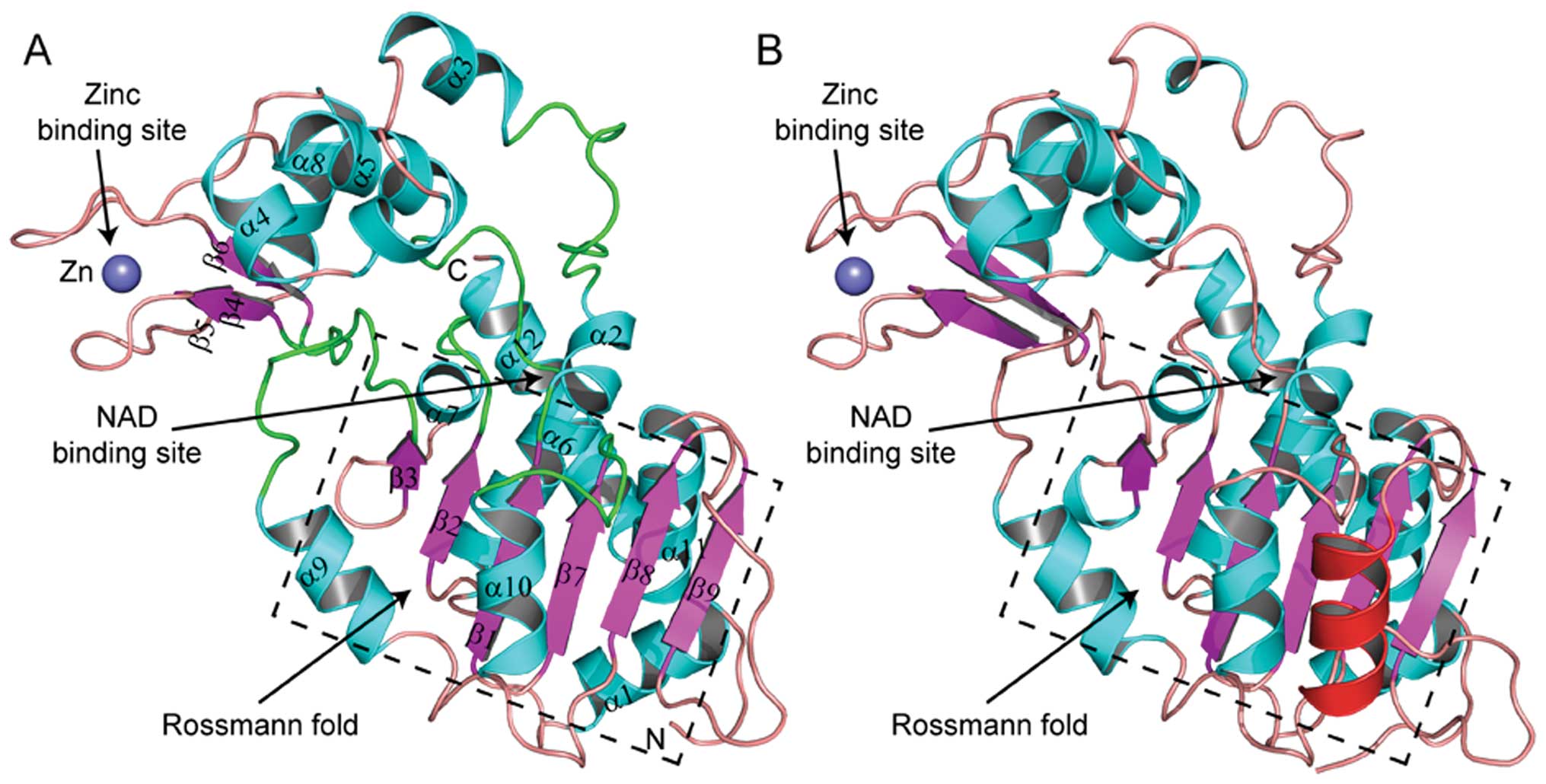
Three-dimensional (3D) model of SsSir2
protein
The homology model of SsSir2 was constructed based
on the X-ray crystal structure of human histone deacetylase SIRT2
(PDB code: 1J8F), which had relatively high sequence identity
(47.7%) with SsSir2, and the manually modified sequence alignment
of SsSir2 and SIRT2. Following the homology modeling and loop
refinement, the best model was selected from 3,000 candidates. The
final model (Fig. 4A) was then
obtained after several rounds of energy minimization to remove bad
contacts between side-chains, and its quality was further validated
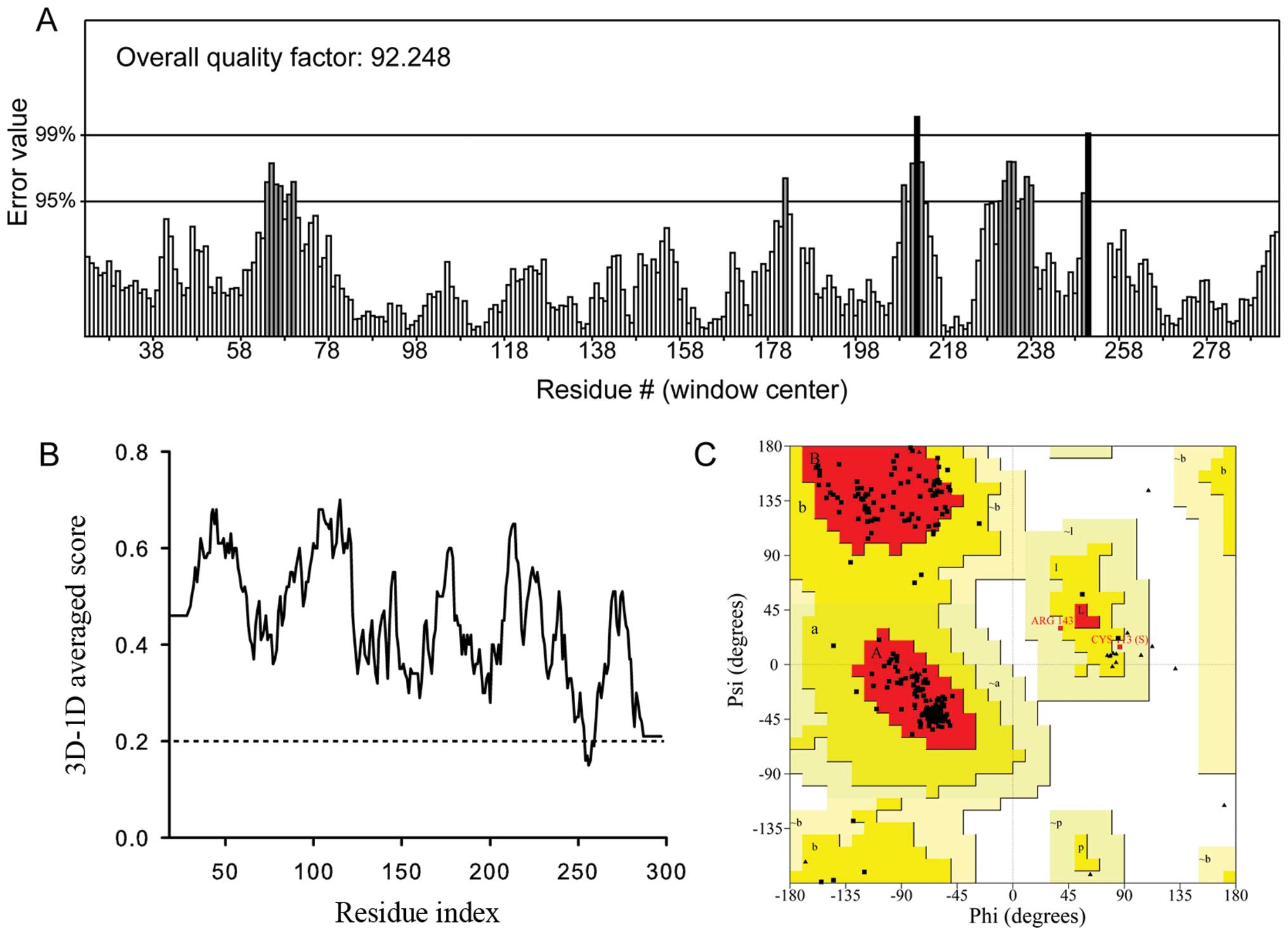
by ERRAT, Verify3D and the Ramachandran plot (Fig. 5). The non-bonded interaction
between different atoms was analyzed by ERRAT. The ERRAT overall
quality factor for the SsSir2 model was 92.248 (Fig. 5A), which was within the range of a
high quality model. The Verify3D results (Fig. 5B) for the SsSir2 model revealed
that 98.22% of the residues had an averaged 3D-1D score of >0.2,
which indicates a well built model since almost all the residues
are valid in their folded conformation. The Ramachandran plot
(Fig. 5C) showed that for the
SsSir2 model, 92.4% residues were in most favored regions, 6.7% in
the additional allowed regions, 0.8% in the generously allowed
regions and none of the residues were in the disallowed region,
which means that this model has a high stereochemical quality.
The SsSir2 model contains 280 amino acids of the
SsSir2 sequence, and the first 17 and the last 145 amino acids were
not built, as these regions were predicted to be unstructured or
loosely folded. SsSir2 includes two domains: a larger domain that
consists of 8 α helix and 6 parallel β sheets and forms a variant
of the Rossmann fold, which present in many NAD(H)/NADP(H) binding
enzymes as prosthetic group, and a small domain that consists of 4
α helix and 3 anti-parallel β sheets and a zinc atom tetrahedrally
coordinated by four cysteine residues (Figs. 3 and 4A and C). The two domains are connected
by four loops (51–61, 98–105, 145–151 and 192–201) (Fig. 4A and C), which are highly
conserved in the Sir2 protein family (Fig. 3). These four loops and other three
conserved loops (43, 126–130 and 222–227) of the large domain form
a large groove, which was supposed to be the NAD binding site
(Fig. 4A and C). By comparing the
structure of SsSir2 and SIRT2, we found that the two structures
were exactly similar, particularly in the NAD binding site,
zinc-binding site and the Rossmann fold, although the amino acids
are not so conserved in these regions. The distinct difference
between SsSir2 and SIRT2 was that the α helix that connects the
fifth (β8) and sixth (β9) β sheets of the Rossmann fold was absent
(Fig. 4A and B).
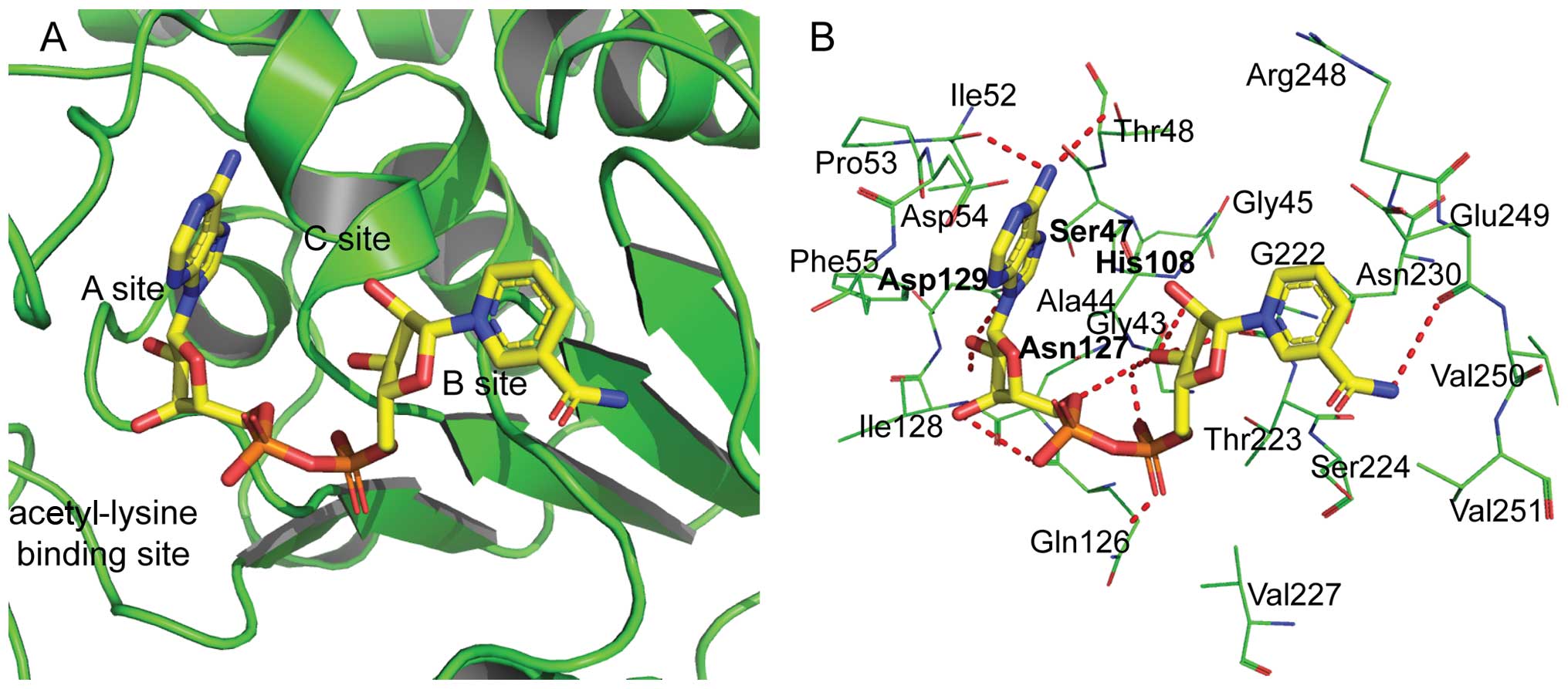
Molecular interaction analysis between
SsSir2 and NAD
To further understand the interaction between SsSir2
and its prosthetic group, NAD was docked into the NAD binding site
of SsSir2 by molecular docking. The optimal binding conformation of
the SsSir2-NAD complex was selected and is presented in Fig. 6. The NAD molecule is bound in a
large groove formed by the conserved loops between the large and
small domain of SsSir2. The NAD binding pocket can be divided into
three regions (Fig. 6A): i) site
A, where the adenine-ribose moiety of NAD is bound; ii) site B,
where the nicotinamide-ribose moiety is bound; and iii) site C,
which is deep inside the pocket and is not near the position of
observed NAD. The detailed interacting residues and hydrogen bonds
are both labeled in Fig. 6B.
Expression of SsSir2 in two stages of S.
schenckii
The mRNA expression of SsSir2 in the different
stages was analyzed by real-time RT-PCR normalized against 18srDNA
levels. The expression was determined as fold increased
2−ΔΔCt levels relative to the stage with the lowest
expression (mycelial) set to 1. The SsSir2 gene was expressed in
two stages of S. schenckii, with higher mRNA levels observed
in the yeast form (1.72-fold). There were significant differences
observed between the mycelial and yeast form (Table II).
| Table IIRelative abundance of differential
gene expression as determined by real-time RT/PCR. |
Table II
Relative abundance of differential
gene expression as determined by real-time RT/PCR.
| cDNA name | Phase | Target CT | 18srDNA CT | ΔCT | ΔΔCT | 2
−ΔΔCT |
|---|
| SsSIR2 | Mycelial | 27.38±0.52 | 20.66±0.25 | 6.7±0.37 | 0±0.37 | 1 |
| Yeast | 29.59±0.16 | 22.09±0.60 | 7.5±0.61 | 0.79±0.61 | 1.72 |
Accession number
The full length of the cDNA sequence and genomic DNA
sequence of the SsSir2 gene were submitted to the GenBank database
under the accession number (JX312330) and (KC304788),
respectively.
Discussion
The Sir2-like protein is the founding member of a
large family of NAD+-dependent deacetylases, the
so-called sirtuins, conserved from bacteria to mammals (8). Sirtuins were grouped into five main
branches designated as classes I–V. Specific amino acid sequence
motifs characterize the different classes of sirtuins. In class III
sirtuins, the GAGISXXXGIPXXR motif is usually GAGISAESGIPTFR,
whereas in class II, it is GAGISTESGIPDYR. Sirtuins of classes I,
IV and V also have GIPD within this motif. In sequences of classes
II, III and U, the PXXXH motif is PNXXH, while in the eukaryotic
class I and IV sequences a T or S usually follows the P. The HG
motif is strictly conserved in all known sirtuins. In class Ia the
HG motif reads CHG, in class Ib it reads AHG and in class Ic (and
in most non-class I sirtuins) it reads LHG. The three residues
located five residues C-terminal to the GTS motif are rather useful
in differentiating between the types of sirtuins. The three
residues usually observed at this position for each class and
subclass are: Ia (PVA or PVS), Ib (PFA), Ic (GVK), V (PAA), II
(SGY), III (PAA), IVa (PXX) and IVb (KKY) (9). In the present study, SsSir2 was
labeled as a class I sirtuin based on the presence of GIPD in the
GAGISXXXGIPXXR motif and PT in the PXXXH motif (Fig. 1). We further identified SsSir2 as
a class Ib sirtuins for its AHG sequence and PFA residues located
five residues C-terminal to the GTS motif, which was also confirmed
by phylogenetic association analysis.
In the absence of an experimentally determined
crystal structure, it is generally recognized that the homology
modeling of proteins is currently the most accurate method for 3D
structure prediction (10).
Homology modeling is based on the assumption that the proteins with
similar sequences might have analogous 3D structures. Thus, the
selection of a suitable template is the first step. In the present
study, SIRT2 was selected as the template for constructing the 3D
model of SsSir2 as it has a 47.7% amino acid sequence identity with
SsSir2. To assess whether the 3D model of SsSir2 is of reasonable
quality, ERRAT, Verify3D and the Ramachandran plot were used and
showed positive results (11).
The results of quality assessment suggested that the model of the
SsSir2 structure was of reasonable quality compared to the crystal
structure of the SIRT2 and sufficient for use in further
experiments.
The NAD molecule is bound in an extensive pocket
between the large and the small domains of the SsSir2 protein
(12). Several important
residues, such as Asn127 and Asp129 identified by the current
docking simulation, have been shown to play an important role in
the histone deacetylase activity of Sir2 family proteins. His108
and Asn127 were shown to guide the substrate to the correct
position with catalytic activity in the catalytic reaction. The
other two conserved amino acids of Ser47 and Asp129 at the bottom
of the protein stabilize the adenine ribose and adenine of
NAD+ which combined with SsSir2, but did not interact
directly with NAD+. These characters of crystal
structure are similar to those reported in the study by Min et
al (12). In our study, as
shown in Fig. 6B, several
hydrogen bonds, which are a key interaction force of protein ligand
binding, were also found between NAD and some important conserved
residues. All of the above properties indicate that the predicted
binding mode was reliable and that the complex was stable.
Sir2 has been proven to be involved in morphogenesis
and phenotypic switching in dimorphic fungi. Rine and Herskowitz
established that sporulation in S. cerevisiae requires the
functional gene products of Sir2 (13). A screen for S. cerevisiae
temperature-sensitive silencing mutants identified a strain with a
point mutation in the Sir2 gene (Ser276 was changed to Cys).
Haploid strains carrying the mutation were severely defective at
mating at 37°C but normal at 25°C (14). In C. albicans, the deletion
of the Sir2 gene produces a dramatic phenotype: variant colony
morphologies arise at frequencies as high as 1 in 10. The
morphologies resemble those described as part of a phenotypic
switching system proposed to contribute to pathogenesis (4). In S. schenckii, the
germination of conidia is a key pathogenicity determinant, as
conidia are infectious propagules. The question remains of whether
SsSir2 has the same function in the phenotypic switching of S.
schenckii similar to the Sir2 ortholog in other dimorphic
fungi. In our previous study, the SsSir2 protein expression level
in yeast cells was found to be 5.47-fold higher than that in the
mycelial phase of S. schenckii (6). In this study, the mRNA expression of
SsSte20 in yeast cells of both ATCC10268 and a clinical S.
schenckii isolate from a patient with fixed sporotrichosis
(data not shown) were higher than in the mycelial ones. These
results suggest that SsSir2 may also play a role in the morphologic
switching of S. schenckii, presumably by transcriptional
silencing of the silent mating type loci.
A variety of environmental signals may affect the
mycelial-yeast transition (15),
and a large body of evidence suggests the existence of several
signaling pathways (16–19), any one of which may be involved in
the mycelial-yeast transition. The ability of S. schenckii
to switch between different phenotypes is an alternative way to
obtain the variability required to survive an uncertain
environment. The mechanisms through which the SsSir2 gene controls
one or more of these pathways remain to be elucidated. It is most
likely that some types of external stress may suppress the Sir2
gene and thereby activate one or more of these signaling pathways
which regulate the mycelial-yeast transition when it may prove
beneficial. This idea has some support from the discovery that gene
silencing in S. cerevisiae (which requires the Sir2 gene) is
inactivated as cells become older (20). Perhaps a different type of stress
would produce a similar inactivation of silencing in S.
schenckii. The detailed functions of SsSir2 require further
investigation. We are currently carrying out further experiments on
generating SsSir2 mutants to investigate its detailed functions in
this important fungal pathogen.
Acknowledgements
This study was partly supported by a grant from the
National Natural Science Foundation of China (no. 81000699).
References
|
1
|
Klein BS and Tebbets B: Dimorphism and
virulence in fungi. Curr Opin Microbiol. 10:314–319. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Soll DR: High-frequency switching in
Candida albicans. Clin Microbiol Rev Apr. 5:183–203.
1992.
|
|
3
|
Pérez-Martín J, Uría JA and Johnson AD:
Phenotypic switching in Candida albicans is controlled by a
SIR2 gene. EMBO J. 18:2580–2592. 1999.
|
|
4
|
Laurenson P and Rine J: Silencers,
silencing and heritable transcriptional changes. Microbiol Rev.
56:543–560. 1992.PubMed/NCBI
|
|
5
|
Ramsey H, Morrow B and Soll DR: An
increase in switching frequency correlates with an increase in
recombination of the ribosomal chromosomes of Candida
albicans strain 3153A. Microbiology. 7:1525–1531. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang ZY, Hou BB, Xin Y and Liu XM:
Protein profiling of the dimorphic pathogenic fungus, Sporothrix
schenckii. Mycopathologia. 173:1–11. 2012. View Article : Google Scholar
|
|
7
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the −DDCT. Method. 25:402–408. 2001.
|
|
8
|
Gasser SM and Cockell MM: The molecular
biology of the SIR proteins. Gene. 279:1–16. 2001. View Article : Google Scholar
|
|
9
|
Frye RA: Phylogenetic classification of
prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res
Commun. 273:793–798. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Y, Dong X and Liu J: Molecular
cloning, expression and molecular modeling of chemosensory protein
from Spodoptera litura and its binding properties with
Rhodojaponin III. PLoS One. 7:1–10. 2012.PubMed/NCBI
|
|
11
|
Kumara R, Kumarb S, Sangwanc S, Yadava IS
and Yadavd R: Protein modeling and active site binding mode
interactions of myrosinase-sinigrin in Brassica juncea- an
in silico approach. J Mol Graph Model. 29:740–746. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Min J, Landry J, Sternglanz R and Xu RM:
Crystal structure of a SIR2 homolog-NAD complex. Cell. 105:269–279.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rine J and Herskowitz I: Four genes
responsible for a position effect on expression from HML and HMR in
Saccharomyces cerevisiae. Genetics. 116:9–22.
1987.PubMed/NCBI
|
|
14
|
Wang CL, Landry J and Sternglanz R: A
yeast Sir2 mutant temperature sensitive for silencing. Genetics.
180:1955–1962. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Floréz AM, Oviedo A, Cardona A and Herrera
M: Molecular cloning and characterization of two hsp 70 homologous
genes from the dimorphic fungus Paracoccidioides
brasiliensis. Biomedica. 23:424–436. 2003.PubMed/NCBI
|
|
16
|
Chen J, Zhou S, Wang Q, Chen X, Pan T and
Liu H: Crk1, a novel Cdc2-related protein kinase, is required for
hyphal development and virulence in Candida albicans. Mol
Cell Biol. 20:8696–8708. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Andaluz E, Calderone R, Reyes G and
Larriba G: Phenotypic analysis and virulence of Candida
albicans LIG4 mutants. Infect Immun. 69:137–147. 2001.
View Article : Google Scholar
|
|
18
|
Sonneborn A, Bockmuhl DP, Gerads M,
Kurpanek K, Sanglard D and Ernst JF: Protein kinase A encoded by
TPK2 regulates dimorphism of Candida albicans. Mol
Microbiol. 35:386–396. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nemecek JC, Wuthrich M and Klein BS:
Global control of dimorphism and virulence in fungi. Science.
312:583–588. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sinclair DA, Mills K and Guarente L:
Molecular mechanisms of yeast aging. Trends Biochem Sci.
23:131–134. 1998. View Article : Google Scholar
|