Introduction
Impaired angiogenic potential and decreased skeletal
muscle capillarization are known to contribute significantly to the
development of cachexia and progressive muscle wasting in several
chronic inflammatory diseases, including chronic obstructive
pulmonary disease (COPD) (1–6).
The underlying mechanisms mediating decreased skeletal muscle
capillarization under these conditions remain poorly
understood.
Several inflammatory markers, part of a systemic
inflammatory response, have been suggested to be important for the
development of cachexia in a number of chronic disorders (7–11)
amongst which, elevated serum levels of the pro-inflammatory
cytokine, tumor necrosis factor (TNF), have been suggested to be of
particular relevance (12). The
potential contribution of TNF to the development of cachexia and
decreased capillarization in a disease state as COPD remains a
matter of debate, due to inconsistency in reports regarding its
serum levels in patients with COPD (13–17). However, elevated circulatory TNF
levels appear to be associated with muscle loss in patients with
confirmed cachexia (16,18), as well as with muscle atrophy and
decreased capillarization in animal models of COPD (19–22). Furthermore, TNF is a potent
catabolic and anti-angiogenic factor in vitro (23–26).
TNF has been demonstrated to promote a catabolic
state in skeletal muscles through the activation of the ubiquitin
proteolytic system (UPS) and the degradation of muscle proteins by
the proteasome (22–24,27). In skeletal muscle, several UPS
members are known to mediate muscle wasting in diverse catabolic
states. These include E3α, atrogin-1/MAFbx, muscle RING-finger
protein 1 (MuRF1), E2(14k) and ubiquitin-specific protease 19
(28,29). In addition, we have recently
reported the overexpression of the E3 ubiquitin ligase, von-Hippel
Lindau (VHL) tumor suppressor and the E2 family member, ubiquitin
conjugating enzyme 2D1 (Ube2D1) in the skeletal muscle of patients
with COPD and in a rodent model (1,30).
In both studies, UPS activation was accompanied by disturbed
hypoxia-angiogenesis signal transduction, reduced capillarization
and muscle fiber atrophy (1,30).
VHL and Ube2D1 regulate muscle angiogenesis and
glycolysis through the regulation of the hypoxia-inducible factor
1-α (HIF1-α), the main cellular oxygen sensor and regulator of
tissue angiogenesis (23,45).
Intracellular HIF1-α levels are kept under tight control in
response to the available oxygen levels (31). Under normoxic conditions, HIF-1α
is maintained at low levels by hydroxylation at the proline
residues 402 and/or 564 by the family of prolyl hydroxylases
(PHD)1–4 and subsequent ubiquitination by VHL and Ube2D1 which
facilitates its proteasomal degradation (31). By contrast, hypoxia inactivates
prolyl hydroxylases and VHL, allowing HIF1-α to stabilize and
become transcriptionally active, thus promoting angiogenesis,
glycolysis and cell survival (31).
In the present study, we aimed to investigate the
effects of TNF stimulation on the regulation of
hypoxia-angiogenesis signal transduction and capillarization in
skeletal muscle using an in vitro model of C2C12 skeletal
myocytes.
Materials and methods
Cell culture
C2C12 cells (Sigma-Aldrich Chemie GmbH, Steinheim,
Germany) were cultured in Dulbecco’s modified Eagle’s medium (DMEM)
(PAA Laboratories, Vienna, Austria) supplemented with 10% foetal
bovine serum (PAA Laboratories), 2 mM L-glutamine (Life
Technologies, Stockholm, Sweden) and 0.1% PEST (50 UI/ml penicillin
and 50 μg/ml streptomycin; Life Technologies) and maintained at
37°C (5% CO2, 21% O2 and 74% N2)
in a CO2 incubator (Binder GmbH, Tuttlingen,
Germany).
For the treatments in all the experiments, C2C12
myoblasts were seeded into collagen-coated six-well plates to a
density of 3×105 cells/well. After reaching
approximatelly 80–90% confluence, differentiation was induced by
shiftting to DMEM containing 2% horse serum, 1 mM L-glutamine and
0.1% PEST. The C2C12 myocytes were considered fully diffrentiated
96 h after the induction of differentiation.
TNF treatment and exposure to
hypoxia
Fully differentiated myocytes were treated with 1–20
ng/ml concentrations of recombinant murine TNF (PeproTech, Rehovot,
Israel) during a time period of 2–72 h. In the experiments
involving exposure to hypoxia, fully differentiated C2C12 myocytes
were treated for 40 h with 10 and 20 ng/ml TNF under normal oxygen
conditions (21% O2, 5% CO2 and 74%
N2) and than deprived of oxygen for an additional 8 h
(1% O2, 5% CO2 and 94% N2) at 37°C
in a hypoxia incubator (Binder GmbH).
RNA extraction and quantitative
polymerase chain reaction (qPCR)
Total RNA was extracted using the Total RNA kit I
(Omega Bio-tek, Norcross, UK). RNA was quantified using a Nanodrop
spectrophotometer (ND-1000; Thermo Fisher Scientific Inc., Uppsala,
Sweden). cDNA (1 μg) was synthesised using a High-Capacity cDNA
Reverse Transcription kit (Life Technologies). The reaction was
performed as per the manufacturer’s instructions using a Uno
Thermoblock Thermal Cycler (Biometra, Göttingen, Germany). qPCR
gene expression analysis was performed on an ABI Prism Sequence
Detection System 7900HT (PE Applied Biosystems, Foster City, CA,
USA). Genes targeted in the expression analysis, including VHL
(Mm00494136_m1), PHD2 (Mm00459770_m1), Ube2D1 (Mm01172638_m1),
vascular endothelial growth factor (VEGF; Mm01281449-m1), atrogin-1
(Mm00429593-m1), Murf-1 (Mm01185221-m1), ubiquitin (Ub;
Mm02525294-m1) and glucose transporter 1 (Glut1; Mm00441480_m1)
were provided as an Assay-on-Demand by PE Applied Biosystems. Gene
expression analysis was normalized to the expression levels of
β2-microglobulin (Mm00446195_m1) which was selected as the most
stable housekeeping control in the analyzed samples. The probes
were labeled using FAM as the reporter dye and TAMRA as the
quencher dye.
Each sample was analyzed in duplicate under the
following conditions: 2 min at 50°C, 10 min at 95°C, 15 sec at 95°C
and 1 min at 60°C. PCR amplification was correlated against a
standard curve. Reactions were performed in MicroAmp Optical
96-well reaction plates (PE Applied Biosystems).
Western blot analysis
Whole cell lysates were prepared using
radioimmunoprecipitation (RIPA) buffer (150 mM NaCl, 1% NP-40, 0.5%
sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS) and 50 nM
Tris, pH 8.0) with a cocktail of protease inhibitors (Sigma-Aldrich
Chemie GmbH). The total protein concentration was measured using
the Micro Bicinchoninic Acid (BCA) Protein Assay kit (Thermo Fisher
Scientific Inc.) - microplate procedure. Equal amounts of total
protein (30–60 μg) were separated under reducing conditions using
8, 10 and 12% SDS-PAGE and transferred onto PVDF membranes
(Amersham/GE Life Sciences, Little Chalfont, UK) in a transblot
electrophoretic transfer cell (Bio-Rad Laboratories, Hercules, CA,
USA). The membranes were probed overnight at 4°C using rabbit
polyclonal anti-HIF1-α in a 1:1,000 dilution (Santa Cruz
Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal anti-VHL
diluted 1:1,000 (Santa Cruz Biotechnology), rabbit polyclonal
anti-VEGFA diluted 1:1,000 (Santa Cruz Biotechnology), rabbit
polyclonal anti-PHD2 diluted 1:2,000 (Santa Cruz Biotechnology),
rabbit polyclonal anti-Ube2D1 diluted 1:1,000 (Abnova Corp.,
Taipei, Taiwan), rabbit polyclonal anti-ubiquitin-activating enzyme
E1 (Ube1) diluted 1:1,000 (Abnova Corp.), rabbit polyclonal
anti-α-tubulin diluted 1:10,000 and rabbit polyclonal
anti-ubiquitin (Sigma-Aldrich Chemie GmbH) then incubated for 1 h
at room temperature with secondary antibody (Sigma-Aldrich Chemie
GmbH). The membranes were developed using an enhanced
chemiluminescence system (Amersham/GE Life Sciences) and exposed to
Hyperfilm enhanced chemiluminescence (Amersham/GE Life Sciences).
Densitometric analysis was performed using the NIH software package
ImageJ (ImageJ 1.46j; NIH, Bethesda, MD, USA).
Immunofluorescence
Immunohistochemical analysis was performed following
a previously described protocol (30) with modifications. In brief, serial
transverse sections (ten-micrometers thick; AMS Biotechnology,
Abingdon, UK) were deparaffinized in xylene and rehydrated in
serial dilutions of ethanol followed by antigen retrieval in
Tris-EDTA (pH 9) buffer using a microwave for 20 min. Following 30
min of permibialization in 0.3% Triton X-100 (AppliChem -
BioChemica, Darmstadt, Germany) solution, the sections were
incubated for 1 h in blocking solution containing 1% BSA and 10%
goat serum. Thereafter, the sections were incubated with primary
antibody raised against VHL, PHD2 (1:100 dilution, rabbit
polyclonal, Santa Cruz Biotechnology), Ube2D1, Ube1 and Ub (1:100
dilution, rabbit polyclonal; Abnova Corp.) overnight at 4°C.
The localization of VHL expression in the C2C12
skeletal muscle myocytes was performed in eight-chambered
immunocytochemical slides (Sarstedt, Nümbrecht, Germany). In brief,
fully differentiated myocytes were fixed with ice-cold
paraformaldehyde (4%) for 15 min; the fixative was removed and
washed three times with PBS. Permeabilization was performed using
0.2% Triton ×100 (AppliChem - BioChemica) solution in PBS for 30
min followed by incubation for 1 h in blocking solution containing
1% BSA and 10% goat serum. The primary antibody in the dilution of
1:100 (VHL, rabbit polyclonal; Santa Cruz Biotechnology) was then
added followed by incubation overnight at 4°C.
Primary antibodies were detected following
incubation with FITC-labeled secondary antibody raised in goat and
directed against rabbit IgG1 (H+L) for 1 h at room temperature.
DAPI was used to visualize the nucleus. As a negative control, the
primary antibody was omitted and the cells were incubated directly
with the secondary antibody. A series of photographs was taken
using a fluorescent microscope (obtained from Olympus, Tokyo,
Japan) and the representative field was presented.
Statistical analysis
The obtained data was normally distributed as
determined by the Anderson-Darling test. The results are presented
as the means ± standard deviation (SD). Data are expressed as the
means ± SD. Statistical comparisons were performed using ANOVA
followed by an independent Student’s t-test. Differences were
considered significant at P<0.05. Statistical analysis was
performed using SPSS v.16 software. The mumber of repetitions (n)
per each experiment was equal to or higher than three.
Results
Increased expression levels of
anti-angiogenic VHL, PHD2 and Ube2D1 proteins in TNF-treated
skeletal muscle myocytes
The stimulation of murine skeletal muscle myocytes
with TNF resulted in a dose- and time-dependant increase in the
expression of VHL protein, which reached a peak after 48 h of
stimulation and at the concentration of 10 ng/ml TNF
(***P<0.001; Fig.
1A–D). In addition, the Ube2D1 protein expression was enhanced
by 1.4-fold following the stimulation of the C2C12 myocytes for 48
h with 10 ng/ml TNF (**P<0.01; Fig. 1C and D). Concurrently, TNF
stimulation induced a dose-dependent increase in PHD2 protein
expression (4-fold increase at 10 ng/ml and 5.3-fold at 20 ng/ml
TNF, ***P<0.01; Fig. 1B
and C). By contrast, TNF stimulation did not significantly
alter the Ube1 expression in the C2C12 myocytes (Fig. 1B and C). The overexpression of
free and conjugated ubiquitin in the TNF-treated myocytes in
response to TNF stimulation was also confirmed by western blot
analysis (Fig. 2A).
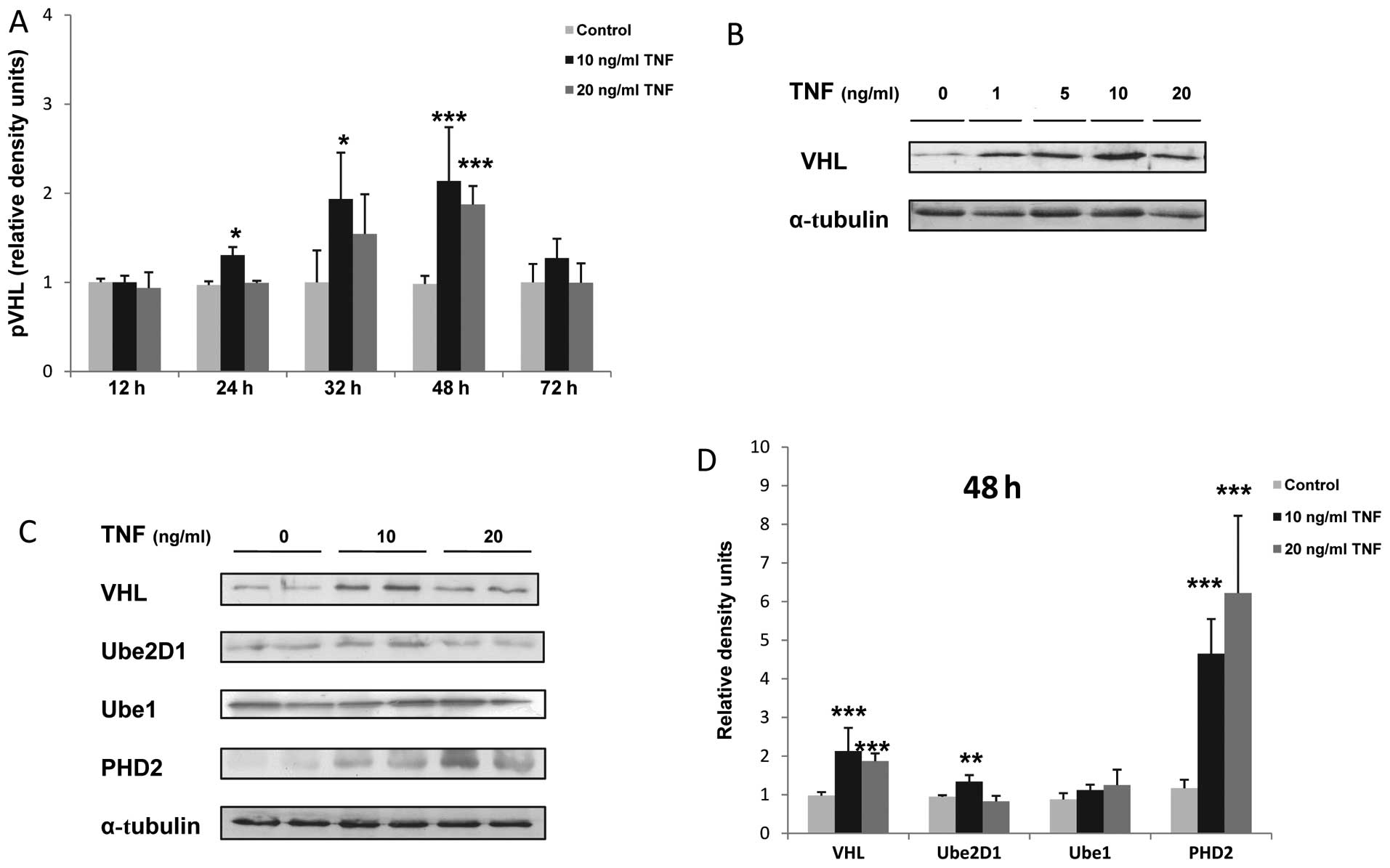 | Figure 1Tumor necrosis factor (TNF) increases
the expression levels of von-Hippel Lindau (VHL), prolyl
hydroxylase (PHD)2 and ubiquitin-conjugating enzyme 2D1 (Ube2D1)
protein in skeletal muscle myocytes. (A) Time-dependant increase in
VHL protein expression; *P<0.05,
***P<0.001, n=3. (B) Dose-dependant increase in VHL
protein expression in response to TNF stimulation. (C)
Representative western blot of VHL, Ube2D1, Ube1 and PHD2
expression after 48 h of TNF stimulation, n≥3. (E) Relative VHL,
Ube2D1, Ube1 and PHD2 density values. α-tubulin was used as the
loading control; *P<0.05, **P<0.01,
***P<0.001, n=3. |
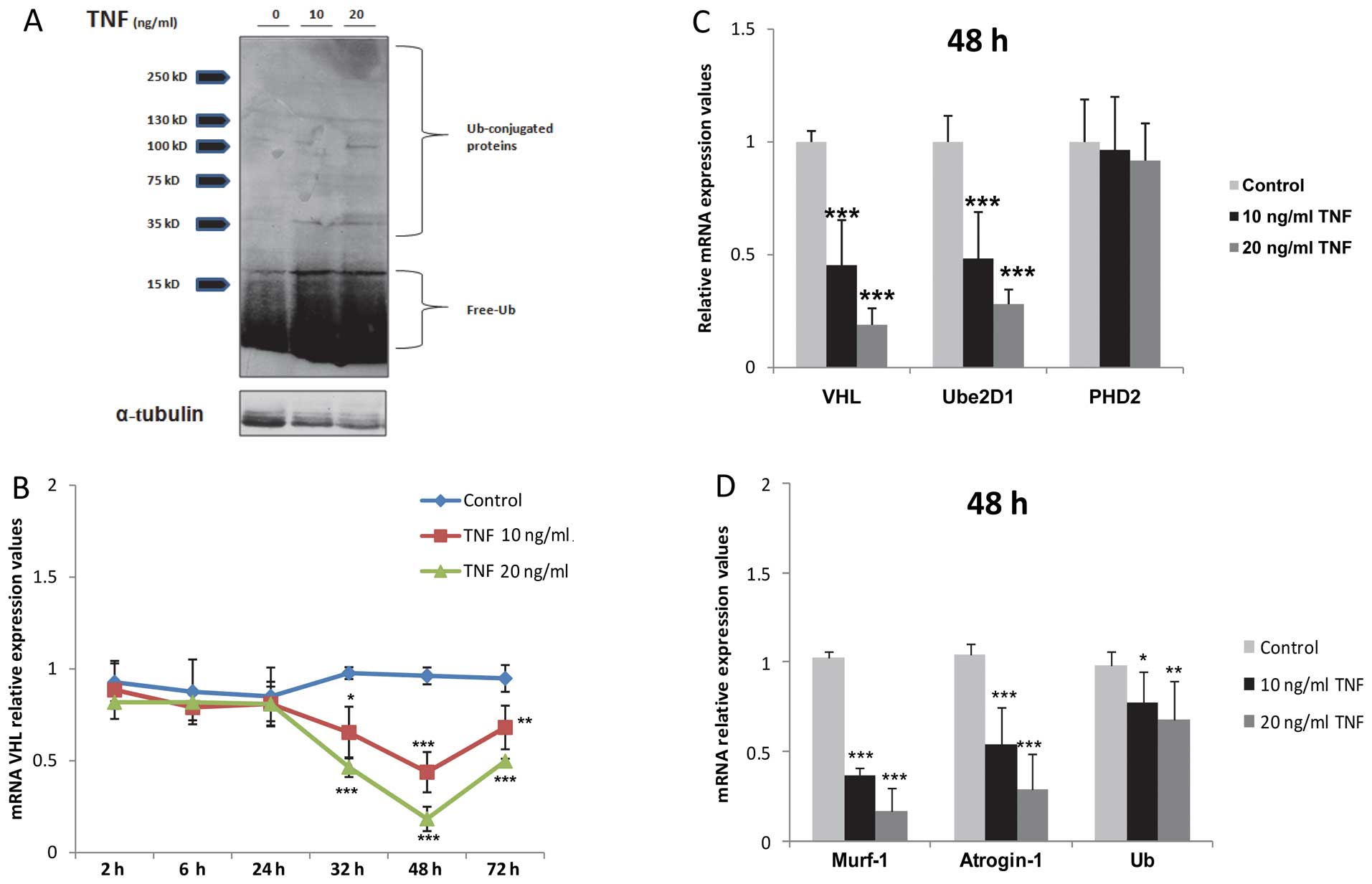 | Figure 2Prolonged tumor necrosis factor (TNF)
stimulation suppresses the transcription of von-Hippel Lindau (VHL)
and other atrophy-mediating genes in skeletal muscle myocytes. (A)
Increased levels of free ubiquitin (Ub) and Ub-conjugated protein
in TNF-treated myocytes. (B) Time curve qPCR analysis of mRNA VHL
expression, *P<0.05, **P<0.01,
***P<0.001, n=3. (C) qPCR analysis of VHL, ubiquitin
conjugating enzyme 2D1 (Ube2D1) and prolyl hydroxylase (PHD)2 mRNA
expression after 48 h of TNF stimulation, ***P<0.001,
n=3. (D) qPCR analysis of atrogin-1, Murf-1 and Ub mRNA expression,
*P<0.05, **P<0.01,
***P<0.001, n=3. Time duration of treatment was 48
h. |
VHL, PHD2 and Ube2D1 protein
overexpression in the TNF-treated myocytes is not a result of an
enhanced transcription
VHL, Ube2D1 and PHD2 protein overexpression in the
TNF-stimulated myocytes was not the result of an enhanced
transcription, as determined by TaqMan PCR assays. Hence, a dose-
and time-dependant downregulation in VHL mRNA expression in the
TNF-treated myocytes, peaking after 48 h of stimulation
(*P<0.05, **P<0.01,
***P<0.001; n=3; Fig.
2B and C) was observed. Similarly, treatment with TNF decreased
Ube2D1 mRNA expression (***P<0.001, n=3; Fig. 2C), while the mRNA expression of
PHD2 was not altered significantly following treatment with TNF
(P>0.05, n=3; Fig. 2C). In
analogy to VHL, our results demonstrated the downregulation of
atrogin-1, Murf-1 and ubiquitin mRNA expression under the same
conditions (*P<0.05, **P<0.01,
***P<0.001; n=3; Fig.
2D).
Nuclear localization of VHL and
associated UPS members in murine skeletal muscle cells
In order to gain a better understanding of how VHL
interacts with its partner proteins and exerts its E3 ligase
activity in skeletal muscle tissue, we assessed the distribution
and localization of VHL, Ube2D1, PHD2, Ube1 and Ub expression in
the murine tibialis anterior muscle. Our results demonstrated a
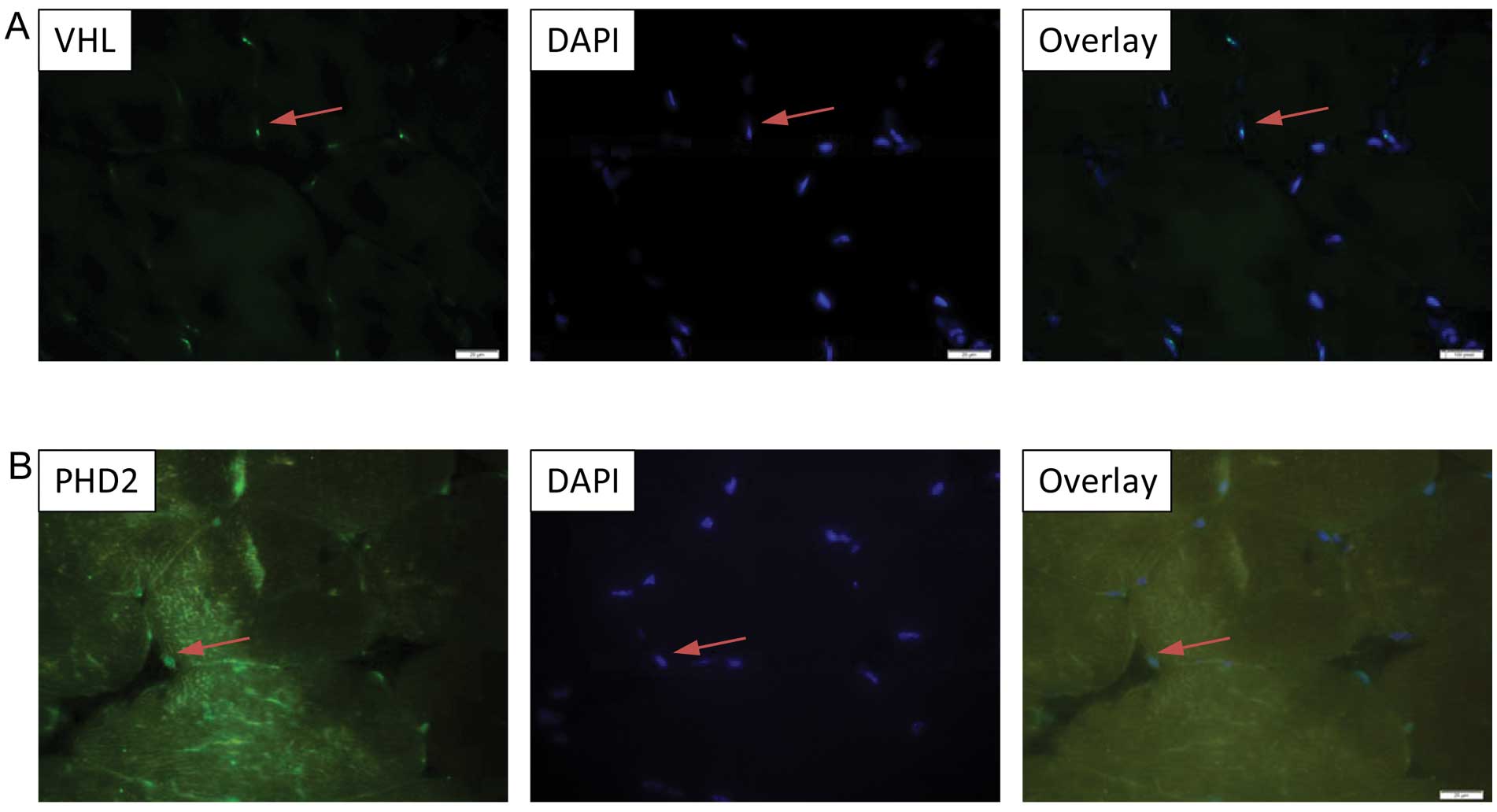
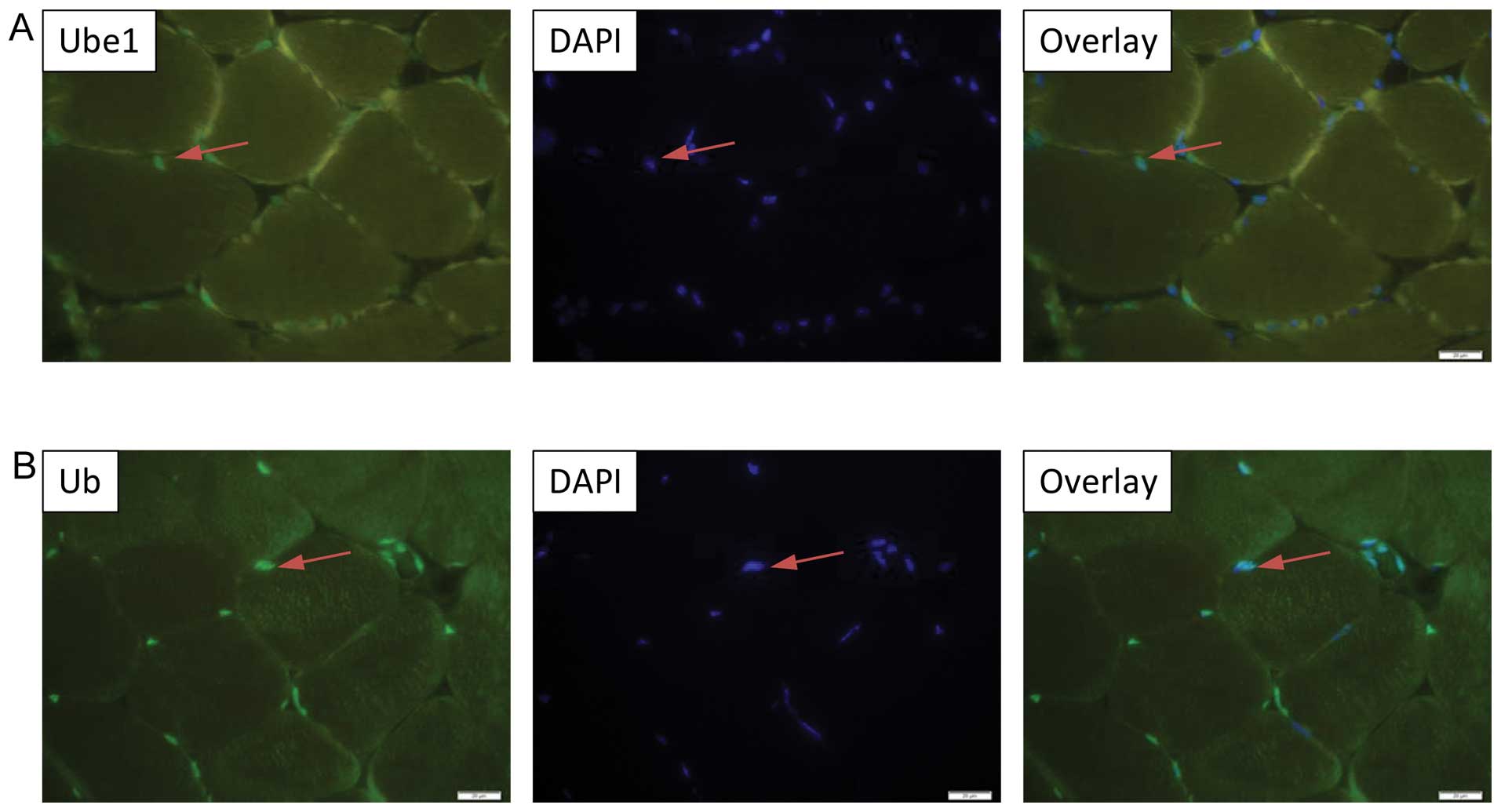
predominant nuclear localization of VHL, PHD2, Ube1 and Ub
expression in the murine tibialis anterior muscle (Figs. 3A and B; 4A and B), while Ube2D1 expression was
not detected following the described protocol. In addition, the
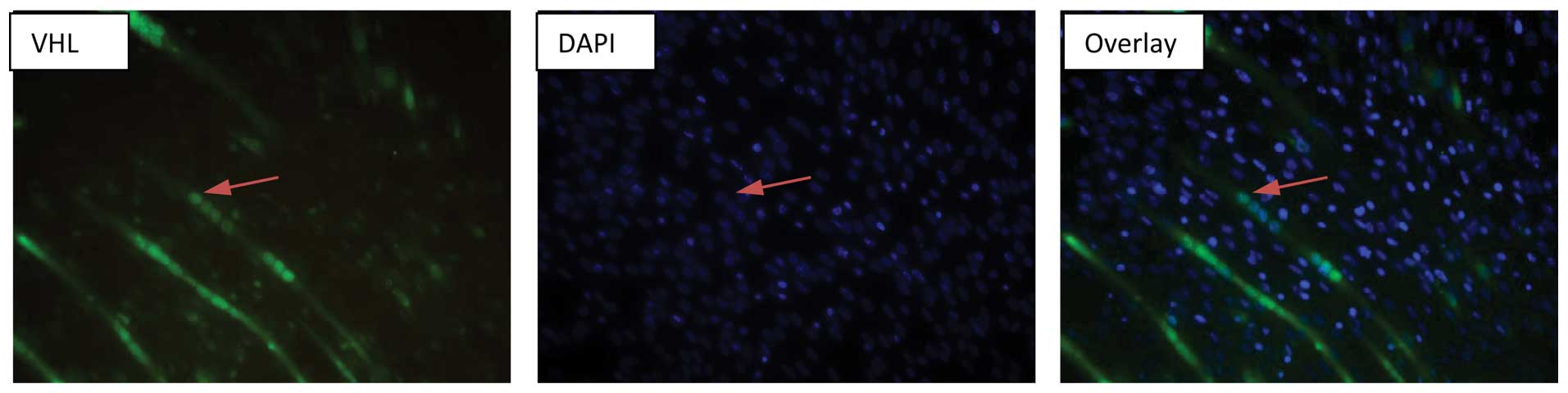
nuclear localization of VHL was been confirmed in the
differentiated C2C12 myocytes by immnucytochemistry (Fig. 5).
TNF disturbs hypoxia-angiogenesis signal
transduction in skeletal muscle myocytes
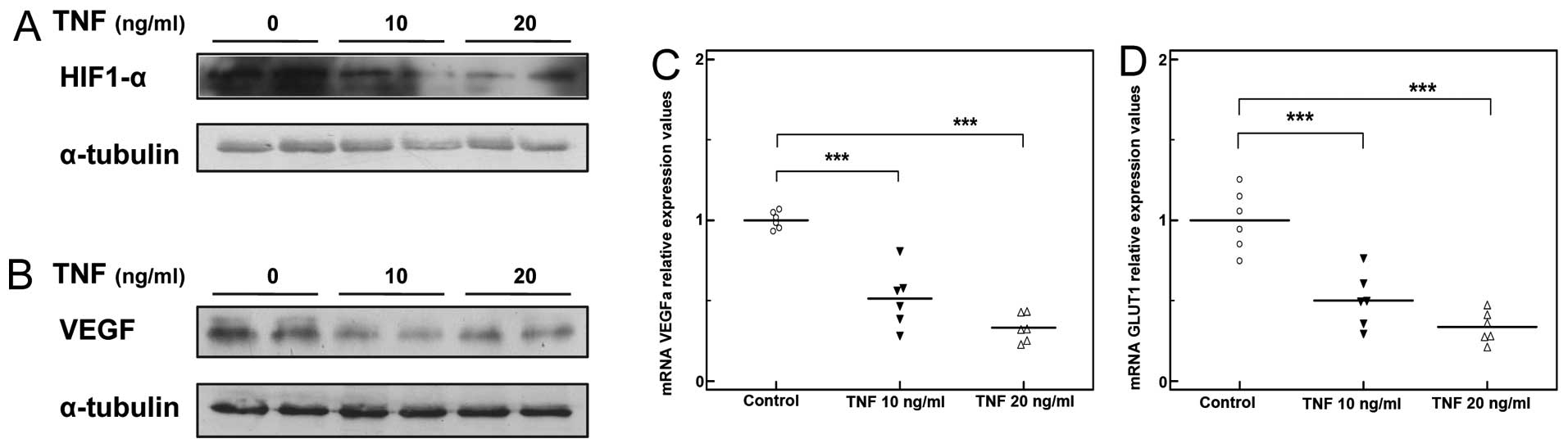
Treatment with TNF induced a moderate decrease in
HIF1-α protein expression in the C2C12 myocytes (Fig. 6A), as well as a significant
decrease in the expression of HIF1-α transcriptional targets,
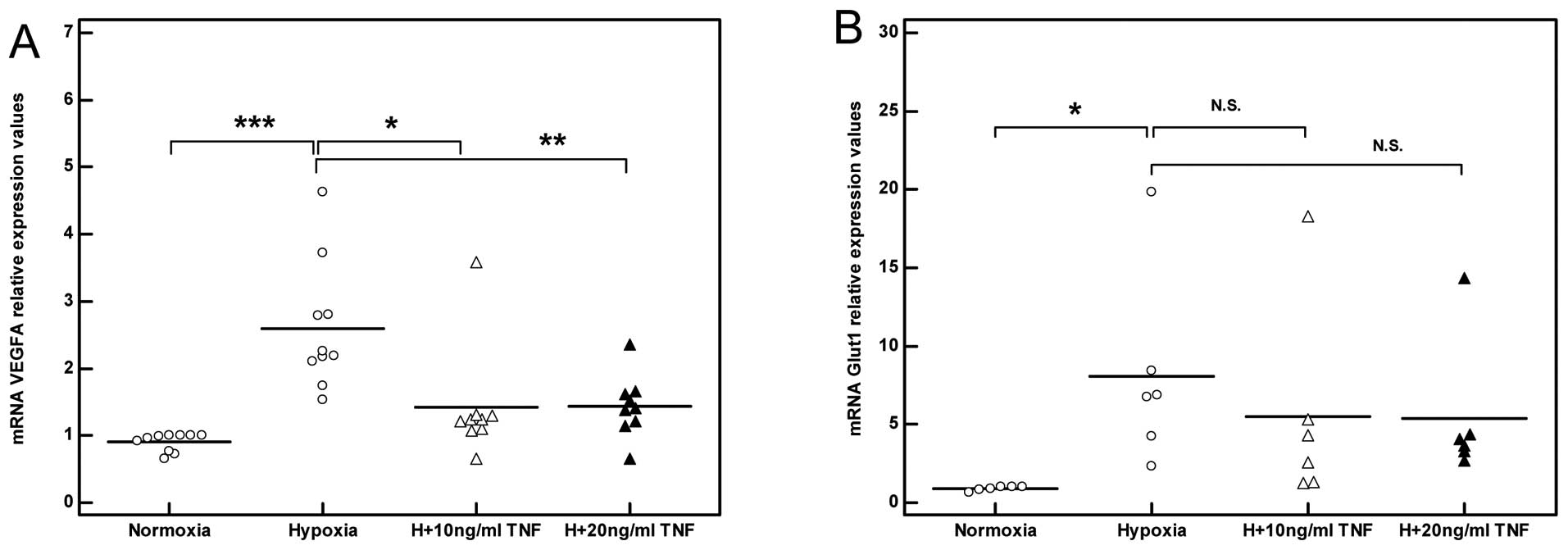
including VEGFA and Glut1 (***P<0.001, n=3; Fig. 6C and D). In line with these
findings, VEGF protein levels were markedly reduced (Fig. 6B).
Hypoxia increases VHL transcript levels
and augments VHL protein expression in TNF-stimulated skeletal
muscle myocytes
The exposure of the myocytes to hypoxia increased
VHL mRNA levels in a time-dependant manner, reaching a peak after 8
h (2-fold, ***P<0.001, n=5; Fig. 7C) and returned to basal levels
after 24 h (Fig. 7C). This
increase was also accompanied by the overexpression of VHL protein
detected after 8 h of exposure to hypoxia (Fig. 7A, B and D). In addition, hypoxia
recovered VHL mRNA expression to the basal levels and enhanced VHL
protein expression in the TNF-stimulated myocytes (Fig. 7B and D).
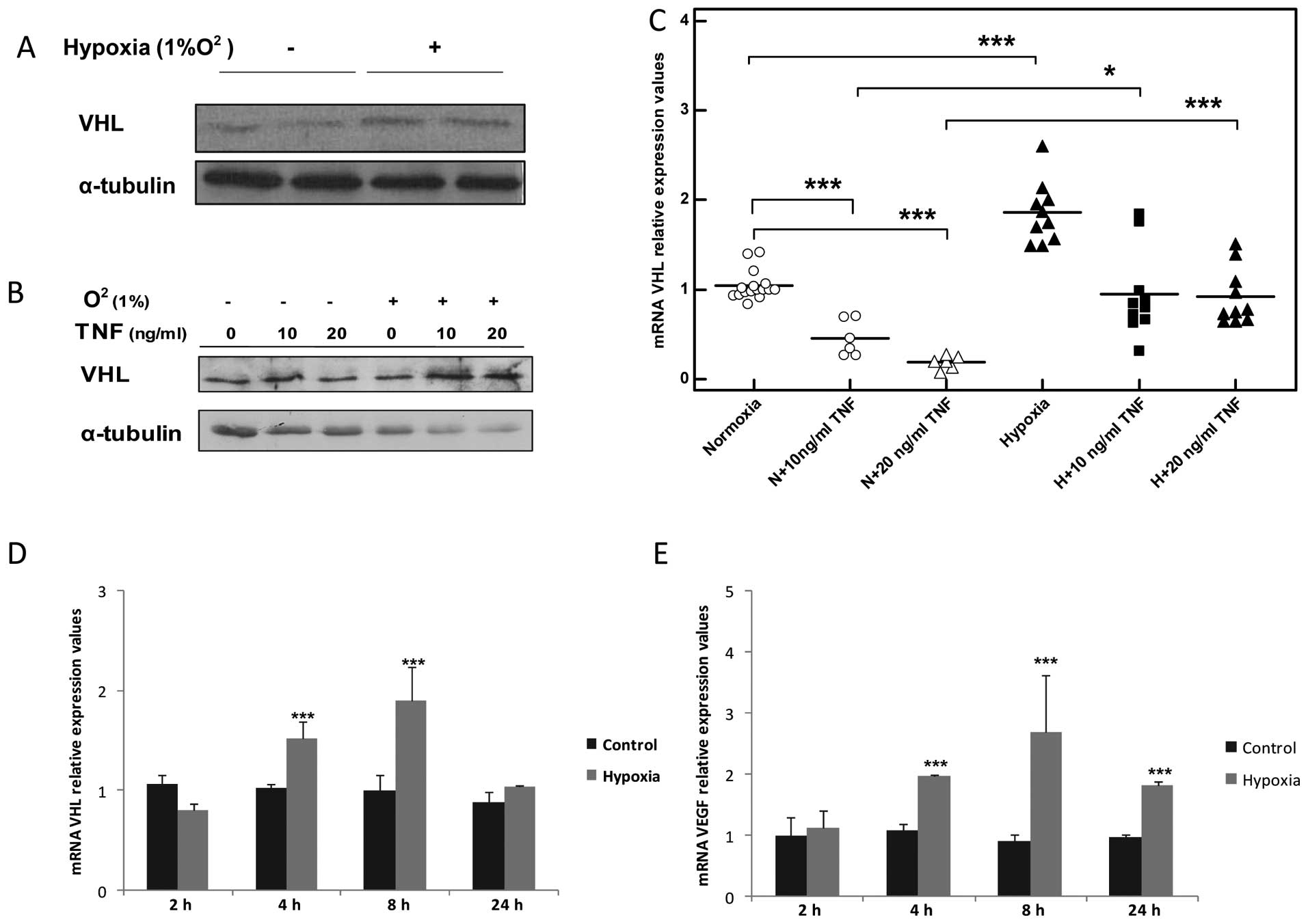 | Figure 7Hypoxia increases the expression of
von-Hippel Lindau (VHL) transcript and augments VHL protein
overexpression in tumor necrosis factor (TNF)-stimulated C2C12
muscle myocytes. (A) Representative western blot of VHL expression
in C2C12 myocytes after 8 h of exposure to hypoxia. Normoxic
conditions (21% O2) are marked with a minus (−) sign.
Hypoxic conditions (1% O2) are marked with a plus sign
(+). (B) TNF augments VHL protein overexpression in TNF-treated
C2C12 myocytes. Normoxic conditions (21% O2) are marked
with a minus (−) sign. Hypoxic conditions (1% O2) are
marked with a plus sign (+). Time duration of exposure to hypoxia
was 8 h, n=3. (C) qPCR analysis of mRNA VHL expression 8 h after
TNF stimulation, *P<0.05, **P<0.01,
***P<0.001, n=5; N, normoxia conditions (21%
O2); H, hypoxia conditions (1% O2). (D) Time
curve representing mRNA VHL expression in C2C12 myocytes exposed to
hypoxia, ***P<0.01, n=3. (E) Time curve representing
mRNA VEGF expression in C2C12 myocytes exposed to hypoxia,
***P<0.01, n=3. |
TNF impairs the angiogenic response of
skeletal muscle myocytes exposed to hypoxia
As expected, exposing skeletal muscle myocytes to
hypoxia induced an angiogenic response mirrored by increased VEGF
transcription which reached a peak level after 8 h (3-fold,
***P<0.001, n=3; Fig.
7E). Stimulation with TNF in the presence of hypoxia failed to
induce a significant angiogenic response to hypoxia as reflected by
the decreased induction of VEGF mRNA levels relative to the
non-stimulated myocytes (Fig.
8A). Concurrently, a trend towards decreased Glut1 induction in
response to hypoxia was also observed in the TNF-stimulated
myocytes (Fig. 8B).
Discussion
We have previously demonstrated enhanced VHL
expression and the disturbance of hypoxia-angiogenesis signal
transduction in skeletal muscle of patients with COPD (1). This observation was further
confirmed in a murine model exposed to cigarette smoke (1,30).
However, the mechanisms mediating VHL overexpression in skeletal
muscle remain unclear. The results of the current study demonstrate
that the pro-inflammatory cytokine, TNF, increases VHL expression
in skeletal muscle myocytes and that this effect is further
augmented in the presence of hypoxia.
TNF has been reported to trigger muscle catabolism
through the NF-κB transcription factor (32), which rapidly enhances the
transcription of E3 ubiquitin ligases, such as atrogin-1/MAFbx and
MuRF1, and increases the levels of free ubiquitin, which in turn
potently mediate the degradation of muscle contractile proteins
(23,27,33,34). By contrast, the results of the
current study demonstrate that TNF increases VHL expression through
a mechanism which is transcription-independent, involving the
enhanced translation/stabilization of the VHL protein. In
accordance with this, TNF has been associated with protein
stabilization and enhanced translation of different members of UPS
through a wide range of post-translational modifications and
diverse signaling pathways (35–38). Moreover, prolonged TNF stimulation
resulted in a significant downregulation in VHL mRNA expression
levels in skeletal muscle myocytes, as well as in the transcript
levels of Ube2D1, atrogin-1, MURF-1 and the ubiquitin (Ub) gene.
These results are in agreement with the studies by Alvarez et
al (37) and Bhatnagar et
al (39), demonstrating the
suppressed transcription of atrophy-mediating UPS genes in C2C12
myocytes in response to prolonged TNF stimulation and excessive UPS
activation. It can be speculated that the excessive
accumulation/stabilization of the VHL protein by TNF treatment may
have induced a feedback mechanism further restricting the
accumulation of VHL protein, as well as an attempt of myocytes to
restrict excessive VHL E3 ligase activity and the degradation of
vital proteins.
Concurrent to the increase in the VHL levels, the
overexpression of PHD2 and Ube2D1 protein, additionally suggested
that TNF treatment enhances E3 ligase activity of the VHL
ubiquitination complex in skeletal muscle myocytes. This was
supported by the decreased protein stability and transcriptional
efficiency of HIF1-α observed in this study. In line with our
results, TNF has been previously reported to enhance VHL-HIF1-α
interaction and increase HIF1-α ubiquitination under normoxic
conditions (40). Furthermore,
TNF has been associated with reduced VEGF expression and signaling
in different cell lines (22,37,38), as well as with a reduction in
muscle VEGF levels (15) and
decreased capillarization in vivo (15,39).
Another highly relevant finding in this study was
that hypoxia regulates VHL expression in skeletal muscle cells.
Hypoxia has previously been reported to enhance VHL gene expression
in non-muscle cell lines as part of a feedback mechanism and HIF1-α
self-regulation (42,43). In agreement with this, we observed
a similar expression pattern between VHL and VEGF, which reached a
peak after 8 h of exposure to hypoxia. This strongly suggests that
under normal conditions, VHL functions to fine-tune HIF1-α
signaling and prevent excessive angiogenic response in skeletal
muscle myocytes. However, our data suggest that this balance is
disturbed in the presence of TNF due to the elevated VHL
expression, which in turn causes the impaired angiogenic adaptation
of skeletal muscle myocytes to hypoxia. This finding is of
particular relevance when viewed in light of our previous findings
of enhanced VHL expression in patients with COPD (1) and mice exposed to cigarette smoke
(30), as well as the reduced
muscle angiogenic potential and systemic inflammatory response
known to occur in patients with COPD (5,6,13,15).
Taken together, the results of the current study
provide evidence that elevated TNF levels disturb
hypoxia-angiogenic signaling in skeletal muscles through a
mechanism that involves increased VHL expression and an excessive
degradation of HIF1-α protein. The current findings provide a
mechanism linking systemic inflammation and impaired angiogenesis
in skeletal muscle. This is of particular relevance in
understanding the mechanisms mediating muscle wasting and cachexia
in patients with chronic inflammatory diseases, such as COPD.
Acknowledgements
This study was supported by the Olle Engkvist
Byggmästare Fund, Sweden (to S.A.-H.) and Örebro University grant
for doctoral students (to V.T.B.).
References
|
1
|
Jatta K, Eliason G, Portela-Gomes GM, et
al: Overexpression of von Hippel-Lindau protein in skeletal muscles
of patients with chronic obstructive pulmonary disease. J Clin
Pathol. 62:70–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jobin J, Maltais F, Doyon JF, et al:
Chronic obstructive pulmonary disease: capillarity and fiber-type
characteristics of skeletal muscle. J Cardiopulm Rehabil.
18:432–437. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prior SJ, McKenzie MJ, Joseph LJ, et al:
Reduced skeletal muscle capillarization and glucose intolerance.
Microcirculation. 16:203–212. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kivela R, Silvennoinen M, Touvra AM, Lehti
TM, Kainulainen H and Vihko V: Effects of experimental type 1
diabetes and exercise training on angiogenic gene expression and
capillarization in skeletal muscle. FASEB J. 20:1570–1572. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gouzi F, Prefaut C, Abdellaoui A, et al:
Blunted muscle angiogenic training-response in COPD patients versus
sedentary controls. Eur Respir J. 41:806–814. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gagnon P, Lemire BB, Dube A, et al:
Preserved function and reduced angiogenesis potential of the
quadriceps in patients with mild COPD. Respir Res. 15:42014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gan WQ, Man SF, Senthilselvan A and Sin
DD: Association between chronic obstructive pulmonary disease and
systemic inflammation: a systematic review and a meta-analysis.
Thorax. 59:574–580. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Petersen AM, Penkowa M, Iversen M, et al:
Elevated levels of IL-18 in plasma and skeletal muscle in chronic
obstructive pulmonary disease. Lung. 185:161–171. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Van Helvoort HA, Heijdra YF, Thijs HM,
Vina J, Wanten GJ and Dekhuijzen PN: Exercise-induced systemic
effects in muscle-wasted patients with COPD. Med Sci Sports Exerc.
38:1543–1552. 2006.
|
|
10
|
Deans C and Wigmore SJ: Systemic
inflammation, cachexia and prognosis in patients with cancer. Curr
Opin Clin Nutr Metab Care. 8:265–269. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morley JE, Thomas DR and Wilson MM:
Cachexia: pathophysiology and clinical relevance. Am J Clin Nutr.
83:735–743. 2006.PubMed/NCBI
|
|
12
|
Delano MJ and Moldawer LL: The origins of
cachexia in acute and chronic inflammatory diseases. Nutr Clin
Pract. 21:68–81. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Piehl-Aulin K, Jones I, Lindvall B,
Magnuson A and Abdel-Halim SM: Increased serum inflammatory markers
in the absence of clinical and skeletal muscle inflammation in
patients with chronic obstructive pulmonary disease. Respiration.
78:191–196. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wagner PD: Possible mechanisms underlying
the development of cachexia in COPD. Eur Respir J. 31:492–501.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Garcia-Rio F, Miravitlles M, Soriano JB,
et al: Systemic inflammation in chronic obstructive pulmonary
disease: a population-based study. Respir Res. 11:632010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pinto-Plata V, Casanova C, Mullerova H, et
al: Inflammatory and repair serum biomarker pattern: association to
clinical outcomes in COPD. Respir Res. 13:712012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tanni SE, Pelegrino NR, Angeleli AY,
Correa C and Godoy I: Smoking status and tumor necrosis
factor-alpha mediated systemic inflammation in COPD patients. J
Inflamm (Lond). 7:292010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eagan TM, Gabazza EC, D’Alessandro-Gabazza
C, et al: TNF-alpha is associated with loss of lean body mass only
in already cachectic COPD patients. Respir Res. 13:482012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Caron MA, Morissette MC, Theriault ME,
Nikota JK, Stampfli MR and Debigare R: Alterations in skeletal
muscle cell homeostasis in a mouse model of cigarette smoke
exposure. PLoS One. 8:e664332013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang K, Wagner PD and Breen EC:
TNF-alpha-mediated reduction in PGC-1alpha may impair skeletal
muscle function after cigarette smoke exposure. J Cell Physiol.
222:320–327. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gosker HR, Langen RC, Bracke KR, et al:
Extrapulmonary manifestations of chronic obstructive pulmonary
disease in a mouse model of chronic cigarette smoke exposure. Am J
Respir Cell Mol Biol. 40:710–716. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Langen RC, Schols AM, Kelders MC, van der
Velden JL, Wouters EF and Janssen-Heininger YM: Muscle wasting and
impaired muscle regeneration in a murine model of chronic pulmonary
inflammation. Am J Respir Cell Mol Biol. 35:689–696. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Garcia-Martinez C, Agell N, Llovera M,
Lopez-Soriano FJ and Argiles JM: Tumour necrosis factor-alpha
increases the ubiquitinization of rat skeletal muscle proteins.
FEBS Lett. 323:211–214. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garcia-Martinez C, Llovera M, Agell N,
Lopez-Soriano FJ and Argiles JM: Ubiquitin gene expression in
skeletal muscle is increased during sepsis: involvement of
TNF-alpha but not IL-1. Biochem Biophys Res Commun. 217:839–844.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Langen RC, Van Der Velden JL, Schols AM,
Kelders MC, Wouters EF and Janssen-Heininger YM: Tumor necrosis
factor-alpha inhibits myogenic differentiation through MyoD protein
destabilization. FASEB J. 18:227–237. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Frater-Schroder M, Risau W, Hallmann R,
Gautschi P and Bohlen P: Tumor necrosis factor type alpha, a potent
inhibitor of endothelial cell growth in vitro, is angiogenic in
vivo. Proc Natl Acad Sci USA. 84:5277–5281. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cao PR, Kim HJ and Lecker SH:
Ubiquitin-protein ligases in muscle wasting. Int J Biochem Cell
Biol. 37:2088–2097. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lecker SH, Solomon V, Price SR, Kwon YT,
Mitch WE and Goldberg AL: Ubiquitin conjugation by the N-end rule
pathway and mRNAs for its components increase in muscles of
diabetic rats. J Clin Invest. 104:1411–1420. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Combaret L, Adegoke OA, Bedard N, Baracos
V, Attaix D and Wing SS: USP19 is a ubiquitin-specific protease
regulated in rat skeletal muscle during catabolic states. Am J
Physiol Endocrinol Metab. 288:E693–E700. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Basic VT, Tadele E, Elmabsout AA, et al:
Exposure to cigarette smoke induces overexpression of von
Hippel-Lindau tumor suppressor in mouse skeletal muscle. Am J
Physiol Lung Cell Mol Physiol. 303:L519–L527. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Semenza GL: Regulation of oxygen
homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda).
24:97–106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li YP, Schwartz RJ, Waddell ID, Holloway
BR and Reid MB: Skeletal muscle myocytes undergo protein loss and
reactive oxygen-mediated NF-kappaB activation in response to tumor
necrosis factor alpha. FASEB J. 12:871–880. 1998.PubMed/NCBI
|
|
33
|
Adams V, Mangner N, Gasch A, et al:
Induction of MuRF1 is essential for TNF-alpha-induced loss of
muscle function in mice. J Mol Biol. 384:48–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pijet B, Pijet M, Litwiniuk A, Gajewska M,
Pajak B and Orzechowski A: TNF-alpha and IFN-s-dependent muscle
decay is linked to NF-kappaB- and STAT-1alpha-stimulated Atrogin1
and MuRF1 genes in C2C12 myotubes. Mediators Inflamm.
2013:1714372013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tong X, Buelow K, Guha A, Rausch R and Yin
L: USP2a protein deubiquitinates and stabilizes the circadian
protein CRY1 in response to inflammatory signals. J Biol Chem.
287:25280–25291. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shukla R, Yue J, Siouda M, et al:
Proinflammatory cytokine TNF-alpha increases the stability of
hepatitis B virus X protein through NF-kappaB signaling.
Carcinogenesis. 32:978–985. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Alvarez B, Quinn LS, Busquets S,
Lopez-Soriano FJ and Argiles JM: Direct effects of tumor necrosis
factor alpha (TNF-alpha) on murine skeletal muscle cell lines.
Bimodal effects on protein metabolism. Eur Cytokine Netw.
12:399–410. 2001.PubMed/NCBI
|
|
38
|
Plaisance I, Morandi C, Murigande C and
Brink M: TNF-alpha increases protein content in C2C12 and primary
myotubes by enhancing protein translation via the TNF-R1, PI3K, and
MEK. Am J Physiol Endocrinol Metab. 294:E241–E250. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bhatnagar S, Panguluri SK, Gupta SK,
Dahiya S, Lundy RF and Kumar A: Tumor necrosis factor-alpha
regulates distinct molecular pathways and gene networks in cultured
skeletal muscle cells. PLoS One. 5:e132622010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou J, Schmid T and Brune B: Tumor
necrosis factor-alpha causes accumulation of a ubiquitinated form
of hypoxia inducible factor-1alpha through a nuclear
factor-kappaB-dependent pathway. Mol Biol Cell. 14:2216–2225. 2003.
View Article : Google Scholar
|
|
41
|
Terasaki H, Kase S, Shirasawa M, et al:
TNF-alpha decreases VEGF secretion in highly polarized RPE cells
but increases it in non-polarized RPE cells related to crosstalk
between JNK and NF-kappaB pathways. PLoS One. 8:e699942013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Turcotte S, Desrosiers RR and Beliveau R:
Hypoxia upregulates von Hippel-Lindau tumor-suppressor protein
through RhoA-dependent activity in renal cell carcinoma. Am J
Physiol Renal Physiol, United States. F338–FP348. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Karhausen J, Kong T, Narravula S and
Colgan SP: Induction of the von Hippel-Lindau tumor suppressor gene
by late hypoxia limits HIF-1 expression. J Cell Biochem.
95:1264–1275. 2005. View Article : Google Scholar : PubMed/NCBI
|