Introduction
Plasma concentrations of high-density lipoprotein
(HDL) cholesterol have been reported to have a strong and
independent inverse association with risk of coronary artery
disease (CAD) events (1). It has
been shown that 30–55% of high-density lipoprotein cholesterol
(HDL-C) concentration variability is determined by genetic factors
(2). Apolipoprotein AI (apoA-I)
and lecithin:cholesterol acyltransferase (LCAT) gene mutations
result in extremely low HDL-C levels (3). LCAT, a plasma enzyme, esterifies
free cholesterol primarily at the surface of the HDL particle, has
a central role in HDL formation and maturation (4), and in the intravascular stage of
reverse cholesterol transport (RCT), the major mechanism by which
HDL modulates the development and progression of atherosclerosis.
In addition to its cholesterol-esterifying activity, LCAT seems to
have a scavenger effect towards low-density lipoprotein (LDL)
oxidation products (5). The human
LCAT gene encompasses 4.2 kb and is localized in the q21–22 region
of chromosome 16 (6). Based on
strict biochemical criteria, homozygotes or compound heterozygotes
for these mutations are classified into the familial LCAT
deficiency (FLD) and fish-eye disease (FED) syndromes (7). In FLD, LCAT plasma is absent or
completely lacks catalytic activity, while in FED, the LCAT mutant
lacks activity on HDL (α-LCAT activity) but esterifies cholesterol
bound to apolipoprotein B-containing lipoproteins (β-LCAT
activity). All reported FLD and FED cases have massive corneal
opacity and markedly reduced plasma HDL levels (3), although the prevalence of CAD may be
higher in FED than in FLD patients (6). Heterozygous carriers of LCAT
mutations may have either low (8–9) or
normal (8) plasma HDL-C levels
without premature CAD. At present, ~60 isolated cases and 70 small
families with partial or complete LCAT deficiency with 86 different
LCAT gene mutations have been described (10). Atherosclerosis susceptibility has
been extensively investigated in FED and FLD patients (10). Studies assessing the association
between the inherited LCAT functional defect and CAD have been
limited to measurements of only some variables, such as the HDL
subclasses (11–12) size (9) and function (13–14), apoA-I and apoA-II plasma
concentrations (9), inflammation
(15), and carotid
intima-media-thickness (cIMT) (15–16). This study aimed to provide more
information on the LCAT-deficient state and its association with
CAD by describing LDL susceptibility to oxidation in addition to
the above-mentioned variables, in a LCAT-deficient patient with
severe premature CAD and his nuclear family.
Materials and methods
Subjects
The proband was a 34 year-old male, with family and
personal history of type 2 diabetes mellitus (DM2), obesity,
hypertension and premature CAD. His cardiovascular history included
unstable angina, coronary bypass surgery, intracoronary placement
of five stents, stent restenosis and myocardial infarction. He had
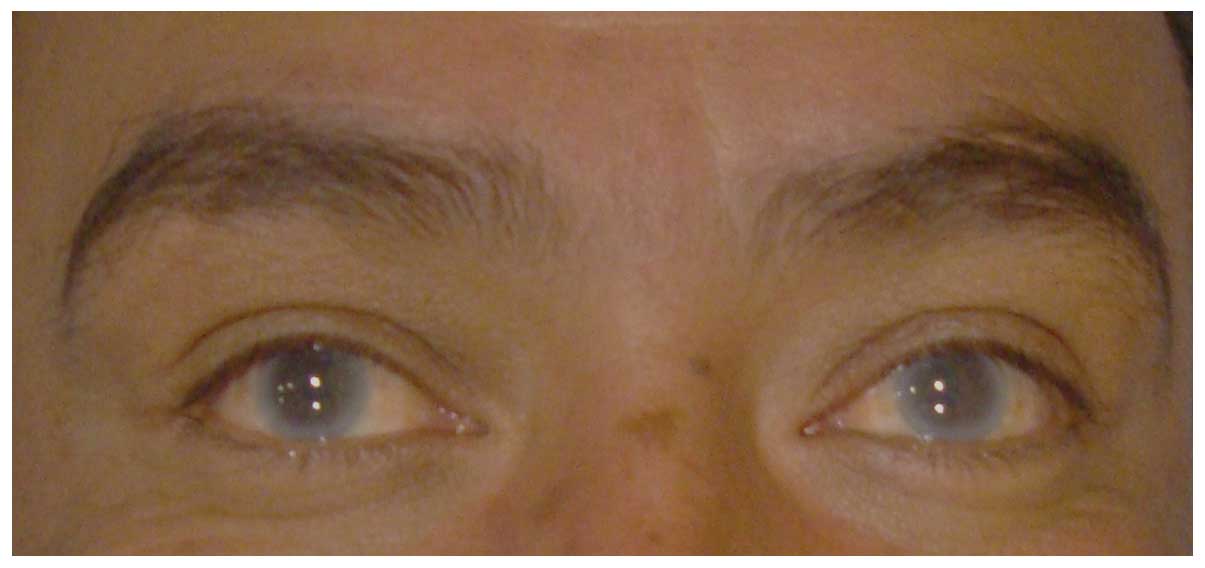
bilateral corneal opacity (Fig.
1) without retinopathy, with no signs of anemia and normal
renal function. First-degree relatives of the proband were invited
to participate in the study and all subjects provided signed
informed consent. The study complies with the Declaration of
Helsinki and was approved by the Ethics Committee of the National
Institute of Cardiology ‘Ignacio Chávez’ (INCICH).
Biochemical measurements
Blood samples were collected after a 10- to 12-h
fast. Plasma glucose, total cholesterol, triglycerides (TG), and
HDL-C were measured using standard enzymatic procedures in a
Hitachi 902 analyzer (Hitachi Ltd., Tokyo, Japan) and LDL
cholesterol levels were estimated. In our laboratory, accuracy and
precision are periodically evaluated by the Centers for Disease
Control and Prevention (Atlanta, GA, USA). Total high-sensitivity
C-reactive protein (hs-CRP), apolipoprotein B (apoB), and apoA-I
levels were determined by immunonephelometry on a BN ProSpec
nephelometer (Dade Behring, Marburg, Germany), with an interassay
coefficient of variation of <6%. Plasma insulin concentrations
were determined by a radioimmunometric assay (Coat-A-Count;
Diagnostic Products, Los Angeles, CA, USA).
Total HDL was isolated from the plasma by sequential
ultracentrifugation in a density of 1.21 g/ml at 4°C in a Beckman
TL-100 ultracentrifuge. The resulting total HDL was dialyzed
against phosphate buffer (pH 7.4), loaded into native 4–25%
polyacrylamide electrophoresis gels and analyzed by automated
densitometry, as previously described (17). The coefficient of variation for
each subclass was <10%.
LCAT activity and the cholesterol esterification
rate (CER) were measured according to the methods of Chen and
Albers (18). LDL susceptibility
to in vitro oxidation was determined by a modification of
the method of Esterbauer (19),
constructing an oxidation kinetics curve to calculate the lag phase
(LP). Intra- and intervariation coefficients for these measurements
were 2.94 and 5.77%, respectively. Serum PON1 activity was
determined photometrically (Beckman DU650, Fullerton, CA, USA)
using paraoxon (Sigma-Aldrich D9286; St. Louis, MO, USA) as a
substrate (20). The capacity of
total isolated HDL to promote cell cholesterol efflux was tested
for each individual. cAMP-stimulated J774 murine macrophages and
Fu5AH hepatoma cells were used to assess ABCA1 and SR-BI-mediated
cholesterol efflux, respectively, following the procedure described
by de la Llera-Moya et al (21–22), with slight modifications. Briefly,
total isolated HDL fractions as estimated by phospholipids (100 μg
of phospholipids/ml) were incubated with 1,2–3H cholesterol labeled
Fu5AH or J774 cells at 37°C for 4 h. The percentage of cholesterol
efflux was calculated comparing radioactivity measurements in the
medium and cells. All the determinations were performed in
triplicate. As an internal control, isolated total HDL was included
in each plate. Intra- and interassay coefficient variations for
this method were 4.9 and 8.1%, respectively.
Radiological studies
Carotid intima-media-thickness measurements were
performed using a MicroMaxx ultrasound device (SonoSite, Inc.,
Bothell, WA, USA), equipped with a 7.0–14 MHz linear-array wideband
transducer to obtain B-mode ultrasound images (23). The common carotid artery, bulb and
internal carotid artery were bilaterally scanned over a length of
10 mm. The mean combined outcome of the six segments was reported.
Computed tomography of the chest and abdomen were performed using a
64-channel multi-detector helical computed tomography system
(Somatom Sensation, Siemens, Munich, Germany) and interpreted by
experienced radiologists. Scans were read to assess and quantify
coronary artery calcification (CAC) score using the Agatston method
(24). Subclinical
atherosclerosis (SA) was defined as the presence of coronary
calcium (CAC score >0).
LCAT gene sequencing
DNA was isolated from peripheral venous blood by a
modified salting out method (25). The amplification mix (50 μl)
included 150–300 ng of genomic DNA, 1 unit of Taq DNA polymerase
(Promega, Madison, WI, USA) in 1X PCR buffer, 5 mM MgCl2
and 0.4 mM of each dNTP. Other conditions for the LCAT gene
amplification are detailed in Table
I. PCR products were purified using the QIAquick PCR
purification kit (Qiagen, Inc., Chatsworth, CA, USA), and
purification products were directly sequenced on an ABI Prism 3100
Sequencer according to the manufacturer’s instructions.
 | Table IConditions for the LCAT gene
amplification. |
Table I
Conditions for the LCAT gene
amplification.
| Exon | Primer
sequence | Primer
concentration μM | DMSO (%) | Initial
denaturation step | No. of cycles | Cycle
conditions | Extension
conditions |
|---|
| 1 |
5′-CGGCAATCTCTGGCCACAACC-3′ (F)
5′-GGCTTATGCAGGGCAGAAGG-3′ (R) | 0.2 | 10 | 94°C for 5 min | 30 | 94°C for 30 sec,
63°C for 30 sec, 72°C for 1 min 16 sec | 72°C, 10 min |
| 2+3 |
5′-GGGTCACGGGGGGAATCCAG-3′ (F)
5′-GGCTTGGGCCATGCCTGCTG-3′ (R) | 0.3 | 10 | 94°C for 5 min | 35 | 94°C for 20 sec,
64°C for 30 sec, 72°C for 40 sec | 72°C, 10 min |
| 4+5 |
5′-GGCTCCAGGCCTGGGTGCTG-3′ (F)
5′-GATAGCACCCCTAGAGGCCACTG-3′ (R) | 0.4 | 20 | 94°C for 5 min | 35 | 94°C for 30 sec,
64°C for 45 sec, 72°C for 30 sec | 72°C, 10 min |
| 6 |
5′-TGAGCCTACACTCAGCAGGTTGTG-3′
(F)
5′-CCCATCTTGCCTCACTGCACACA-3′ (R) | 0.3 | 7.5 | 94°C for 5 min | 35 | 94°C for 30 sec,
61°C for 45 sec, 72°C for 45 sec | 72°C, 10 min |
Results
Mutational analysis
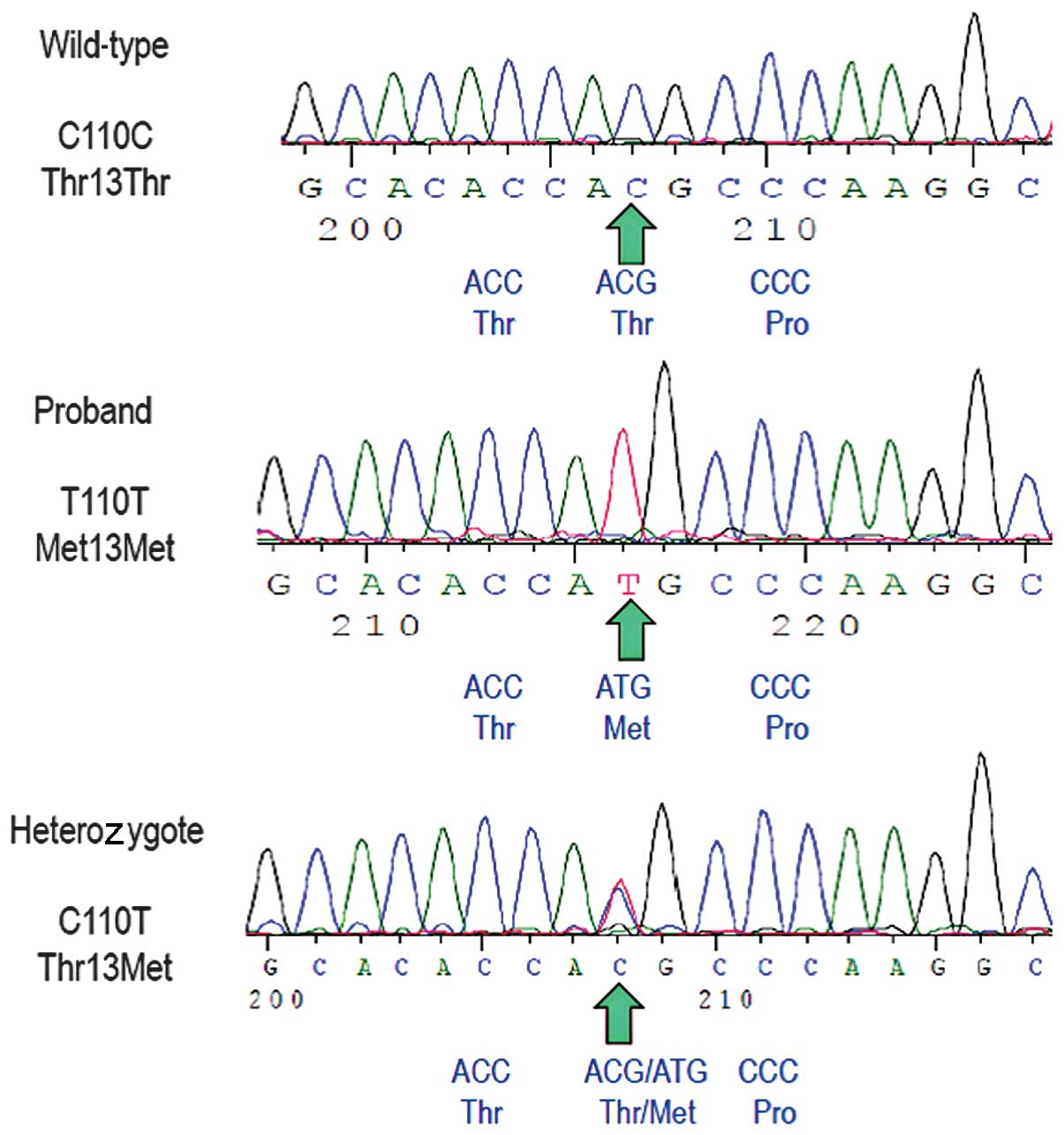
The sequence of all six exons of the LCAT gene
revealed the proband was homozygous for a c.110C>T transition in
exon 1, a previously described mutation (Fig. 2) (26). The predicted translation of the
mutant allele resulted in a missense non-conservative amino acid
substitution of threonine for methionine at position 37 of the LCAT
protein (Thr37Met). No other sequence variations were identified.
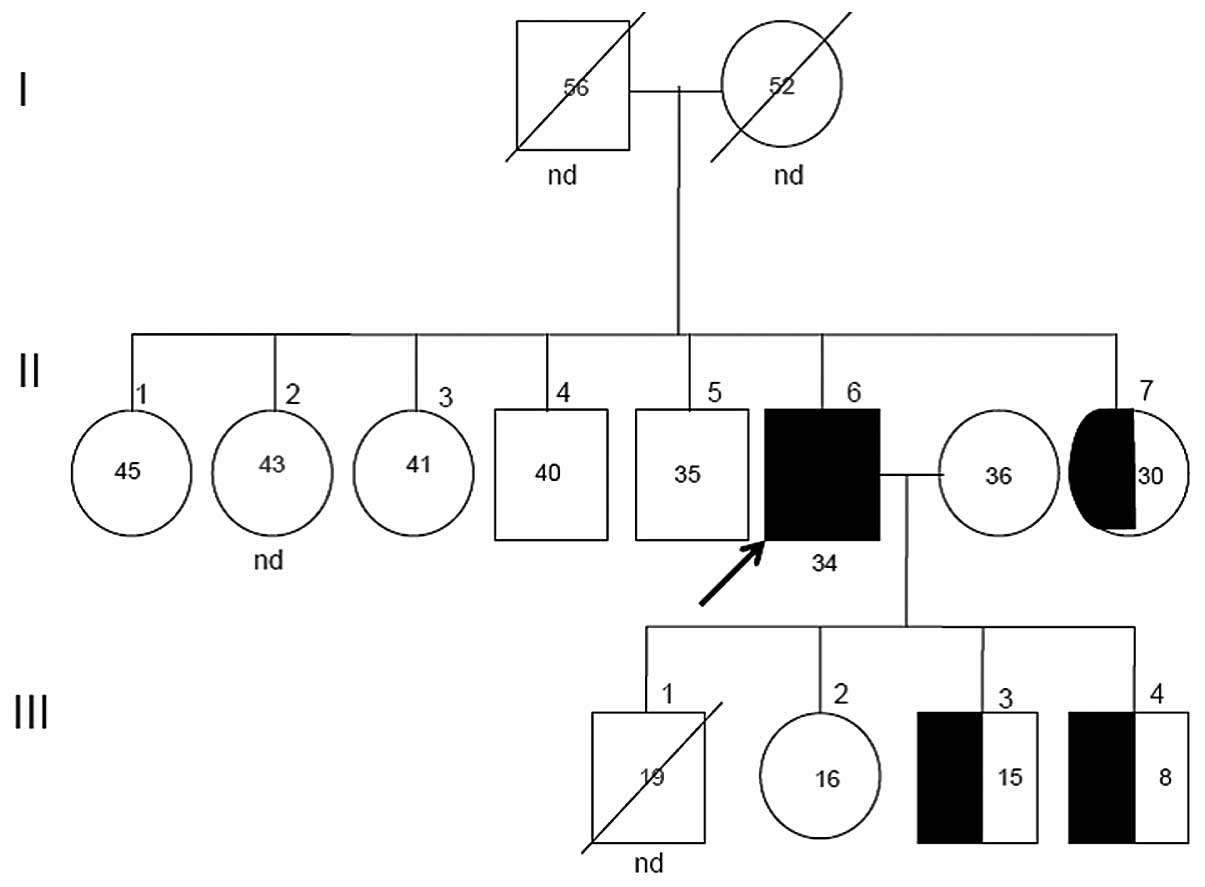
The segregation of the mutant allele in this family is shown in
Fig. 3. Two sisters (II-1 and
II-3), two brothers (II-4 and II-5) and the daughter (III-2) of the
proband were found to have wild-type genotypes, while one of the
probands sisters (II-7) and his two sons (III-3- and III-4) were
found to be heterozygous for the Thr37Met mutation.
Biochemical analysis
HDL-C levels were markedly low in the proband (2
mg/dl), very low in all the heterozygous subjects (16–18 mg/dl) and
low in non-carriers of the mutation as compared to a group of
previously reported healthy male subjects (Table II) (27). It should be noted that all but one
of the the proband’s siblings had HDL-C levels <40 mg/dl,
commonly associated with high TG levels (>150mg/dl). Compared
with insulin and hs-CRP levels observed in healthy control
subjects, several family members had increased insulin and hs-CRP
(>2.0 mg/dl) levels. All the individuals with the exception of
the proband’s daughter (III-2) were overweight or obese, which may
explain the increased levels of TG, insulin and hs-CRP. The proband
and one of his sisters (II-3) had abnormal fasting glucose, and one
of his brothers (II-4) had DM2. As expected, LCAT activity was
lowest in the proband, (2.1 nmol/ml/h), followed by the
heterozygous subjects and individuals with wild-type genotypes. The
proband also showed lower CER values. LDL susceptibility to
oxidation analysis in vitro revealed that the LP was 70%
shorter in the proband as compared to the mean LP estimated in
healthy controls, indicating increased LDL oxidability, whereas LP
duration was not different in the heterozygous and family
non-mutation carriers, but was shorter in family members as
compared to the healthy controls.
| Table IIClinical and laboratory data of
patient, family members and healthy subjects. |
Table II
Clinical and laboratory data of
patient, family members and healthy subjects.
| ID subject | Proband | Healthy
subjectsa n=20 |
|---|
|
|---|
| II-1 | II-3 | II-4 | II-5 | II-6 | II-7 | III-2 | III-3 | III-4 |
|---|
| Genotype | CC | CC | CC | CC | TT | CT | CC | CT | CT | - |
| Age (years) | 45 | 41 | 40 | 35 | 34 | 30 | 16 | 15 | 8 | 53±8.5 |
| BMI
(kg/m2) | 39 | 33 | 25 | 31 | 28 | 33 | 23 | 25 | 27 | 26±2.5 |
| Corneal
opacity | − | − | − | − | + | − | − | − | − | − |
| Presence of
diabetes | − | − | + | − | + | − | − | − | − | − |
| TC (mg/dl) | 148 | 159 | 210 | 157 | 141 | 169 | 157 | 174 | 153 | 183±33 |
| HDL-C (mg/dl) | 32 | 30 | 51 | 34 | 2 | 16 | 42 | 18 | 17 | 59±18 |
| LDL-C (mg/dl) | 100 | 92 | 133 | 93 | 43 | 127 | 103 | 108 | 95 | 108±30 |
| TG (mg/dl) | 98 | 233 | 156 | 186 | 597 | 161 | 73 | 297 | 255 | 100±27 |
| Apo A-I
(mg/dl) | 116 | 115 | 163 | 140 | 45 | 76 | 126 | 91 | 95 | 167±38 |
| Apo B (mg/dl) | 94 | 92 | 146 | 92 | 172 | 121 | 100 | 109 | 103 | 82±17 |
| Glucose
(mg/dl) | 92 | 106 | 181 | 91 | 104 | 85 | 79 | 90 | 84 | 87±6 |
| Insulin
(μU/ml) | 12 | 11 | nd | 10 | 4.9 | 15 | 1.2 | 10 | 7.3 | 9±6 |
| hs-CRP (mg/l) | 2.3 | 5.7 | 0.65 | 3.4 | 8.4 | 4.0 | 0.5 | 1.4 | 4.8 | 1.2±0.8 |
| LCAT activity
(nmol/ml/h) | 15 | 12 | nd | 12 | 2.1 | 8 | 12 | 9 | 10 | - |
| CER
(nmol/ml/h) | 26 | 26 | nd | 26 | 9 | 17 | 18 | 24 | 21 | - |
| LP (min) | 36 | 40 | 33 | 43 | 19 | 47 | 32 | 37 | 46 | 62±5 |
HDL subclasses and cholesterol
efflux
Table III shows
the distribution of HDL subclasses for all the participants. The
proband showed an absence of large HDL, while heterozygous mutation
carriers showed reduced proportions of large HDL levels
(HDL2b and HDL2a) and increased small HDL
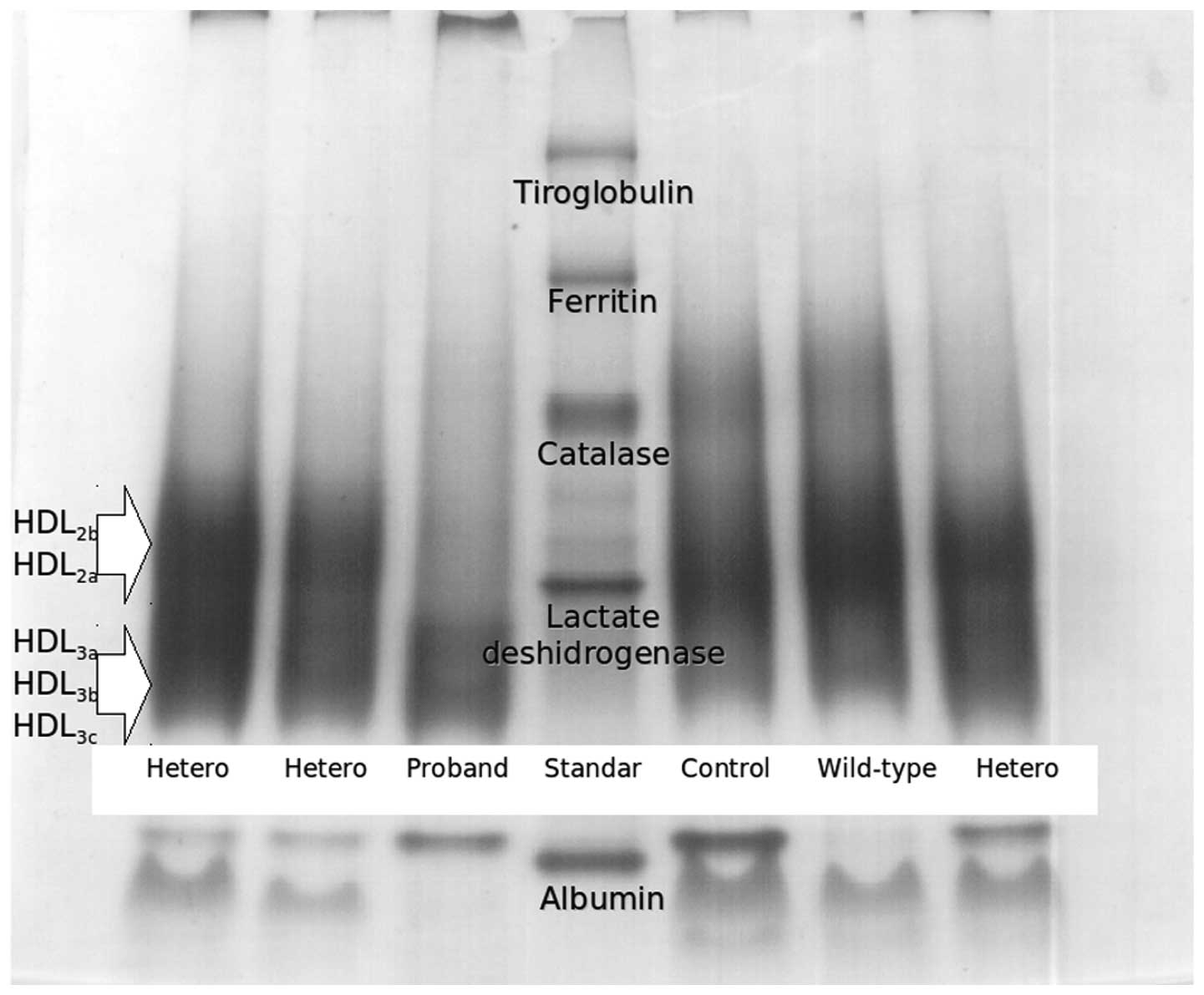
(HDL3c) as compared to healthy subjects. Fig. 4 shows an electrophoretogram of
HDL, where the proband showed an absence of large subclasses and
higher proportion of small HDL subclasses. Compared with healthy
control subjects, the HDL size was lower in the heterozygous
subjects and proband had the smallest HDL size. PON1 activity was
low in the proband and in three siblings not carrying the mutation
(II-1, II-4, II-5), while the heterozygous family members showed
normal values. cAMP-stimulated J774 murine macrophages and Fu5AH
hepatoma cells were used to assess the ABCA1 and SR-BI-mediated
cholesterol efflux capacity from total isolated HDL of all the
participants. While ABCA-1 mediated cholesterol efflux to total
isolated HDL was similar among all subjects independently of the
presence of LCAT mutation, SRB1-mediated cholesterol efflux from
the proband was less than half of that observed in healthy controls
and heterozygous participants (Table III).
| Table IIIHDL subclass distribution,
paraoxonase activity and cholesterol efflux of patient, family
members and healthy subjects. |
Table III
HDL subclass distribution,
paraoxonase activity and cholesterol efflux of patient, family
members and healthy subjects.
| ID subject | Proband | Healthy
subjectsa n=20 |
|---|
|
|---|
| II-1 | II-3 | II-4 | II-5 | II-6 | II-7 | III-2 | III-3 | III-4 |
|---|
| Genotype | CC | CC | CC | CC | TT | CT | CC | CT | CT | - |
| HDL subclass
distribution |
| HDL2a,
% | 17 | 22 | 18 | 17 | 0 | 14 | 20 | 13 | 13 | 21±2.9 |
| HDL2b,
% | 9 | 13 | 12 | 10 | 0 | 10 | 14 | 10 | 7 | 16±4.6 |
| HDL2,
% | 26 | 35 | 31 | 26 | 0 | 24 | 34 | 24 | 20 | 37±7.2 |
| HDL3a,
% | 29 | 31 | 25 | 30 | 15 | 28 | 28 | 27 | 27 | 24±2.2 |
| HDL3b,
% | 26 | 21 | 23 | 25 | 30 | 25 | 23 | 25 | 26 | 24±2.8 |
| HDL3c,
% | 19 | 13 | 22 | 18 | 55 | 23 | 15 | 25 | 27 | 15±4.9 |
| HDL3,
% | 74 | 65 | 69 | 74 | 100 | 76 | 66 | 76 | 80 | 63±7.2 |
| HDL size (nm) | 8.6 | 8.8 | 8.7 | 8.6 | 7.8 | 8.5 | 8.8 | 8.5 | 8.4 | 9±0.2 |
| PON1 activity
(nmol/ml/min) | 77 | 321 | 33 | 98 | 75 | 328 | 341 | 296 | 328 | 338±112b |
| Cholesterol efflux
to total isolated HDL |
| ABCA1-mediated
(%) | 3.2 | 3.8 | 3.5 | 2.6 | 2.7 | 3.1 | 2.6 | 3.9 | 3.5 | - |
| SRB1-mediated
(%) | 4 | 6 | 7 | 8 | 2.5 | 7 | 7 | 5 | 7 | 5.6±1.4 |
Intima media thickness results
As shown in Table
IV, cIMT was above the 50th percentile for age and gender in
the proband, in one of his heterozygous sons (III-4) and in one of
his wild-type brothers (II-4), while carotid plaque was present
only in the proband and in the wild-type brother. Calcified
coronary artery (CAC>0), as a measure of subclinical
atherosclerosis, was present in brother II-4 and in one of his sons
(III-3) (Table IV).
| Table IVCoronary artery calcium, carotid
intima-media-thickness and plaque of patient and his family
members. |
Table IV
Coronary artery calcium, carotid
intima-media-thickness and plaque of patient and his family
members.
| Proband |
|---|
|
|
|---|
| ID subject | II-1 | II-3 | II-4 | II-5 | II-6 | II-7 | III-2 | III-3 | III-4 |
|---|
| Genotype | CC | CC | CC | CC | TT | CT | CC | CT | CT |
| Coronary artery
calcium (Agatston) | 0 | 0 | 141.5 | 0 | − | − | − | 2.9 | 0 |
| Carotid intimal
media thickness (mm) | 0.65 | 0.58 | 0.68a | 0.63 | 0.78a | − | − | 0.43 | 0.49a |
| Carotid plaque | − | − | + | − | + | − | − | − | − |
Discussion
The present study assessed increased LDL oxidability
and structural and functional abnormalities of HDL particles from
the second youngest LCAT-deficient patient with markedly severe
premature CAD studied thus far. The case was a 34-year-old man with
an adverse cardiovascular risk profile, multivessel coronary
disease, corneal opacity, very low HDL-C levels, normal renal
function, and no signs of anemia. Although his parents were
apparently not consanguineous, he was found to be homozygous for a
previously described mutation in the LCAT gene (c.110T in exon 1)
(26). Since CER and LCAT
activity were detectable in the proband, he could not be classified
as either FLD or FED according to current biochemical criteria
(7,28). LCAT is a critical enzyme in HDL
metabolism. Individuals with loss-of-function mutations in the two
alleles show an inability to form mature HDL particles with a
cholesteryl ester core and rapid catabolism of circulating apoA-I
and apoA-II (29). As a result,
these patients have very low HDL-C levels and several
physicochemical and functional HDL abnormalities.
In a large series of LCAT-deficient subjects,
Calabresi et al (12)
demonstrated that HDL particle size distribution in carriers of two
mutant LCAT alleles is characterized by the lack of particles in
the HDL2 size range and the presence of a single
HDL3 subpopulation of particles, with an average size
smaller than that of control HDL3. This is in agreement
with our findings of reduced large HDL and increased small HDL
subclasses, and lower HDL size in the homozygous and the
heterozygous subjects when compared with healthy control subjects.
PON1 activity is associated with HDL in human serum (30), and inhibits the accumulation of
oxidized lipids (31), thus
preventing oxidative modifications of LDL and the conversion of
this lipoprotein into particles with atherogenic properties
(32). While preserved PON1
activity has been found in HDL-deficient states (14,33,34), in our patient, PON1 activity was
reduced 78% compared with the healthy control subjects (Table III). The interindividual
variability in serum PON1 activity seems to be regulated by genetic
SNPs, environmental (tobacco, alcohol, fatty acids) (35) and biochemical (HDL-C and total
cholesterol levels) (36) factors
that may increase or reduce PON1 activity, which may explain the
low activity found in the proband and some of the family
members.
While ABCA1-mediated cholesterol efflux to total
isolated HDL was preserved in mutation carriers (Table III), SRB1-mediated efflux to
total isolated HDL was reduced ~50% in the proband relative to the
efflux of the healthy control group. These results are in line with
a previous study reporting that serum from LCAT gene mutation
carriers is similar to control serum in the ability to promote
cholesterol efflux via ABCA1, but less efficient than serum from
control subjects in promoting SRB1-mediated cell cholesterol efflux
(37).
LDL are heterogeneous in size, density, lipid
composition, and possibly atherogenicity. Compared to LDL from
control subjects, LDL particles from carriers of two mutant LCAT
alleles are smaller (12), have
an abnormal composition (poor in cholesteryl esters and rich in TG
and phospholipids) (38), and are
more easily oxidized. Oxidation results in a cytotoxic LDL with
pro-inflammatory and atherogenic properties. In addition to
abnormalities in HDL-related characteristics, LDL particles from
the proband showed a 70% reduction in the LP compared to the
average LP of normal LDL from healthy subjects, indicating that
despite his low LDL-C levels (43 mg/dl), these lipoproteins were
highly susceptible to oxidation. Reduced PON1 and LCAT activities
are likely to cause lower scavenger activity towards LDL oxidation
products (13,31), which together with a potential
abnormal LDL composition, may explain the high LDL oxidability
found in this patient.
Plasma concentrations of HDL-C have been shown to
have a strong and independent inverse association with risk of CAD
(1). Nevertheless, some monogenic
disorders with extremely low HDL-C levels, such as the apoA-IMilano
mutation, Tangier disease and LCAT deficiency, are not generally
associated with accelerated atherosclerosis. More than 86 defects
in LCAT gene have been identified (10), and for several decades conflicting
findings on the association of LCAT deficiency with atherosclerosis
have been reported in humans and experimental animals (7,39).
Thus, some (15,40) but not all studies (16), have found that reduced LCAT
activity is associated with increased cIMT and with the presence of
CAD (10). A recent study
reported that compared with 80 matched healthy controls, 12
individuals with two loss-of-function LCAT mutations and 28
heterozygous carriers with one mutation did not have increased cIMT
as compared to 80 matched healthy controls, and instead there was a
strong suggestion for a gene dose-dependent reduction in cIMT
(16). These findings suggest
that LCAT deficiency is not associated with increased
cardiovascular risk. In the present study, the proband had severe
and premature CAD, HDL abnormalities including very low HDL-C and
apoA-I levels, absence of large HDL, increased small HDL, smaller
HDL size, reduced PON1 activity, low SRB1-mediated cholesterol
efflux, increased LDL susceptibility to oxidation, as well as
increased TG, apoB and hsCRP levels. All these abnormalities have
been associated with increased CAD risk (1,3,27–28,41).
However, these metabolic alterations may also occur
in overweight patients with DM2, hypertension, and CAD (27,42), that were present in this patient,
and could have opposed the antiatherogenic effect of low
circulating levels of HDL previously described in other
LCAT-deficient patients (5,13,37).
In conclusion, these findings suggest that the
adverse coronary risk profile and not the LCAT defect may be
responsible for the CAD found in our proband, and the early
atherosclerosis observed in the two heterozygotes and in the
wild-type family members.
Acknowledgements
We would like to thank all family members
investigated whose enthusiasm and helpfulness enabled us to perform
this study. We also acknowledge Nélyda G. González-Lara for
technical assistance.
References
|
1
|
Gotto AM Jr and Brinton EA: Assessing low
levels of high-density lipoprotein cholesterol as a risk factor in
coronary heart disease: a working group report and update. J Am
Coll Cardiol. 43:717–724. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Breslow JL: Familial disorders of high
density lipoprotein metabolism. The Metabolic and Molecular Bases
of Inherited Disease. Scriver CR, Beaudet AL, Sly WS and Valle D:
7th editon. McGraw-Hill Publishing; New York: pp. 2031–2052.
1995
|
|
3
|
Assmann G, von Eckardstein A and Funke H:
High density lipoproteins, reverse transport of cholesterol, and
coronary artery disease. Insights from mutations. Circulation.
87(Suppl 4): III28–III34. 1993.PubMed/NCBI
|
|
4
|
Glomset JA: The plasma
lecithins:cholesterol acyltransferase reaction. J Lipid Res.
9:155–167. 1968.PubMed/NCBI
|
|
5
|
Howlader ZH, Kamiyama S, Shirakawa H, et
al: Detoxification of oxidized LDL by transferring its oxidation
product(s) to lecithin:cholesterol acyltransferase. Biochim Biophys
Res Commun. 291:758–763. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jonas A: Lecithin cholesterol
acyltransferase. Biochim Biophys Acta. 1529:245–256. 2000.
View Article : Google Scholar
|
|
7
|
Kuivenhoven JA, Pritchard H, Hill J, et
al: The molecular pathology of lecithin: cholesterol
acyltransferase (LCAT) deficiency syndromes. J Lipid Res.
38:191–205. 1997.PubMed/NCBI
|
|
8
|
Frohlich J, McLeod R, Pritchard PH, et al:
Plasma lipoprotein abnormalities in heterozygotes for familial
lecithin: cholesterol acyltransferase deficiency. Metabolism.
37:3–8. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kasid A, Rhyne J, Zeller K, et al: A novel
TC deletion resulting in Pro(260)-Stop in the human LCAT gene is
associated with a dominant effect on HDL-cholesterol.
Atherosclerosis. 156:127–132. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kunnen S and Van Eck M:
Lecithin:cholesterol acyltransferase: old friend or foe in
atherosclerosis? J Lipid Res. 53:1783–1799. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Asztalos BF, Schaefer EJ, Horvath KV, et
al: Role of LCAT in HDL remodeling: investigation of LCAT
deficiency states. J Lipid Res. 48:592–599. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Calabresi L, Pisciotta L, Costantin A, et
al: The molecular basis of lecithin:cholesterol acyltransferase
deficiency syndromes: a comprehensive study of molecular and
biochemical findings in 13 unrelated Italian families. Arterioscler
Thromb Vasc Biol. 25:1972–1978. 2005. View Article : Google Scholar
|
|
13
|
Charlton-Menys V, Pisciotta L, Durrington
PN, et al: Molecular characterization of two patients with severe
LCAT deficiency. Nephrol Dial Transplant. 22:2379–2382. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bérard AM, Clerc M, Brewer B Jr and
Santamarina-Fojo S: A normal rate of cellular cholesterol removal
can be mediated by plasma from a patient with familial
lecithin-cholesterol acyltransferase (LCAT) deficiency. Clin Chim
Acta. 314:131–139. 2001.PubMed/NCBI
|
|
15
|
Hovingh GK, Hutten BA, Holleboom AG, et
al: Compromised LCAT function is associated with increased
atherosclerosis. Circulation. 112:879–884. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Calabresi L, Baldassarre D, Castelnuovo S,
et al: Functional lecithin: cholesterol acyltransferase is not
required for efficient atheroprotection in humans. Circulation.
120:628–635. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Medina-Urrutia A, Juarez-Rojas JG,
Martínez-Alvarado R, et al: High-density lipoprotein subclasses
distribution and composition in Mexican adolescents with low HDL
cholesterol and/or high triglyceride concentrations, and its
association with insulin and C-reactive protein. Atherosclerosis.
201:392–397. 2008. View Article : Google Scholar
|
|
18
|
Chen CH and Albers JJ: Characterization of
proteoliposomes containing apoprotein A-I: a new substrate for the
measurement of lecithin: cholesterol acyltransferase activity. J
Lipid Res. 23:680–691. 1982.PubMed/NCBI
|
|
19
|
Posadas-Sánchez R, Posadas-Romero C,
Zamora-González J, et al: LDL size and susceptibility to oxidation
in experimental nephrosis. Mol Cell Biochem. 220:61–68. 2001.
|
|
20
|
Eckerson HW, Wyte CM, LA and la Du BN: The
human serum paraoxonase/arylesterase polymorphism. Am J Hum Genet.
35:1126–1138. 1983.PubMed/NCBI
|
|
21
|
De la Llera-Moya M, Atger V, Paul JL, et
al: A cell system for screening human serum for ability to promote
cellular cholesterol efflux. Relations between serum components and
efflux, esterification, and transfer. Arterioscler Thromb Vasc
Biol. 14:1056–1065. 1994.PubMed/NCBI
|
|
22
|
De la Llera-Moya M, Drazul-Schrader D,
Asztalos BF, et al: The ability to promote efflux via ABCA1
determines the capacity of serum specimens with similar
high-density lipoprotein cholesterol to remove cholesterol from
macrophages. Arterioscler Thromb Vasc Biol. 30:796–801.
2010.PubMed/NCBI
|
|
23
|
Stein JH, Korcarz CE, Hurst RT, et al;
American Society of Echocardiography Carotid Intima-Media Thickness
Task Force. Use of carotid ultrasound to identify subclinical
vascular disease and evaluate cardiovascular disease risk: a
consensus statement from the American Society of Echocardiography
Carotid Intima-Media Thickness Task Force. Endorsed by the Society
for Vascular Medicine. J Am Soc Echocardiogr. 21:93–111. 2008.
|
|
24
|
Mautner GC, Mautner SL, Froehlich J, et
al: Coronary artery calcification: assessment with electron beam CT
and histomorphometric correlation. Radiology. 192:619–623. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miller SA, Dykes DD and Polesky HF: A
simple salting out procedure for extracting DNA from human
nucleated cells. Nucleic Acids Res. 16:12151988. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Argyropoulos G, Jenkins A, Klein RL, et
al: Transmission of two novel mutations in a pedigree with familial
lecithin:cholesterol acyltransferase deficiency: structure-function
relationships and studies in a compound heterozygous proband. J
Lipid Res. 39:1870–1876. 1998.
|
|
27
|
Posadas-Sánchez R, Posadas-Romero C,
Mendoza-Pérez E, et al: Cholesterol efflux and metabolic
abnormalities associated with low
high-density-lipoprotein-cholesterol and high triglycerides in
statin-treated coronary men with low density
lipoprotein-cholesterol <70 mg/dl. Am J Cardiol. 5:636–641.
2012.PubMed/NCBI
|
|
28
|
Santamarina-Fojo S, Hoeg JM, Assmann G, et
al: Lecithin Cholesterol Acyltransferase Deficiency and Fish Eye
Disease. The Metabolic and MolecularBases of Inherited Diseases.
Scriver CR, Beaud, Sly WS, Valle D, et al: McGraw-Hill Publishing;
New York: pp. 2817–2833. 2001
|
|
29
|
Rader DJ, Ikewaki K, Duverger N, et al:
Markedly accelerated catabolism of apolipoprotein A-II (ApoA-II)
and high density lipoproteins containing ApoA-II in classic
lecithin: cholesterol acyltransferase deficiency and fish-eye
disease. J Clin Invest. 93:321–330. 1994. View Article : Google Scholar
|
|
30
|
Kelso GJ, Stuart WD, Richter RJ, et al:
Apolipoprotein J is associated with paraoxonase in human plasma.
Biochemistry. 33:832–839. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mackness MI, Arrol S and Durrington PN:
Paraoxonase prevents accumulation of lipoperoxides in low-density
lipoprotein. FEBS Lett. 286:152–154. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Watson AD, Berliner JA, Hama SY, et al:
Protective effect of high density lipoprotein associated
paraoxonase. Inhibition of the biological activity of minimally
oxidized low density lipoprotein. J Clin Invest. 96:2882–2891.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mackness MI, Walker CH and Carlson LA: Low
A-esterase activity in serum of patients with fish-eye disease.
Clin Chem. 33:587–588. 1987.PubMed/NCBI
|
|
34
|
Huang Y, von Eckardstein A, Wu S, et al: A
plasma lipoprotein containing only apolipoprotein E and with gamma
mobility on electrophoresis releases cholesterol from cells. Proc
Natl Acad Sci USA. 91:1834–1838. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Durrington PN, Mackness B and Mackness MI:
The hunt for nutritional and pharmacological modulators of
paraoxonase. Arterioscler Thromb Vasc Biol. 22:1248–1250. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ferré N, Camps J, Fernández-Ballart J, et
al: Regulation of serum paraoxonase activity by genetic,
nutritional, and lifestyle factors in the general population. Clin
Chem. 49:1491–1497. 2003.PubMed/NCBI
|
|
37
|
Calabresi L, Favari E, Moleri E, et al:
Functional LCAT is not required for macrophage cholesterol efflux
to human serum. Atherosclerosis. 204:141–146. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nishiwaki M, Ikewaki K, Bader G, et al:
Human lecithin:cholesterol acyltransferase deficiency: in vivo
kinetics of low-density lipoprotein and lipoprotein-X. Arterioscler
Thromb Vasc Biol. 26:1370–1375. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Savel J, Lafitte M, Pucheu Y, et al: Very
low levels of HDL cholesterol and atherosclerosis, a variable
relationship-a review of LCAT deficiency. Vasc Health Risk Manag.
8:357–361. 2012.PubMed/NCBI
|
|
40
|
Duivenvoorden R, Holleboom AG, van den
Bogaard B, et al: Carriers of lecithin cholesterol acyltransferase
gene mutations have accelerated atherogenesis as assessed by
carotid 3.0-T magnetic resonance imaging. J Am Coll Cardiol.
58:2481–2487. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
El Harchaoui K, Arsenault BJ, Franssen R,
et al: High-density lipoprotein particle size and concentration and
coronary risk. Ann Intern Med. 150:84–93. 2009.PubMed/NCBI
|
|
42
|
Syvänne M, Castro G, Dengremont C, et al:
Cholesterol efflux from Fu5AH hepatoma cells induced by plasma of
subjects with or without coronary artery disease and
non-insulin-dependent diabetes: importance of LpA-I:A-II particles
and phospholipid transfer protein. Atherosclerosis. 127:245–253.
1996.
|