Introduction
Huntington’s disease (HD) is an autosomal dominant
neurodegenerative disorder caused by an abnormal polyglutamine
expansion within the protein huntingtin (1). Despite similar expression levels
throughout the brain, mutant huntingtin selectively targets
striatal neurons, with the cerebral cortex being affected later in
the disease (2). Although
selective striatal cell death is a prominent feature of HD, the
underlying mechanism of striatal susceptibility remains to be
clarified. Mitochondrial dysfunction has been reported to
contribute to the pathogenesis of HD (3) and may underlie the selective
neuronal degeneration (4).
Administration of 3-nitropropionic acid (3NP) in
rodents and non-human primates has provided useful experimental
models for HD (5,6). 3NP is an irreversible inhibitor of
the mitochondrial complex II, which causes energy impairment and
replicates most of the clinical and pathophysiological
characteristics of HD, including spontaneous choreiform and
dystonic movements, as well as the selective degeneration of
striatum (6,7). 3NP has been reported to trigger the
generation of superoxide radicals, secondary excitotoxicity and
apoptosis (7,8). It has been reported that the c-Jun
N-terminal kinase (JNK)/c-Jun signaling pathway plays an important
role in 3NP-induced striatal degeneration (5).
Heat shock proteins (HSPs) are considered important
protective effectors against a variety of cellular stresses
(9,10). HSPs suppress protein misfolding by
assisting misfolded proteins in the refolding process. For example,
the overexpression of HSP 70 reduces the toxic accumulation of
abnormal polyglutamine proteins and suppresses cell death in a
variety of cellular models of polyglutamine diseases including HD
(7,11–13). In addition, HSP 70 has been
reported to block several steps of the apoptotic cascade such as
upstream from mitochondria, release of cytochrome c and
apoptosis-inducing factor (AIF), nuclear import of AIF, activation
of procaspase-9 and -3, and even downstream of active caspase-3
(10,14–19).
Geldanamycin (GA) is a benzoquinone ansamycin
antibiotic that inhibits the function of HSP 90 by binding to the
ADP/ATP-binding pocket of the protein (20). HSP 90 client proteins play
important roles in the regulation of the cell cycle, cell growth,
survival, apoptosis, angiogenesis and oncogenesis (20). HSP 90 is a major repressor of the
heat shock transcription factor 1 (HSF1), a major transcription
factor of HSPs (21). Upon
binding to HSP 90, GA induces the expression of HSP 70 through the
action of HSF1 (21,22). It has been reported that GA
activates a heat shock response and inhibits huntingtin aggregation
in a cell culture model of HD (23).
The present study was conducted to examine whether
GA attenuated 3NP-induced striatal damage and the underlying
mechanism involved. GA exhibited an increased expression of HSP 70
and significantly suppressed 3NP-induced apoptosis, reactive oxygen
species (ROS) generation, and JNK activation.
Materials and methods
Cell culture
The immortalized striatal progenitor cell line
(STHdhQ7), which expresses endogenous wild-type
huntingtin, was obtained from Dr Marcy E. MacDonald and maintained
in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10%
(v/v) FBS, 10 U/ml of penicillin (all from Gibco, Invitrogen,
Carlsbad, CA, USA) at 33°C in humidified air with 5%
CO2.
Cell viability assay
Striatal cells were plated in 6-well culture plates
(Greiner Bio-One Inc., Longwood, FL, USA) and incubated at 33°C
under 5% CO2, and 95% humidified air incubator. The
cells were incubated with GA for 4 h prior to treatment with 3NP
for another 24 h. After washing with PBS, 0.6 mg/ml
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
was added (100 μl/well) and incubated for 2 h at 33°C. MTT solution
(40 μl) was then removed from each well and replaced with 500 μl of
dimethyl sulfoxide (DMSO). The plates were incubated for 1 h at
33°C. Absorbance readings were taken at 570 nm using a Multiskan Ex
microtitre plate reader (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). Data are expressed as % MTT reduction compared to a 100%
signal from non-transfected cells.
Lactate dehydrogenase leakage (LDH)
assay
Striatal cells were exposed to 3NP (10 μM) overnight
at 33°C for 24 h. After exposure to 3NP and GA, the medium was
centrifuged at 250 × g for 10 min to harvest the cell culture media
and the cell-free supernatant was obtained for the LDH activity
assay using a commercial LDH detection kit (Roche Diagnostics Gmbh
Mannheim, Germany) according to the manufacturer’s
instructions.
Western blotting
Striatal cells were washed with PBS three times and
lysed by PRO-PREP protein extraction solution (Intron
Biotechnology, Inc., Gyeonggi, Korea), and sonicated on ice.
Protein concentrations of the homogenates were measured using the
BCA method (Sigma-Aldrich, St. Louis, MO, USA) and diluted to a
final concentration of 2 mg/ml with 2× reducing stop buffer (0.25 M
Tris-HCl, pH 6.8, 5 mM EDTA, 5 mM EGTA, 25 mM dithiothreitol, 2%
SDS, 10% glycerol, and bromophenol blue as the tracking dye). Equal
amounts of proteins were separated on 8–12% SDS-polyacrylamide gels
and transferred to a Hybond PVDF transfer membrane (GE Healthcare,
Amersham, UK). The membranes were blocked in 5% skim milk in TBST
(20 mM Tris-HCl, pH 7.6, 137 mM NaCl, 0.05% Tween-20) for 30 min at
room temperature and sequentially incubated with an appropriate
antibody; anti-HSP 90 monoclonal antibody (1:1,000), anti-HSP 70
monoclonal antibody (1:1,000) (both from Stressgen Biotechnologies,
Victoria, BC, Canada), anti-cleaved caspase-3 and total caspase-3
polyconal antibody (1:1,000), anti-Cleaved PARP and total PARP
polyconal antibody (1:1,000) (Cell Signaling Technology, Inc.,
Danvers, MA, USA), anti-IκB-α monoclonal antibody (Santa Cruz
Biotechnology Inc., Santa Cruz, CA, USA), total JNK and P-JNK
polyconal antibody (1:1,000), anti-c-Jun and P-c-Jun polyconal
antibody (1:1,000) (both from Cell Signaling Technology, Inc.) and
β-actin monoclonal antibody (1:2,500; Sigma-Aldrich) in the same
buffer overnight at 4°C. After thoroughly washing with TBST, the
membranes were washed three times with TBST and incubated with
HRP-conjugated goat anti-rabbit IgG for polyclonal antibodies, or
with HRP-conjugated goat anti-mouse IgG (1:2,500; Jackson
ImmunoResearch Laboratories, West Grove, PA, USA) for 2 h at room
temperature. The membranes were rinsed three times for 30 min with
TBST, followed by four quick rinses with distilled water, and
developed with the enhanced chemiluminescence method (GE
Healthcare).
FACS assay
Striatal cells were collected with a cell scraper,
washed twice with cold PBS and then resuspended in 1× binding
buffer at a concentration of 1×106 cells/ml. One hundred
microliters of the solution (1×105 cells) were
transferred to a 5 ml culture tube and 5 μl of Annexin V-PE and 5
μl of 7-AAD were added. The mixture was gently vortexed, incubated
for 15 min at room temperature in the dark, and 400 μl of binding
buffer was added to each tube. The stained cells were analyzed by
flow cytometry (BD Model FACScan, BD Biosciences, Franklin Lakes,
NJ, USA).
DAPI staining
For DNA fragmentation studies, striatal cells were
cultured in 6-well culture plates and treated with 3NP, GA and
GA+3NP for 24 h. After washing with PBS, the striatal cells were
fixed using 1% paraformaldehyde in PBS for 20 min, permeabilized
using 0.1% Triton X-100 for 5 min, and stained with DAPI (1 μg/ml)
(Invitrogen Life Technologies, Carlsbad, CA, USA) for 20 min. All
steps were carried out at room temperature. Representative data
were obtained by using confocal microscopy (Carl Zeiss, Inc.,
Thornwood, NY, USA).
Measurement of intracellular ROS
production
The striatal cells were cultured on 18 mm round
coverslip at 33°C, 5% CO2. After 1 day, the coverslips
were transferred into the 12-well plates and the media were changed
(no phenol red media) for 1 h stabilization. After washing, each
sample was stained by 10 μM H2DCFDA (H2DCFDA, Molecular Probe cat.
no. D-399, stocked 50 mM in DMSO) for 10 min under culture
conditions. The coverslips were washed three times with DMEM (no
phenol red). Coverslips were located in the chamber, and media were
added, and cells were immediately observed by confocal laser
scanning microscopy (Olympus, Tokyo, Japan).
Immunocytochemistry
The effect of 3NP into the nuclear translocation of
NF-κB was examined by immunofluorescence assay using confocal
microscopy. Following treatment, the cells were fixed with 4% PFA
diluted in PBS for 20 min at room temperature and incubated for 10
min with 0.1% Triton X-100. After washing with PBS, the plates were
preincubated with PBS, prior to incubation with 10% normal goat
serum for 1 h to reduce the background at room temperature,
followed by incubation with the anti-NF-κB antibody in PBS
containing 10% normal goat serum overnight at 4°C. The plates were
rinsed and incubated with an anti-rabbit TRITC-conjugated antibody
(Invitrogen Life Technologies) for 2 h at room temperature. After
washing, the nuclei were stained with Hoechst 33258 (100 mM)
(Invitrogen Life Technologies) for 10 min and mounted under
coverslips at room temperature. Representative data were obtained
by using confocal microscopy (Carl Zeiss, Inc.). The digitally
stored images were combined and shown with the accompanying
software and Adobe Photoshop 4.0.
Statistical analysis
Data were presented as means ± SE obtained from at
least three independent experiments. The statistical difference was
analyzed by one-way ANOVA with Tukey’s post-hoc test using SPSS
software 12K (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered statistically significant.
Results
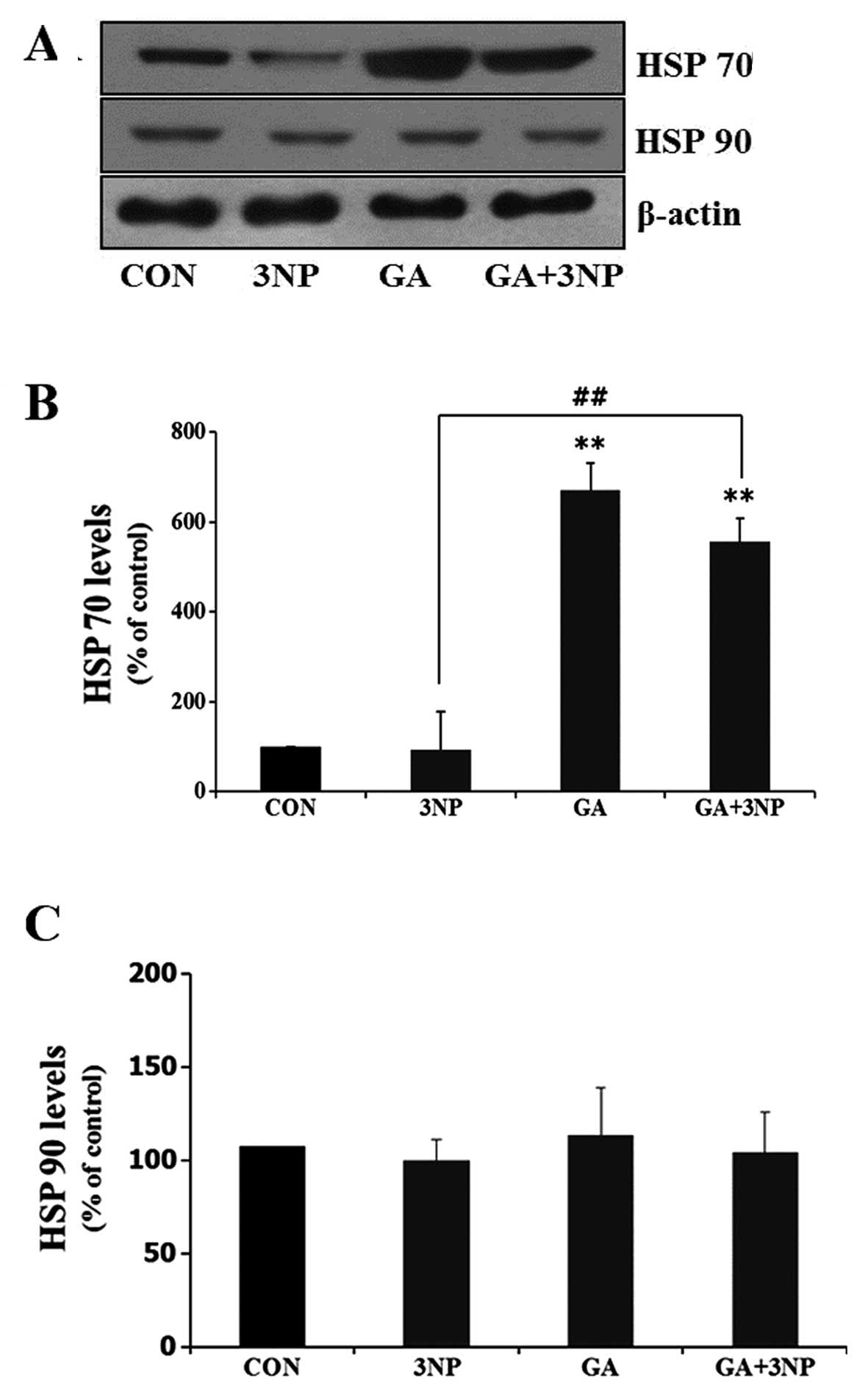
GA results in the increased expression of
HSP 70 and attenuates 3NP-induced apoptosis in striatal cells
To examine the effect of GA, an HSP 90 inhibitor, in
the expression of HSPs, the expression levels of HSP 70 and HSP 90
were examined. GA resulted in the increased expression of HSP 70
(Fig. 1A). However, the
expression level of HSP 90 was not significantly changed with GA,
which inhibits the function of HSP 90 by binding to the
ADP/ATP-binding pocket of the protein.
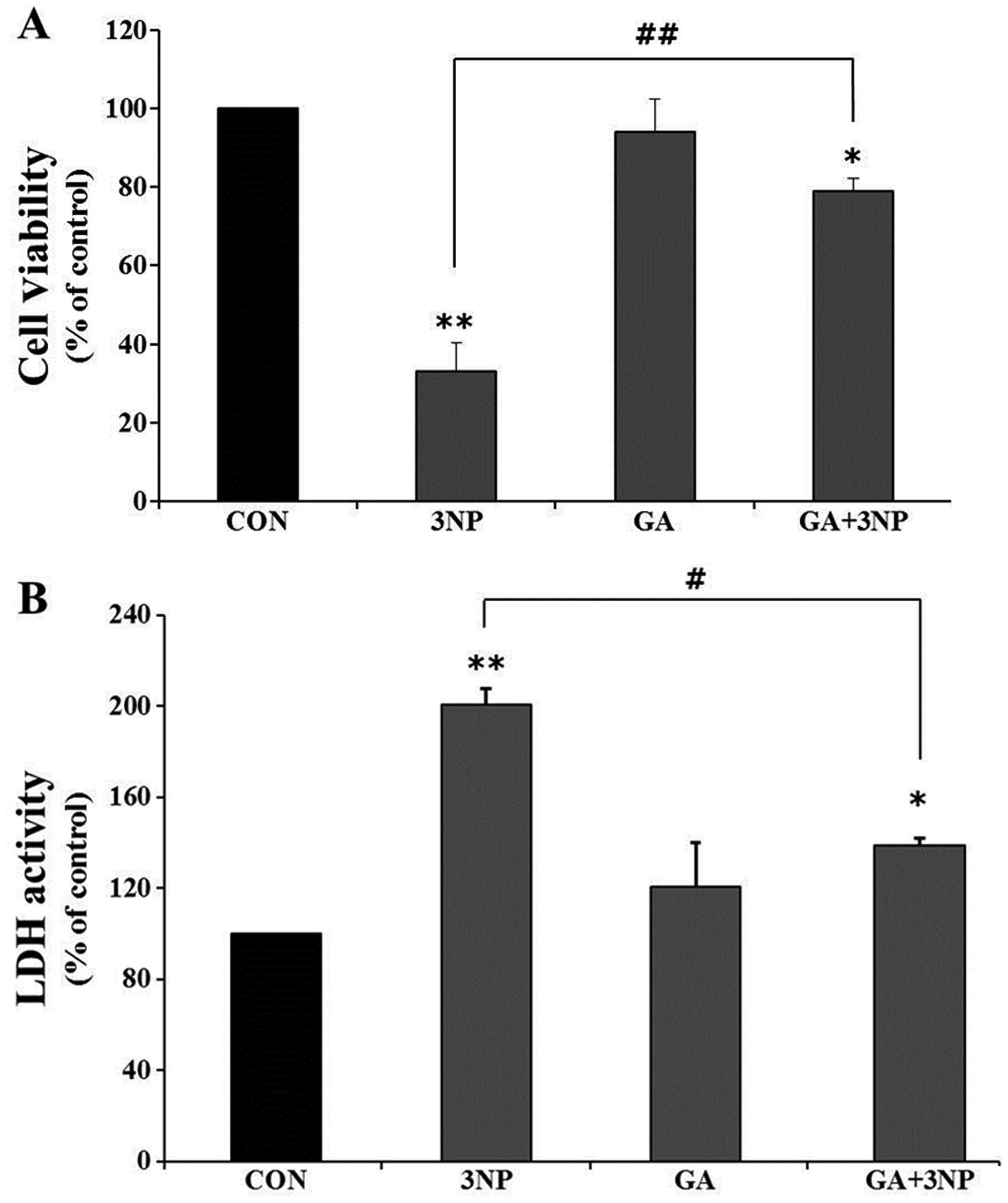
In order to examine the effect of GA on the
viability of 3NP-stimulated striatal cells, the cells were treated
with 3NP in the absence or presence of GA. Significant striatal
cell death was observed with 3NP treatment in MTT and LDH assays
(Fig. 2A and B). However, GA
significantly attenuated 3NP-induced striatal cell death. In
addition, the number of positive cells of 7-AAD and FITC, which
indicate dead cells, was significantly reduced with GA in the FACS
analysis (Fig. 2Ca and b).
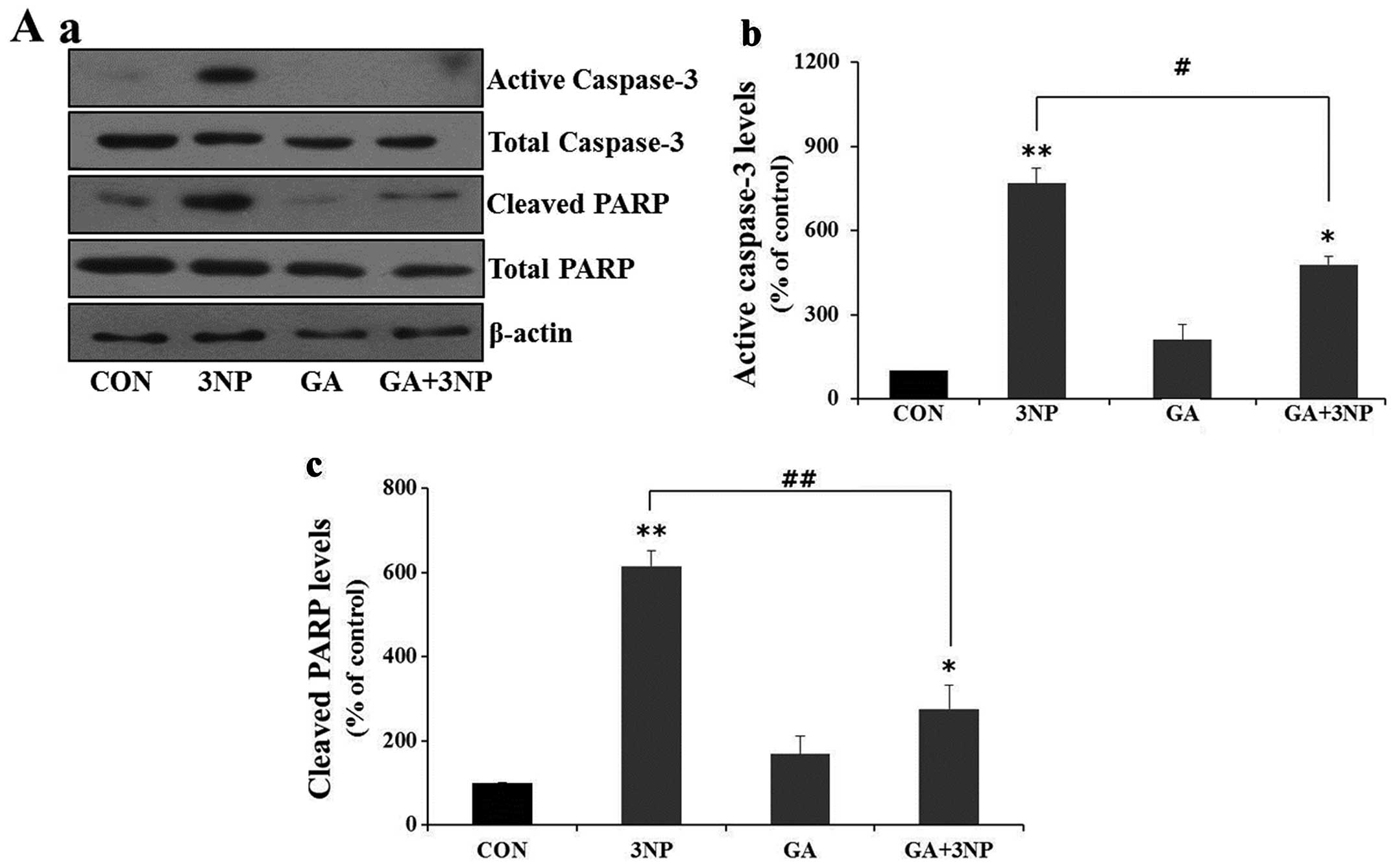
Given the fact that 3NP induces striatal cell death
in the present study, we examined whether 3NP induces apoptosis and
whether GA attenuated 3NP-induced striatal cell death. Treatment of
3NP resulted in the cleavage of caspase-3 and PARP, which indicate
the activation of apoptosis (Fig.
3A). GA significantly attenuated 3NP-induced production of
active caspase-3 and cleaved PARP in striatal cells (Fig. 3A). In addition, GA significantly
reduced the number of 3NP-induced apoptotic nuclei (Fig. 3B).
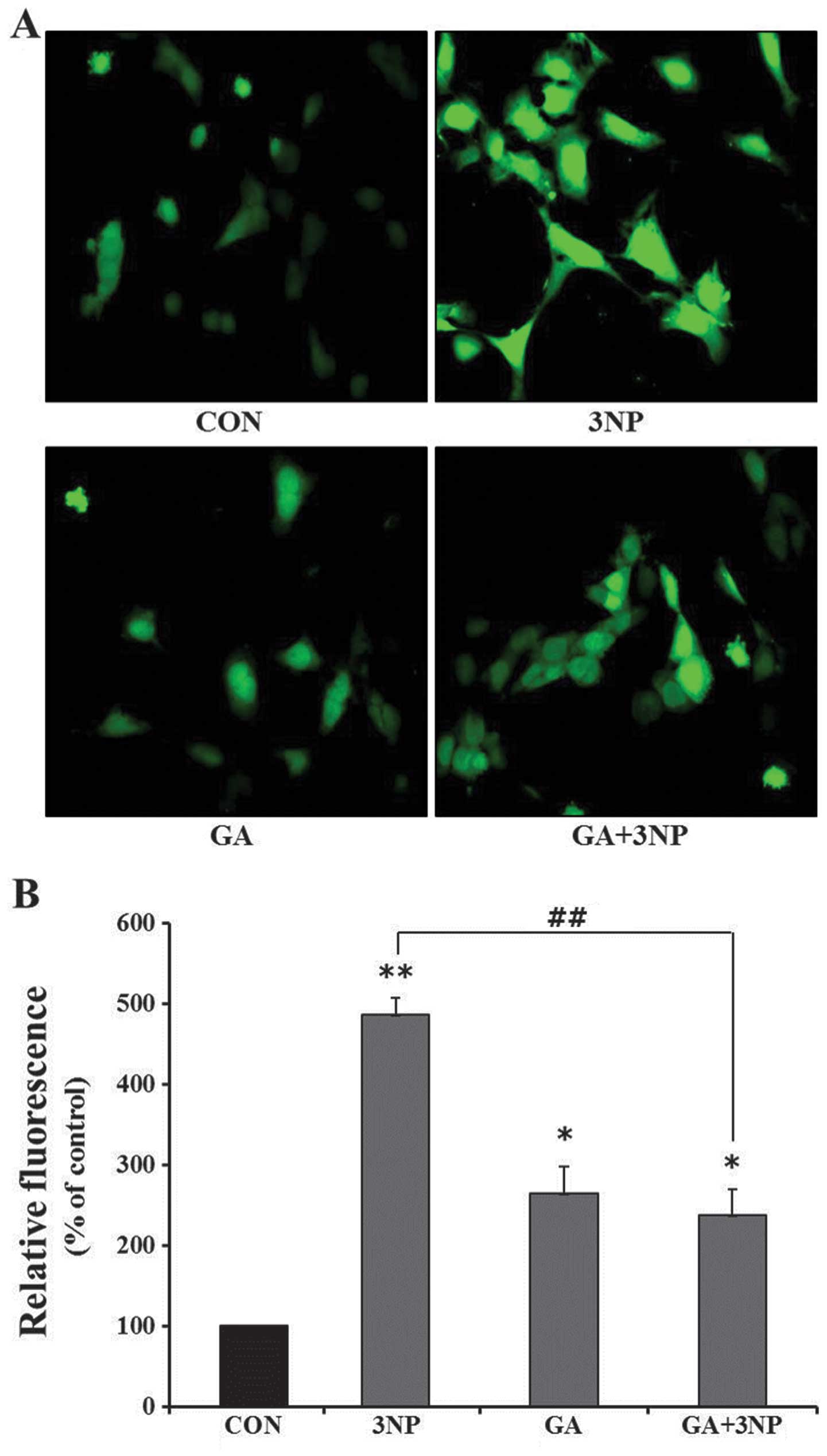
GA attenuates 3NP-induced ROS
production
We have previously reported that the overexpression
of HSF1 significantly attenuated 3NP-induced ROS production in
striatal cells (7). To
investigate the effects of GA in 3NP-induced ROS production, the
intracellular ROS generation was measured in the absence or
presence of GA in 3NP-challenged striatal cells. Treatment of 3NP
resulted in the production of a considerable amount of the
intracellular ROS in striatal cells. GA significantly attenuated
3NP-induced ROS production, albeit not completely (Fig. 4). Fig. 4A shows a representative confocal
image of intracellular level of ROS and Fig. 4B shows quantitative analysis of
ROS production. The result demonstrates that GA protects cells by
inhibiting the production of ROS in 3NP-challenged striatal
cells.
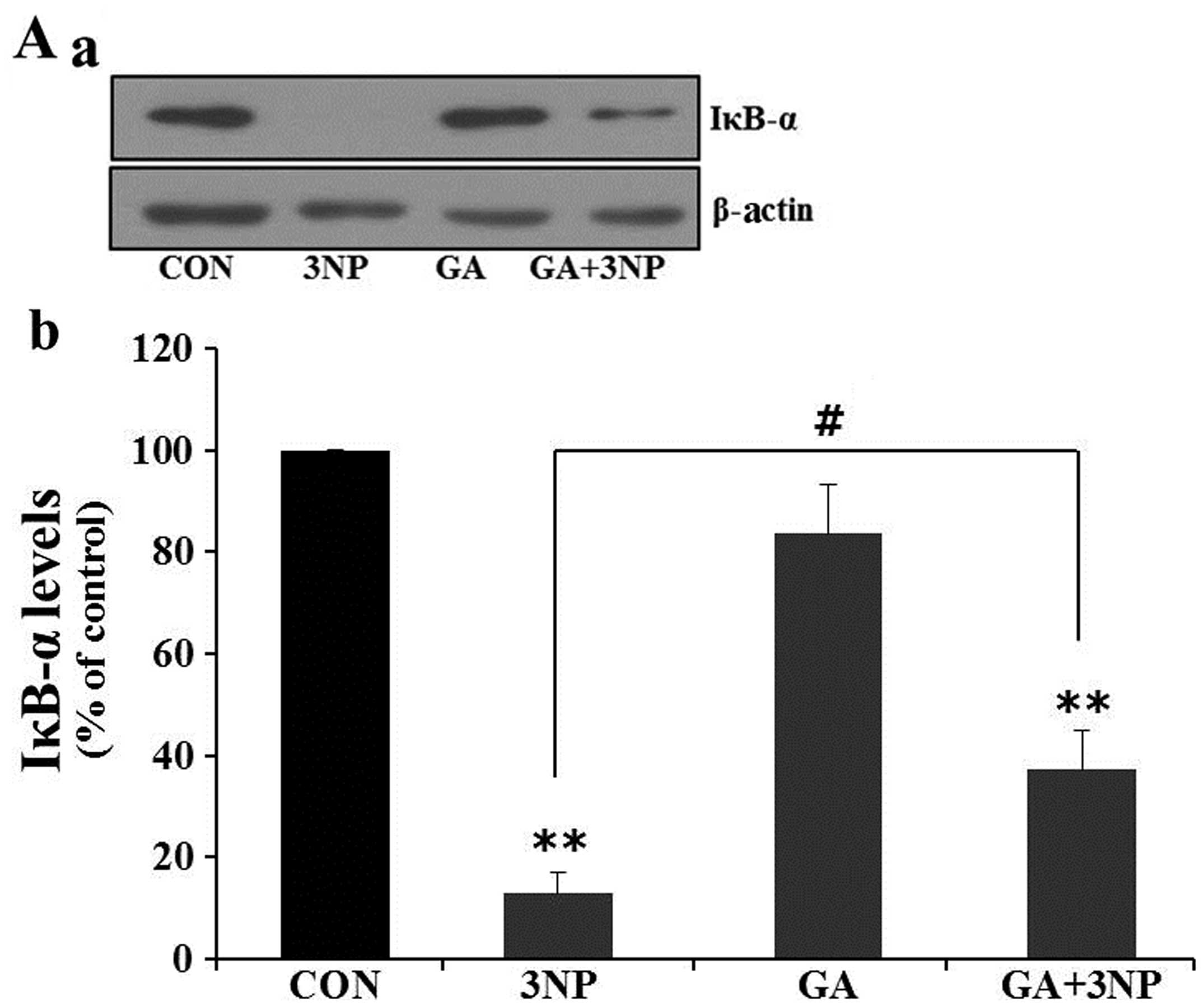
GA attenuates 3NP-induced IκB degradation
and nuclear translocation of NF-κB
It has been reported that ROS facilitates cell death
by inducing inflammatory responses via the activation of
NF-κB-mediated transcription (24–26). IκB inhibits the nuclear
translocation of NF-κB by retaining it in cytoplasm. Under stress
conditions, IκB proteins are rapidly degraded by the proteasome,
and released NF-κB translocates into the nucleus to activate
apoptotic pathways. To examine whether GA has an impact on NF-κB
transcription, the effects of GA on IκB degradation and the nuclear
translocation of NF-κB were examined. Treatment of 3NP markedly
depleted intracellular IκB in striatal cells. However, GA
significantly attenuated 3NP-induced IκB degradation (Fig. 5A). To confirm whether the nuclear
translocation of NF-κB was affected by change at the intracellular
level of IκB, intracellular localization of NF-κB was examined with
immunocytochemistry. Treatment of 3NP obviously increased the
nuclear translocation of p65 subunit of NF-κB (Fig. 5B). Geldanamycin significantly
attenuated the nuclear translocation of NF-κB in 3NP-challeged
striatal cells (Fig. 5Ba and
b).
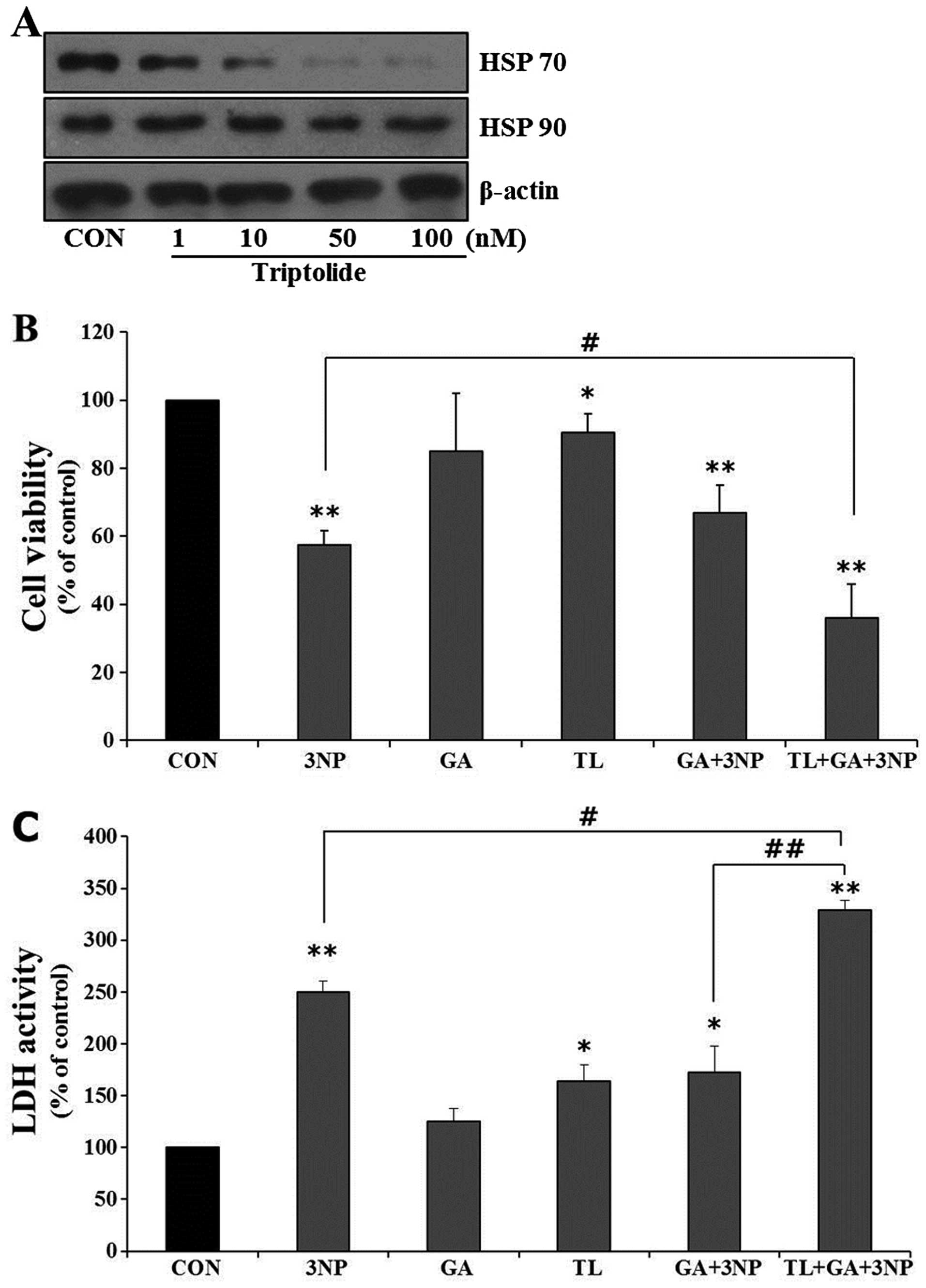
Triptolide (TL) abrogates GA-mediated HSP
70 expression
Given the hypothesis that HSP 70 plays an important
role in GA-mediated cellular protection in 3NP-challenged striatal
cells, the effect of GA was examined in the absence of HSP 70
expression. The depletion of HSP 70 expression was achieved with
TL, which inhibits the endogenous HSP 70 gene expression (27). TL downregulated HSP 70 expression
in a concentration-dependent manner in striatal cells without
affecting HSP 90 expression (Fig.
6A). To investigate the role of TL in GA-mediated cellular
protection, cell viability was examined. TL showed negligible
cytotoxicity at a concentration range used in the present study.
However, TL significantly abolished GA-mediated cellular protection
against 3NP treatment (Fig. 6B and
C). The results strongly suggested that HSP 70 plays an
essential role in GA-mediated cellular protection in 3NP-challenged
striatal cells.
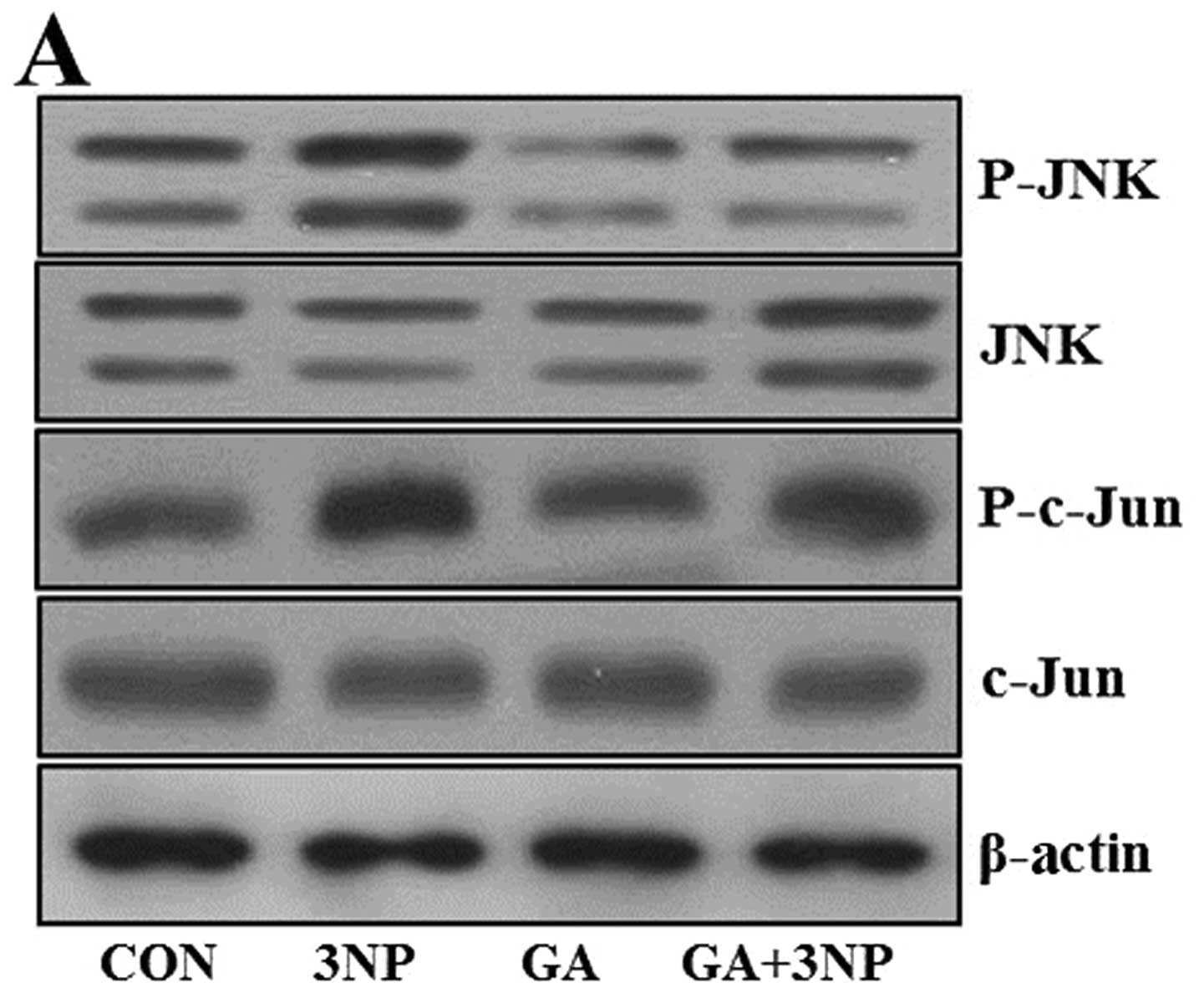
GA inhibits 3NP-induced JNK and c-Jun
phosphorylation
It has been previously reported that activation of
JNK plays a key role in 3NP-induced striatal neurodegeneration
(5). Therefore, the effects of GA
on 3NP-induced JNK activation and subsequent c-Jun phosphorylation
were examined in the present study. Treatment of 3NP resulted in
the activation of JNK (Fig. 7A).
However, GA significantly attenuated JNK phosphorylation in
3NP-challenged striatal cells (Fig.
7A). In addition, GA significantly suppressed 3NP-induced c-Jun
phosphorylation in striatal cells. The data strongly demonstrate
that the JNK/c-Jun signaling pathway is an important molecular
pathway in 3NP-induced striatal damage and that GA exerts a
cellular protective effect through suppression of the JNK/c-Jun
pathway.
Discussion
The present study has demonstrated that GA
significantly suppressed 3NP-induced apoptotic cell death and ROS
generation through the expression of HSP 70 in a striatal cell
model. GA also significantly attenuated the 3NP-induced activation
of the JNK/c-Jun signaling pathway.
HSPs have been reported to attenuate protein
aggregation and neurodegeneration in HD models (28,29). HSP 70 expression was increased in
oxidative stress conditions, which is presumably considered a
cellular compensatory mechanism against oxidative stresses
(14,30–33). It has been reported that inducers
of HSP 70 significantly suppressed toxicity of mutant huntingtin in
a C. elegans model (34).
Recently, we observed that the overexpression of HSF1 resulted in a
significantly increased expression of HSP 70 and attenuated
apoptosis in 3NP-challenged striatal cells (7). In addition, inhibition of HSP 70
function with methylene blue significantly abolished HSF1-mediated
protection against 3NP-induced striatal damage (7), suggesting that HSP 70 plays a major
role in HSF1-mediated anti-apoptotic action in striatal cells. In
the present study, inhibition of HSP 70 gene expression with TL
also significantly attenuated GA-mediated protection in striatal
cells, confirming that HSP 70 plays an essential role in cellular
protection in 3NP-challenged striatal cells.
HSP 90 is constitutively expressed in mammalian
cells and plays an essential role in facilitating the proper
folding, maturation, and activity of its client proteins (35), which eventually regulates a
variety of cellular events including cell survival, apoptosis, and
oncogenesis (20). HSP 90
associates with its client proteins in an ATP-dependent manner
(36,37). GA specifically interferes with
this association by occupying the ATP-binding pocket of HSP 90 and
dissociates client proteins from the protein (38–40). HSP 90 has also been reported to be
a major repressor of HSF1 (21).
It has been reported that GA induces the expression of HSPs such as
HSP 70 and HSP 40 and inhibits huntingtin aggregation in a cell
culture model of HD (23). In
that study, HSPs significantly attenuated the mutant
huntingtin-induced toxicity and the number of mutant huntingtin
aggregates. However, it has also been reported that HSPs did not
affect the aggregate formation of mutant huntingtin (41), suggesting that HSPs may exert its
protective actions independent of suppression of aggregate
formation. Therefore, more studies are necessary to elucidate the
exact mechanism by which HSPs exert cytoprotection in the
pathogenesis of HD. In the present study, GA exhibited an increased
expression of HSP 70 and a significant suppression of apoptotic
cell death ROS production in 3NP-challenged striatal cells. GA has
also been reported to protect against MPTP-induced dopaminergic
neurotoxicity through the induction of HSP 70 in an animal model
(42).
Mitochondrial dysfunctions have been reported to be
involved in the pathogenesis of HD (43). Individuals with HD showed a
decreased mitochondrial enzyme activities in the striatum (44,45) and mitochondria from HD patients
were shown to be more sensitive to apoptosis (46). Mitochondrial toxin 3NP produces
selective striatal lesions (47,48) and clinical features of HD such as
choreiform movements and locomotor dysfunction (49,50). 3NP is known to induce striatal
neurodegeneration via the activation of the JNK/c-Jun signaling
pathway (5). Activation of JNK
occurred progressively and selectively in the striatum and c-Jun
activation was followed in the same striatal region (5). Furthermore, overexpression of the
dominant negative form of c-Jun completely abolished 3NP-induced
striatal neurodegeneration, indicating that the JNK/c-Jun signaling
pathway is an important molecular event in 3NP-induced striatal
degeneration (5). In the present
study, significantly increased phosphorylation of JNK and c-Jun was
observed with 3NP treatment in striatal cells, confirming that
JNK/c-Jun signaling pathway is crucial in 3NP-induced striatal
toxicity.
HSP 70 has been reported to block JNK activation and
prevent apoptosis in response to protein-damaging and physiological
stimuli (51–53). HSP 70 modulates the activity of
JNK through direct binding to the protein (54) and that HSP 70 deficiency results
in activation of the JNK signaling pathway (55). Recently, it has been reported that
HSP 70 inhibits the JNK signaling pathway and subsequently prevents
Bax-mediated apoptosis (56). In
the present study, inhibition of HSP 70 gene expression with TL
significantly abolished GA-mediated striatal survival against
3NP-induced cell death, suggesting that HSP 70 may be a major
mediator for the suppression of JNK/c-Jun activation in striatal
cells. However, more studies are necessary to clearly understand
the mechanism by which geldanamycin-induced HSP 70 inhibits the
JNK/c-Jun signaling pathway in 3NP-challenged striatal cells.
Taken together, the present study clearly
demonstrates that GA exerts anti-apoptotic properties such as
suppression of apoptosis and JNK/c-Jun signaling in 3NP-challenged
striatal cells presumably through the expression of HSP 70,
suggesting that GA may be a valuable therapeutic agent to increase
the intracellular level of HSP 70, which plays a beneficial role in
the pathogenesis of HD.
Acknowledgements
The authors would like to thank Dr Marcy E.
MacDonald for the generous gift of the striatal cell line. This
study was supported by the Basic Science Research Program through
the National Research Foundation of Korea (NRF) funded by the
Ministry of Education, Science and Technology (C1010000-01-01).
References
|
1
|
Andrew SE, Goldberg YP, Kremer B, et al:
The relationship between trinucleotide (CAG) repeat length and
clinical features of Huntington’s disease. Nat Genet. 4:398–403.
1993.
|
|
2
|
Vonsattel JP and DiFiglia M: Huntington
disease. J Neuropathol Exp Neurol. 57:369–384. 1998. View Article : Google Scholar
|
|
3
|
Beal MF: Mitochondria take center stage in
aging and neurodegeneration. Ann Neurol. 58:495–505. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oliveira JM, Jekabsons MB, Chen S, et al:
Mitochondrial dysfunction in Huntington’s disease: the
bioenergetics of isolated and in situ mitochondria from transgenic
mice. J Neurochem. 101:241–249. 2007.
|
|
5
|
Garcia M, Vanhoutte P, Pages C, Besson MJ,
Brouillet E and Caboche J: The mitochondrial toxin 3-nitropropionic
acid induces striatal neurodegeneration via a c-Jun N-terminal
kinase/c-Jun module. J Neurosci. 22:2174–2184. 2002.PubMed/NCBI
|
|
6
|
Brouillet E, Condé F, Beal MF and Hantraye
P: Replicating Huntington’s disease phenotype in experimental
animals. Prog Neurobiol. 59:427–468. 1999.
|
|
7
|
Choi YJ, Om JY, Kim NH, et al: Heat shock
transcription factor-1 suppresses apoptotic cell death and ROS
generation in 3-nitropropionic acid-stimulated striatal cells. Mol
Cell Biochem. 375:59–67. 2013.PubMed/NCBI
|
|
8
|
Dedeoglu A, Ferrante RJ, Andreassen OA,
Dillmann WH and Beal MF: Mice overexpressing 70-kDa heat shock
protein show increased resistance to malonate and 3-nitropropionic
acid. Exp Neurol. 176:262–265. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takayama S, Reed JC and Homma S:
Heat-shock proteins as regulators of apoptosis. Oncogene.
22:9041–9047. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Beere HM, Wolf BB, Cain K, et al:
Heat-shock protein 70 inhibits apoptosis by preventing recruitment
of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2:469–475.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cummings CJ, Mancini MA, Antalffy B,
DeFranco DB, Orr HT and Zoghbi HY: Chaperone suppression of
aggregation and altered subcellular proteasome localization imply
protein misfolding in SCA1. Nat Genet. 19:148–154. 1998. View Article : Google Scholar
|
|
12
|
Kobayashi Y, Kume A, Li M, et al:
Chaperones Hsp70 and Hsp40 suppress aggregate formation and
apoptosis in cultured neuronal cells expressing truncated androgen
receptor protein with expanded polyglutamine tract. J Biol Chem.
275:8772–8778. 2000. View Article : Google Scholar
|
|
13
|
Wyttenbach A, Swartz J, Kita H, et al:
Polyglutamine expansions cause decreased CRE-mediated transcription
and early gene expression changes prior to cell death in an
inducible cell model of Huntington’s disease. Hum Mol Genet.
10:1829–1845. 2001.PubMed/NCBI
|
|
14
|
Mosser DD, Caron AW, Bourget L,
Denis-Larose C and Massie B: Role of the human heat shock protein
hsp70 in protection against stress-induced apoptosis. Mol Cell
Biol. 17:5317–5327. 1997.PubMed/NCBI
|
|
15
|
Jäättelä M, Wissing D, Kokholm K, Kallunki
T and Egeblad M: Hsp70 exerts its anti-apoptotic function
downstream of caspase-3-like proteases. EMBO J. 17:6124–6134.
1998.PubMed/NCBI
|
|
16
|
Mosser DD, Caron AW, Bourget L, et al: The
chaperone function of hsp70 is required for protection against
stress-induced apoptosis. Mol Cell Biol. 20:7146–7159. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ravagnan L, Gurbuxani S, Susin SA, et al:
Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat
Cell Biol. 3:839–843. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gurbuxani S, Schmitt E, Cande C, et al:
Heat shock protein 70 binding inhibits the nuclear import of
apoptosis-inducing factor. Oncogene. 22:6669–6678. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Koga F, Xu W, Karpova TS, McNally JG,
Baron R and Neckers L: Hsp90 inhibition transiently activates Src
kinase and promotes Src-dependent Akt and Erk activation. Proc Natl
Acad Sci USA. 103:11318–11322. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schulte TW, Akinaga S, Soga S, et al:
Antibiotic radicicol binds to the N-terminal domain of Hsp90 and
shares important biologic activities with geldanamycin. Cell Stress
Chaperones. 3:100–108. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zou J, Guo Y, Guettouche T, Smith DF and
Voellmy R: Repression of heat shock transcription factor HSF1
activation by HSP90 (HSP90 complex) that forms a stress-sensitive
complex with HSF1. Cell. 94:471–480. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bagatell R, Paine-Murrieta GD, Taylor CW,
et al: Induction of a heat shock factor 1-dependent stress response
alters the cytotoxic activity of hsp90-binding agents. Clin Cancer
Res. 6:3312–3318. 2000.PubMed/NCBI
|
|
23
|
Sittler A, Lurz R, Lueder G, et al:
Geldanamycin activates a heat shock response and inhibits
huntingtin aggregation in a cell culture model of Huntington’s
disease. Hum Mol Genet. 10:1307–1315. 2001.PubMed/NCBI
|
|
24
|
Bian X, McAllister-Lucas LM, Shao F, et
al: NF-kappa B activation mediates doxorubicin-induced cell death
in N-type neuroblastoma cells. J Biol Chem. 276:48921–48929. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bubici C, Papa S, Pham CG, Zazzeroni F and
Franzoso G: The NF-kappaB-mediated control of ROS and JNK
signaling. Histol Histopathol. 21:69–80. 2006.PubMed/NCBI
|
|
26
|
Dumont A, Hehner SP, Hofmann TG, Ueffing
M, Dröge W and Schmitz ML: Hydrogen peroxide-induced apoptosis is
CD95-independent, requires the release of mitochondria-derived
reactive oxygen species and the activation of NF-kappaB. Oncogene.
18:747–757. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Westerheide SD, Kawahara TL, Orton K and
Morimoto RI: Triptolide, an inhibitor of the human heat shock
response that enhances stress-induced cell death. J Biol Chem.
281:9616–9622. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wyttenbach A: Role of heat shock proteins
during polyglutamine neurodegeneration: mechanisms and hypothesis.
J Mol Neurosci. 23:69–96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Muchowski PJ and Wacker JL: Modulation of
neurodegeneration by molecular chaperones. Nat Rev Neurosci.
6:11–22. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ciocca DR, Clark GM, Tandon AK, Fuqua SA,
Welch WJ and McGuire WL: Heat shock protein hsp70 in patients with
axillary lymph node-negative breast cancer: prognostic
implications. J Natl Cancer Inst. 85:570–574. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dive C and Hickman JA: Drug-target
interactions: only the first step in the commitment to a programmed
cell death? Br J Cancer. 64:192–196. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Seo JS, Park YM, Kim JI, et al: T cell
lymphoma in transgenic mice expressing the human Hsp70 gene.
Biochem Biophys Res Commun. 218:582–587. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wei YQ, Zhao X, Kariya Y, Teshigawara K
and Uchida A: Inhibition of proliferation and induction of
apoptosis by abrogation of heat-shock protein (HSP) 70 expression
in tumor cells. Cancer Immunol Immunother. 40:73–78. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Haldimann P, Muriset M, Vigh L and
Goloubinoff P: The novel hydroxylamine derivative NG-094 suppresses
polyglutamine protein toxicity in Caenorhabditis elegans. J
Biol Chem. 286:18784–18794
|
|
35
|
Neckers L: Hsp90 inhibitors as novel
cancer chemotherapeutic agents. Trends Mol Med. 8:S55–S61. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Panaretou B, Prodromou C, Roe SM, et al:
ATP binding and hydrolysis are essential to the function of the
Hsp90 molecular chaperone in vivo. EMBO J. 17:4829–4836. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Grenert JP, Johnson BD and Toft DO: The
importance of ATP binding and hydrolysis by hsp90 in formation and
function of protein heterocomplexes. J Biol Chem. 274:17525–17533.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schneider C, Sepp-Lorenzino L, Nimmesgern
E, et al: Pharmacologic shifting of a balance between protein
refolding and degradation mediated by Hsp90. Proc Natl Acad Sci
USA. 93:14536–14541. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Connell P, Ballinger CA, Jiang J, et al:
The co-chaperone CHIP regulates protein triage decisions mediated
by heat-shock proteins. Nat Cell Biol. 3:93–96. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu W, Marcu M, Yuan X, Mimnaugh E,
Patterson C and Neckers L: Chaperone-dependent E3 ubiquitin ligase
CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad
Sci USA. 99:12847–12852. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wacker JL, Huang SY, Steele AD, et al:
Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar
aggregates in a mouse model of Huntington’s disease. J Neurosci.
29:9104–9114. 2009.
|
|
42
|
Shen HY, He JC, Wang Y, Huang QY and Chen
JF: Geldanamycin induces heat shock protein 70 and protects against
MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem.
280:39962–39969. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Beal MF: Energetics in the pathogenesis of
neurodegenerative diseases. Trends Neurosci. 23:298–304. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gu M, Gash MT, Mann VM, Javoy-Agid F,
Cooper JM and Schapira AH: Mitochondrial defect in Huntington’s
disease caudate nucleus. Ann Neurol. 39:385–389. 1996.
|
|
45
|
Browne SE, Bowling AC, MacGarvey U, et al:
Oxidative damage and metabolic dysfunction in Huntington’s disease:
selective vulnerability of the basal ganglia. Ann Neurol.
41:646–653. 1997.
|
|
46
|
Sawa A, Wiegand GW, Cooper J, et al:
Increased apoptosis of Huntington disease lymphoblasts associated
with repeat length-dependent mitochondrial depolarization. Nat Med.
5:1194–1198. 1999. View
Article : Google Scholar
|
|
47
|
Beal MF, Brouillet E, Jenkins BG, et al:
Neurochemical and histologic characterization of striatal
excitotoxic lesions produced by the mitochondrial toxin
3-nitropropionic acid. J Neurosci. 13:4181–4192. 1993.
|
|
48
|
Hamilton BF and Gould DH: Nature and
distribution of brain lesions in rats intoxicated with
3-nitropropionic acid: a type of hypoxic (energy deficient) brain
damage. Acta Neuropathol. 72:286–297. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Brouillet E, Hantraye P, Ferrante RJ, et
al: Chronic mitochondrial energy impairment produces selective
striatal degeneration and abnormal choreiform movements in
primates. Proc Natl Acad Sci USA. 92:7105–7109. 1995. View Article : Google Scholar
|
|
50
|
Palfi S, Ferrante RJ, Brouillet E, et al:
Chronic 3-nitropropionic acid treatment in baboons replicates the
cognitive and motor deficits of Huntington’s disease. J Neurosci.
16:3019–3025. 1996.PubMed/NCBI
|
|
51
|
Gabai VL, Meriin AB, Yaglom JA, Volloch VZ
and Sherman MY: Role of Hsp70 in regulation of stress-kinase JNK:
implications in apoptosis and aging. FEBS Lett. 438:1–4. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Meriin AB, Yaglom JA, Gabai VL, et al:
Protein-damaging stresses activate c-Jun N-terminal kinase via
inhibition of its dephosphorylation: a novel pathway controlled by
HSP72. Mol Cell Biol. 19:2547–2555. 1999.PubMed/NCBI
|
|
53
|
Kyriakis JM and Avruch J: Protein kinase
cascades activated by stress and inflammatory cytokines. Bioessays.
18:567–577. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Park HS, Lee JS, Huh SH, Seo JS and Choi
EJ: Hsp72 functions as a natural inhibitory protein of c-Jun
N-terminal kinase. EMBO J. 20:446–456. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lee JS, Lee JJ and Seo JS: HSP70
deficiency results in activation of c-Jun N-terminal Kinase,
extracellular signal-regulated kinase, and caspase-3 in
hyperosmolarity-induced apoptosis. J Biol Chem. 280:6634–6641.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li H, Liu L, Xing D and Chen WR:
Inhibition of the JNK/Bim pathway by Hsp70 prevents Bax activation
in UV-induced apoptosis. FEBS Lett. 584:4672–4678. 2010. View Article : Google Scholar : PubMed/NCBI
|