Introduction
Breast cancer is the most prevalent malignancy and
was expected to be the second leading cause of cancer-related
mortality among women in the United States in 2013 (1). Triple-negative breast cancer (TNBC),
accounting for approximately 15% of breast cancer cases, is an
immunohistochemically-defined phenotype and is estrogen receptor
(ER)-negative, progesterone receptor (PR)-negative and is also
negative for HER2 expression (2).
Currently, TNBC remains a tremendous clinical challenge due to its
aggressive behavior and relatively unfavorable prognosis (3). It is not only associated with an
early relapse and a high risk of visceral and central nervous
system metastases, but also with an early mortality during the
first 3–5 years of follow up (4–7).
Since TNBC is refractory to endocrine therapies, such as tamoxifen
and aromatase inhibitors, as well as current HER2-targeted
therapies, such as trastuzumab, chemotherapy remains the mainstay
of treatment for patients with TNBC (2). Therefore, the improvement of
chemotherapeutic efficacy is of particular importance to the
optimal management of TNBC.
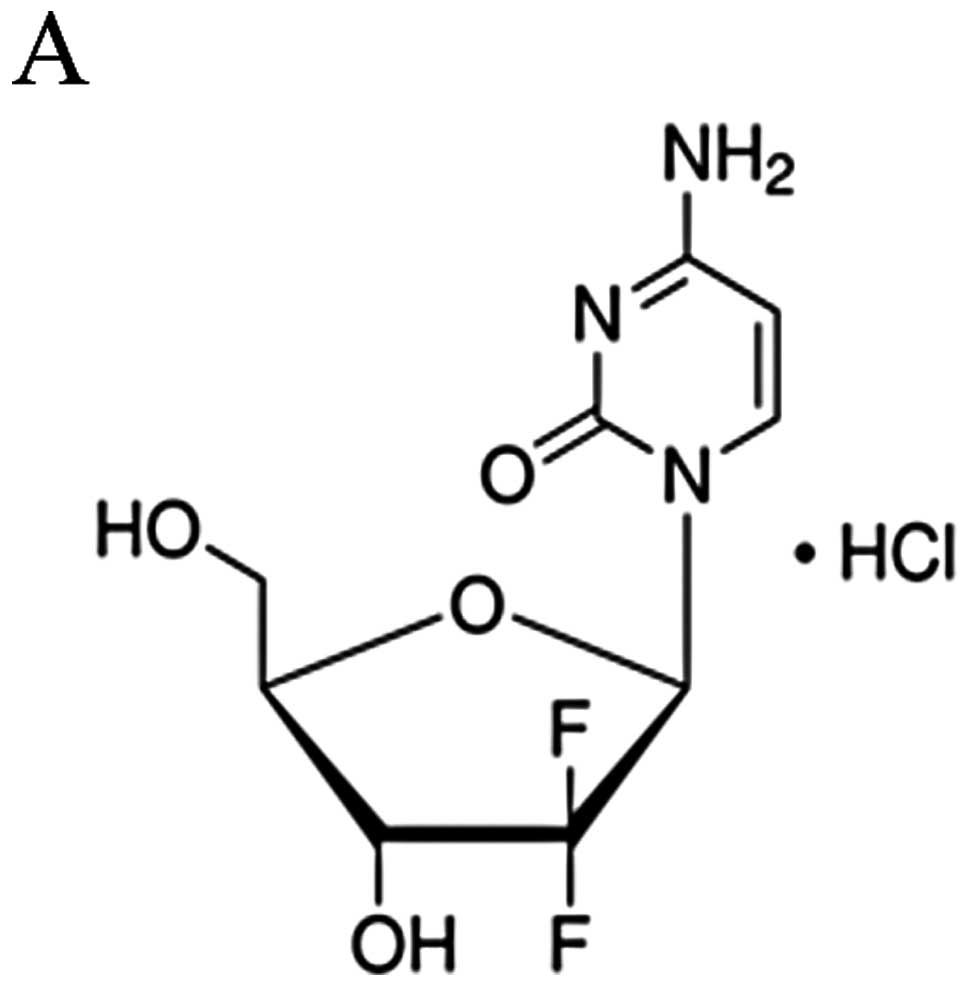
Gemcitabine is a deoxycytidine analogue, which
competes with deoxycytidine triphosphate for incorporation into
DNA, and thus inhibits DNA synthesis mainly in its triphosphate
form (8). It has been used in a
wide spectrum of malignancies, both as a single agent and in
combination with other cytotoxic drugs. As a single agent, the
response rates of gemcitabine range from 14 to 37% as a first-line
therapy for advanced breast cancer and 12–30% in patients
previously treated with anthracyclines and/or taxanes (9). In combination with taxanes and/or
anthracyclines as doublets or triplets, gemcitabine has been
determined as a first-line therapy in clinical practice for the
treatment of patients with metastatic breast cancer, achieving
overall response rates ranging from 39 to 75% (10–12). However, intrinsic and acquired
resistance to gemcitabine is common, whereas the molecular
mechanisms underlying chemoresistance to gemcitabine are not yet
fully understood (13).
Autophagy is an evolutionarily well-conserved
lysosomal degradation pathway that maintains cytoplasmic
homeostasis by eliminating protein aggregates and damaged
organelles (14). It is
characterized by the formation of double-membraned autophagosomes
that engulf cytoplasmic components and eventually fuse with
lysosomes to form autolysosomes for the degradation of the contents
(15). Considering the role of
autophagy in helping cells to adapt to starvation or unfavorable
stress conditions (e.g., hypoxia, radiation therapy and cytotoxic
drugs), autophagy is usually identified as a cellular pro-survival
mechanism (16). By contrast,
under certain circumstances, autophagy may also lead to cell death
directly, causing autophagic cell death or type II programmed cell
death (PCD; type II PCD), which is distinct from apoptosis (type I
PCD) or necrosis (type III PCD) (17–20).
As regards the connections between the apoptotic and
autophagic processes, a complex interplay between them has come to
light, taking into account that numerous genes, such as p53, the
death-associated protein kinase (DAPK) family and Bcl-2 family
members, are shared between these two response machineries. This
suggests that apoptosis and autophagy jointly seal the fate of
cells (21).
A number of studies have demonstrated that
gemcitabine induces autophagy in pancreatic cancer cells and the
modulation of autophagy may improve the efficacy of gemcitabine
(22–25). However, whether autophagy is
associated with gemcitabine treatment for TNBC remains unknown. In
the current study, we aimed to explore whether gemcitabine
treatment induces autophagy in triple-negative MDA-MB-231 breast
cancer cells, and establish the function of such inducible
autophagy in the therapeutic efficacy of gemcitabine. Moreover, the
possible interplay between autophagy and apoptosis in
triple-negative MDA-MB-231 cells is also explored.
Materials and methods
Reagents and antibodies
The chemicals used in the present study were the
following: gemcitabine (G6423), chloroquine diphosphate salt
(C6628) and monodansylcadaverine (MDC; 30432) (all from
Sigma-Aldrich, St. Louis, MO, USA). The Annexin V FITC Apoptosis
Detection kit (88-8005) and the propidium iodide (PI) staining
solution (00-6990) were from eBioscience, Inc. (San Diego, CA,
USA). The primary antibodies were the following: anti-LC3B antibody
produced in rabbit (L7543; Sigma-Aldrich), beclin 1 antibody
(NB500-249; Novus Biologicals, Littleton, USA), anti-Bax rabbit
polyclonal antibody (AB20073a), anti-Bcl-2L1 rabbit polyclonal
antibody (AB20306a) (both from Life Science Products &
Services), anti-p53 (WBL, K0181-3) and mTOR rabbit antibody (#2972;
Cell Signaling Technology, Danvers, MA, USA). The secondary
antibodies, HRP-conjugated goat anti-rabbit (sc-2357) and goat
anti-mouse IgG (sc-2371), were obtained from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell culture
The monolayer culture of MDA-MB-231 cells [obtained
from the American Type Culture Collection (ATCC), Manassas, VA,
USA] was maintained in L-15 medium supplemented with 10% fetal
bovine serum, 100 μg/ml streptomycin and 100 U/ml penicillin in a
humid incubator without CO2 at 37°C.
MTT assay
Cells seeded in 96-well plates were allowed to
attach overnight, and treated with various concentrations of the
reagents for the indicated periods of time. Subsequently, 20 μl MTT
(5 mg/ml) were added to each well for 4 h of incubation. The
supernatant was then removed and 150 μl DMSO were added to each
well to dissolve the blue-purple formazan crystals. The plates were
agitated for 15 min. Absorbance values at 490 nm were detected with
a model 680 MicroPlate Reader (Bio-Rad, Hercules, CA, USA).
Flow cytometry with Annexin V-FITC
(AV-FITC)/PI staining
The cells were seeded at 4×105 cells/well
in 6-well plates and allowed to attach overnight. The cells were
then treated for the indicated periods of time and were then
collected and dyed with AV-FITC and PI according to the
instructions of the manufacturer. Cell viability was analyzed by
flow cytometry (BD FACSCalibur; BD Biosciences, San Jose, CA,
USA).
Western blot analysis
The cells were treated for the indicated periods of
time. Both the detached and attached cells were collected, and
whole cell lysates were prepared. Approximately 30 μg total
proteins were separated by SDS-PAGE with a proper concentration and
transferred onto PVDF membranes (Bio-Rad). The PVDF membranes were
blocked by 5% non-fat dry milk for 90 min at room temperature, and
were then incubated with primary antibodies overnight with light
agitation at 4°C and secondary antibodies for 2 h at room
temperature. Visualization was carried out with
electrochemiluminescence (ECL; Bio-Rad).
Visualization of MDC-labeled
vacuoles
The cells were cultured on coverslips in 24-well
plates and treated for the indicated periods of time. Subsequently,
fresh medium-containing MDC (50 μmol/l) was added to the plates for
15 min. Subsequently, the cells were fixed using a freshly-prepared
4% formaldehyde solution for 10 min at room temperature. The
specimens were analyzed under a BX61W1-FV1000 confocal microscope
(Olympus, Tokyo, Japan).
EGFP-LC3 plasmid transfection
The EGFP-LC3 plasmids were transiently transfected
into the cells using Lipofectamine 2000™ (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the instructions of
the manufacturer. After 24 h, the cells were treated with the
chemicals for the indicated periods of time, and then fixed with 4%
formaldehyde solution for 10 min at room temperature. The
fluorescence of EGFP was analyzed using a BX61W1-FV1000 confocal
microscope (Olympus). The number of LC3-II+ puncta was
counted manually in confocal images from random fields to quantify
the EGFP-LC3-tagged autophagosomes.
Transmission electron microscopy
(TEM)
The treated cells were fixed for 1 h in ice-cold 2.5
glutaraldehyde and 1% osmium tetroxide for a further 1 h. The
samples were stained with 4% uranyl acetate for 30 min, dehydrated
through a graded series of ethanol solutions (ranging from 50 to
100%) and embedded in Epon resin. Ultrathin sections (120 nm) were
post-stained with uranyl acetate and lead citrate, and were then
examined under a TECNAI 10 transmission electron microscope
(Philips, Amsterdam, The Netherlands) at 60 kV.
RNA interference
The cells were transfected with either scrambled
siRNA or beclin 1-targeting siRNA using interfering siRNA
transfection reagent (PolyPlus Transfection SA, Illkirch, France)
according to the recommendations of manufacturer. The si-BECN1
targeting sequence was UUC AAC ACU CUU CAG CUC AUC AUC C and the
scrambled siRNA sequence was UUC UCC GAA CGU GUC ACG UTT. All
siRNAs were obtained from GenePharma, Ltd. (Shanghai, China).
Statistical analyses
To determine whether the difference between 2 groups
was statistically significant, Student’s t-tests were performed.
The significance level was set at P≤0.05.
Results
Gemcitabine decreases cell viability and
induces apoptosis in MDA-MB-231 cells
The MDA-MB-231 cells were treated with gemcitabine
at various concentrations, and cell viability was assessed with by
MTT assay. The results revealed that cell viability (% of the
control) gradually decreased with the increasing concentrations of
gemcitabine (Fig. 1B). The half
maximal inhibitory concentration (IC50) of 48 h of
treatment with gemcitabine in the MDA-MB-231 cells was 1.2 μg/ml.
In this study, 1 μg/ml was selected as the treatment concentration
in the subsequent experiments. Flow cytometry with AV-FITC/PI
staining was used to determine the apoptotic rate of the MDA-MB-231
cells following treatment with gemcitabine for different periods of
time. The AV-FITC-positive cells were defined as apoptotic and
double-negative cells were considered viable. The percentage of
AV-FITC-positive cells increased with the increasing duration of
gemcitabine treatment (Fig.
1C).
Gemcitabine induces mTOR-independent
autophagy in triple-negative MDA-MB-231 cells
To evaluate gemcitabine-induced autophagy, we first
tested the conversion of LC3-I into LC3-II in the MDA-MB-231 cells.
As shown in Fig. 2A, gemcitabine
induced the conversion of LC3-I into LC3-II in a time-dependent
manner. Compared with that at 24 h, the ratio of LC3-II/β-actin at
48 h was decreased, possibly due to the fact that autophagy is a
highly dynamic, multi-step process. We further examined the
autophagic flux by monitoring the protein level of LC3-II relative
to β-actin after 24 h of treatment with gemcitabine with or without
chloroquine (CQ) (Fig. 2B).
LC3-II protein expression was markedly increased following
treatment with CQ, and the relative level of LC3-II vs. actin was
significantly higher than that observed with CQ treatment alone
following treatment with gemcitabine plus CQ. No apparent change in
the mTOR protein levels was observed following treatment with
various concentrations of gemcitabine, indicating that the
gemcitabine-induced autophagy in MDA-MB-231 cells was possibly
mTOR-independent (Fig. 2C).
 | Figure 2Gemcitabine induces mTOR-independent
autophagy in triple-negative MDA-MB-231 cells. (A) The conversion
of Lc3-I (18 KDa) into LC3-II (16 KDa) was assayed in cells treated
with gemcitabine (1 μg/ml) for 0 h (untreated control) or 12, 24
and 48 h. (B) Cells were treated with gemcitabine, chloroquine (CQ;
5 μM; added prior to treatment with gemcitabine for 1 h), or
gemcitabine plus CQ for 0 h (control) or 24 h, then the conversion
of Lc3-I into LC3-II was assessed. (C) The levels of mTOR protein
were assessed following treatment with gemcitabine (0, 0.5 and 1, 2
μg/ml) for 24 h. (D) Cells exposed to gemcitabine, CQ, or
gemcitabine plus CQ for 0 h (control) or 24 h were treated with
monodansylcadaverine (MDC) (50 μmol/l) for 15 min and the samples
were examined under a confocal microscope. (E) Cells transiently
transfected with EGFP-LC3 plasmids were exposed to gemcitabine, CQ,
or gemcitabine plus CQ for 0 h (control) or 24 h. A confocal
microscope was used to measure EGFP fluorescence distribution. The
punctate EGFP-LC3 was indicative of autophagosomes. The number of
LC3-II+ puncta was counted in at least 75 cells from
random fields and the fold change was calculated by normalizing to
the amount of the control group. (F) Cells were treated with
gemcitabine for 0 (control) or 24 h. The ultrastructural
characteristics of the cells were analyzed by transmission electron
microscopy (TEM). Autophagical vacuoles with typical double-layer
membrane containing remnants of organelles are highlighted by white
arrows. *P<0.05;**P<0.01. Gem,
gemcitabine; Ctrl, control. |
MDC is a selective fluorescent dye used to observe
for autophagic vacuoles. As shown in Fig. 2D, gemcitabine treatment increased
the number of bright blue puncta. Moreover, the number of
MDC-labeled puncta was markedly increased and the average puncta
volume was significantly larger following treatment with
gemcitabine plus CQ. The transient transfection of EGFP-LC3
plasmids was performed to further determine gemcitabine-induced
autophagy in the MDA-MB-231 cells. The number of EGFP-LC3 puncta
per cell in the group treated with gemcitabine plus CQ was markedly
higher than that observed in the cells treated with gemcitabine
alone or CQ alone (Fig. 2E), thus
indicating the increased autophagic flux level in triple-negative
MDA-MB-231 cells treated with gemcitabine. Finally, TEM confirmed
that numerous autophagic vacuoles with a typical double-layer
membrane containing organelle remnants were present in the
gemcitabine-treated MDA-MB-231 cells as opposed to the untreated
(control) cells (Fig. 2F). Our
results indicated that gemcitabine treatment induce autophagy in
TNBC cells.
Gemcitabine-induced autophagy plays a
cytoprotective role in triple-negative MDA-MB-231 cells
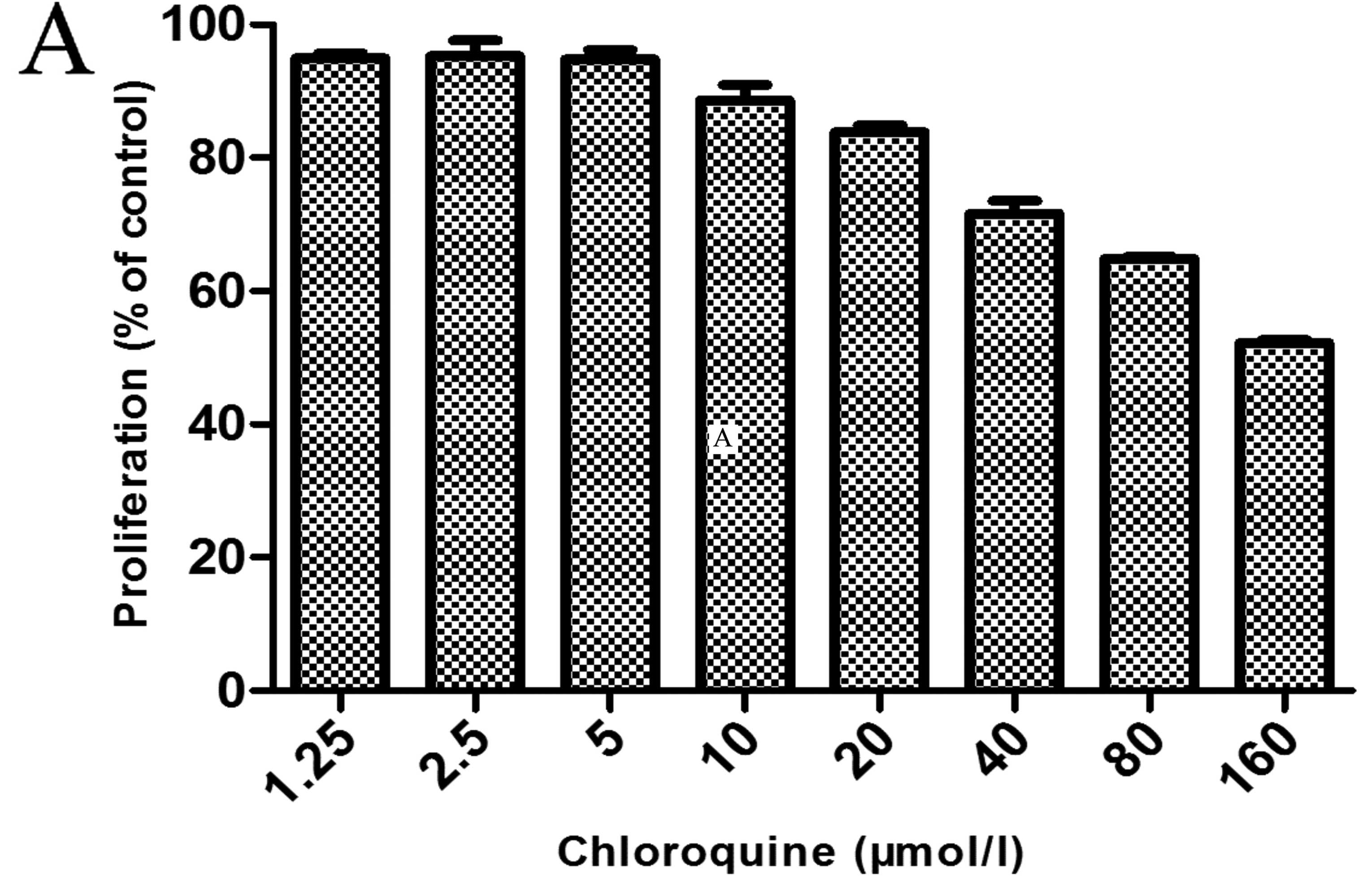
As shown by the results of MTT assay, CQ treatment
alone (5 μM) had little toxic effect on the MDA-MB-231 cells
(Fig. 3A). However, the
inhibition of autophagy by CQ significantly enhanced the inhibitory
effects of gemcitabine on cell viability at 12 and 48 h (Fig. 3B). Flow cytometry revealed that
after the cells were treated with gemcitabine plus CQ, the number
of apoptotic markedly increased compared with the cells treated
with gemcitabine alone (Fig.
3C).
We further inhibited autophagy by silencing the
beclin 1 gene, as beclin 1 is one of the essential
autophagy-related genes (Atgs) (26). Transfection with beclin
1-targeting siRNA resulted in a marked reduction in beclin 1
protein expression, while the scrambled siRNA-transfected group
showed no change (Fig. 4A). As
shown in Fig. 4B, beclin 1
knockdown markedly reduced the viability of cells treated with
gemcitabine at 12 and 48 h. Of note, si-BECN1 alone also inhibited
cell viability, thus indicating that beclin 1 plays a positive role
in the growth of TNBC cells. As shown by flow cytometry, treatment
with scrambled siRNA plus gemcitabine produced results that were
comparable to those that were obtained by treatment with
gemcitabine alone, while treatment with gemcitabine plus si-BECN1
led to a more significant increase in the percentage of apoptotic
cells (Fig. 4C). These results
demonstrate that beclin 1-mediated autophagy induced by gemcitabine
contributes to TNBC cell growth and survival.
Autophagy inhibition by CQ shifts the
expression levels of p53 and Bcl-2 family proteins in favor of
promoting apoptosis
The p53 and Bcl-2 family member proteins play a
critical role in the intrinsic apoptotic pathway, which regulates
apoptosis by modulating mitochondrial outer membrane integrity
(27).
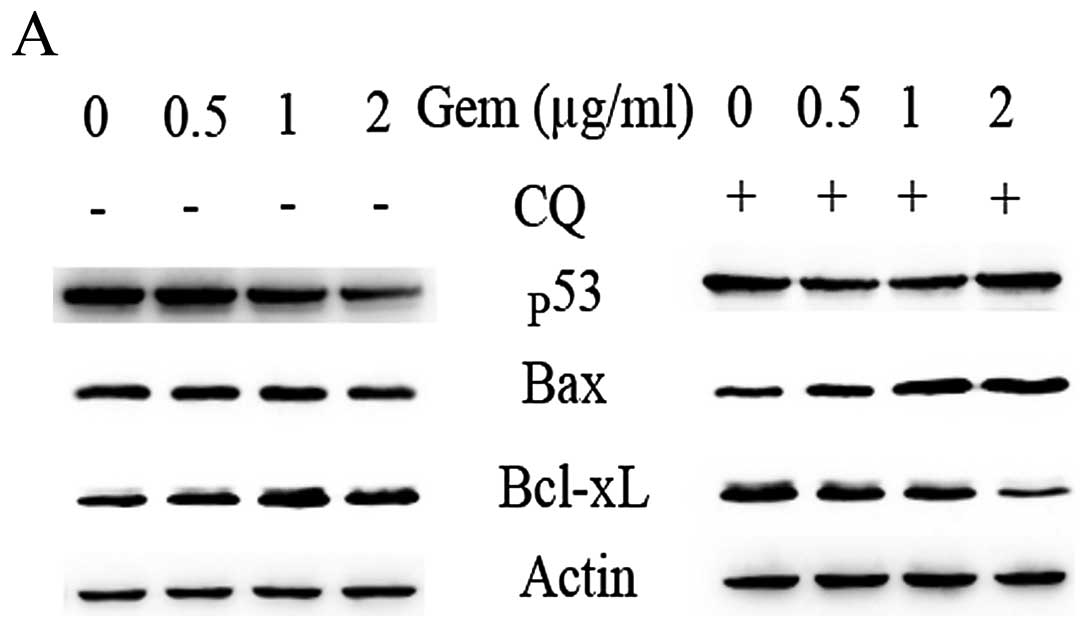
We investigated the protein levels of p53, the
anti-apoptotic Bcl-xL gene and the pro-apoptotic Bax gene. As shown
by our results, the Bcl-xL protein levels increased, while the p53
and Bax protein levels decreased in a concentration-dependent
manner following treatment with gemcitabine (Fig. 5A, left panel). Following treatment
with gemcitabine plus the autophagic inhibitor, CQ, the Bcl-xL
protein levels markedly decreased with a concomitant increase in
the p53 and pro-apoptotic Bax levels in a concentration-dependent
manner, which was in contrast to what was observed following
treatment with gemcitabine alone (Fig. 5A, right panel). We further
assessed the relative Bax/Bcl-xL ratio following treatment with
gemcitabine with or without CQ. With the increasing gemcitabine
concentration, the relative Bax/Bcl-xL ratio slightly decreased,
while it markedly increased following treatment with CQ plus
gemcitabine (Fig. 5B).
Discussion
A number of studies have demonstrated that
anti-cancer therapy induces autophagy in cancer cells, while there
is an ongoing debate on the association between autophagy and
therapeutic efficacy (28). A
series of studies have reported that chemotherapeutic drugs induce
autophagy, promoting the survival of cancer cells (29–31), thus suggesting that autophagy is
involved in the development of resistance to chemotherapy.
CQ has been extensively used as an autophagic
inhibitor that prevents the acidification of the lysosome, thus
blocking the late stage of the autophagy process (32). It is noteworthy to mention that
some clinical trials testing CQ-mediated autophagy inhibition,
using hydroxychloroquine (HCQ) combined with ixabepilone for
metastatic breast cancer and CQ for ductal carcinoma in
situ, have been carried out (http://www.clinicaltrials.gov/ct2/show/results/NCT00765765)
or are ongoing (http://www.clinicaltrials.gov/ct2/show/NCT01023477);
this indicates that chloroquine is clinically applicable. Our
results demonstrated that the addition of CQ to gemcitabine
treatment greatly enhanced the inhibitory effect on cell viability
and increased cell apoptosis in the MDA-MB-231 cells. Our data
support the therapeutic potential of CQ in combination with
gemcitabine for enhancing the efficacy of gemcitabine in TNBC.
Beclin 1 is an essential Atg that signals the onset
of the autophagy process (33).
In our study, the silencing of beclin 1 significantly enhanced the
inhibitory effect of gemcitabine on the growth and viability of
MDA-MB-231 cells, indicating that beclin 1-mediated autophagy plays
a cytoprotective role in TNBC cells. Of note, treatment with
si-BECN1 alone markedly inhibited the viability of MDA-MB-231
cells, indicating that the beclin 1 gene plays a positive role in
the growth of TNBC cells. By contrast, the study by Liang et
al (26) produced different
results. They evaluated the effects of beclin 1 on the growth of
ER+ MCF-7 cells before and after stably transfecting
MCF-7 cells with a vector containing flag-beclin 1. They also
evaluated cell proliferation and tumor formation in nude mice with
MCF-7 tumors. The number of MCF-7 beclin 1 clones was significantly
lower compared with the control group. Therefore, the authors
concluded that beclin 1 acts as a negative regulator of mammary
cell growth and tumorigenesis (26). Thus, it can be hypothesized that
the effects of beclin 1 on the growth of breast cancer cells may
depend on the status of steroid hormone receptors.
There are multiple connections between the apoptotic
and autophagic processes (21).
In this study, we demonstrated that pre-treatment with CQ (to
inhibit autophagy) prior to gemcitabine treatment shifted the p53
protein level and the Bax/Bcl-xL ratio in favor of promoting
apoptosis. This indicates that gemcitabine-induced autophagy
inhibits the apoptotic process to a certain extent, and that the
inhibition of the induced autophagy may promote apoptosis,
resulting in an improved efficacy of gemcitabine in TNBC cells. Our
results also suggest that the inhibitory effect of autophagy on
apoptosis is another mechanism behind autophagy-induced
cytoprotection other than the basic cellular functions of autophagy
in triple-negative MDA-MB-231 cells.
In conclusion, the present study clearly
demonstrates that gemcitabine induces mTOR-independent autophagy in
triple-negative MDA-MB-231 cells and suggests that
gemcitabine-induced autophagy plays an active role in the
refractory response of TNBC cells to gemcitabine. Our results also
indicate that the downregulation of cellular apoptotic levels by
autophagy constitutes another mechanism responsible for
autophagy-induced cytoprotection in gemcitabine-exposed TNBC cells.
Thus, the combination of an autophagic inhibitor with gemcitabine
may be a promising approach to promote greater therapeutic efficacy
in patients with TNBC. However, more preclinical trials are
required to further determine the positive effects of autophagic
inhibitors on gemcitabine treatment in patients with TNBC.
Acknowledgements
We gratefully acknowledge the help of Professor Wang
Xiaojian (The Institute of Molecular Immunology, Zhejiang
University, Hangzhou, China) for kindly providing the reagents.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2
|
Irvin WJ Jr and Carey LA: What is
triple-negative breast cancer? Eur J Cancer. 44:2799–2805. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bauer KR, Brown M, Cress RD, Parise CA and
Caggiano V: Descriptive analysis of estrogen receptor
(ER)-negative, progesterone receptor (PR)-negative, and
HER2-negative invasive breast cancer, the so-called triple-negative
phenotype: a population-based study from the California cancer
Registry. Cancer. 109:1721–1728. 2007. View Article : Google Scholar
|
|
4
|
Lin NU, Claus E, Sohl J, Razzak AR,
Arnaout A and Winer EP: Sites of distant relapse and clinical
outcomes in patients with metastatic triple-negative breast cancer:
high incidence of central nervous system metastases. Cancer.
113:2638–2645. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Haffty BG, Yang Q, Reiss M, et al:
Locoregional relapse and distant metastasis in conservatively
managed triple negative early-stage breast cancer. J Clin Oncol.
24:5652–5657. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dent R, Trudeau M, Pritchard KI, et al:
Triple-negative breast cancer: clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kassam F, Enright K, Dent R, et al:
Survival outcomes for patients with metastatic triple-negative
breast cancer: implications for clinical practice and trial design.
Clin Breast Cancer. 9:29–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang P, Chubb S, Hertel LW, Grindey GB
and Plunkett W: Action of 2′,2′-difluorodeoxycytidine on DNA
synthesis. Cancer Res. 51:6110–6117. 1991.
|
|
9
|
Silvestris N, D’Aprile M, Andreola G,
Locopo N, Marini L, Crucitta E, De Lena M and Lorusso V: Rationale
for the use of gemcitabine in breast cancer (Review). Int J Oncol.
24:389–398. 2004.PubMed/NCBI
|
|
10
|
Passardi A, Massa I, Zoli W, et al: Phase
II study of gemcitabine, doxorubicin and paclitaxel (GAT) as
first-line chemotherapy for metastatic breast cancer: a
translational research experience. BMC Cancer. 6:762006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
O’Shaughnessy JA, Pluenneke R, Sternberg
J, Khandelwal P, Ilegbodu D and Asmar L: Phase II trial of weekly
docetaxel/gemcitabine as first-line chemotherapy in patients with
locally recurrent or metastatic breast cancer. Clin Breast Cancer.
6:505–510. 2006.
|
|
12
|
Tomao S, Romiti A, Tomao F, et al: A phase
II trial of a biweekly combination of paclitaxel and gemcitabine in
metastatic breast cancer. BMC Cancer. 6:1372006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hernández-Vargas H, Rodríguez-Pinilla SM,
Julián-Tendero M, et al: Gene expression profiling of breast cancer
cells in response to gemcitabine: NF-kappaB pathway activation as a
potential mechanism of resistance. Breast Cancer Res Treat.
102:157–172. 2007.PubMed/NCBI
|
|
14
|
Mizushima N and Komatsu M: Autophagy:
renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rubinsztein DC, Codogno P and Levine B:
Autophagy modulation as a potential therapeutic target for diverse
diseases. Nat Rev Drug Discov. 11:709–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kroemer G and Levine B: Autophagic cell
death: the story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baehrecke EH: Autophagy: dual roles in
life and death? Nat Rev Mol Cell Biol. 6:505–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kroemer G and Jäättelä M: Lysosomes and
autophagy in cell death control. Nat Rev Cancer. 5:886–897. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gozuacik D and Kimchi A: Autophagy and
cell death. Curr Top Dev Biol. 78:217–245. 2007. View Article : Google Scholar
|
|
21
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Donohue E, Thomas A, Maurer N, et al: The
autophagy inhibitor verteporfin moderately enhances the antitumor
activity of gemcitabine in a pancreatic ductal adenocarcinoma
model. J Cancer. 4:585–596. 2013. View
Article : Google Scholar
|
|
23
|
Pardo R, Lo Ré A, Archange C, et al:
Gemcitabine induces the VMP1-mediated autophagy pathway to promote
apoptotic death in human pancreatic cancer cells. Pancreatology.
10:19–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mukubou H, Tsujimura T, Sasaki R and Ku Y:
The role of autophagy in the treatment of pancreatic cancer with
gemcitabine and ionizing radiation. Int J Oncol. 37:821–828.
2010.PubMed/NCBI
|
|
25
|
Papademetrio DL, Cavaliere V, Simunovich
T, et al: Interplay between autophagy and apoptosis in pancreatic
tumors in response to gemcitabine. Target Oncol. Apr 16–2013.(Epub
ahead of print).
|
|
26
|
Liang XH, Jackson S, Seaman M, et al:
Induction of autophagy and inhibition of tumorigenesis by Beclin 1.
Nature. 402:672–676. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jain MV, Paczulla AM, Klonisch T, et al:
Interconnections between apoptotic, autophagic and necrotic
pathways: implications for cancer therapy development. J Cell Mol
Med. 17:12–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li J, Hou N, Faried A, Tsutsumi S and
Kuwano H: Inhibition of autophagy augments 5-fluorouracil
chemotherapy in human colon cancer in vitro and in vivo model. Eur
J Cancer. 46:1900–1909. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu D, Yang Y, Liu Q and Wang J:
Inhibition of autophagy by 3-MA potentiates cisplatin-induced
apoptosis in esophageal squamous cell carcinoma cells. Med Oncol.
28:105–111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun WL, Chen J, Wang YP and Zheng H:
Autophagy protects breast cancer cells from epirubicin-induced
apoptosis and facilitates epirubicin-resistance development.
Autophagy. 7:1035–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Carew JS, Medina EC, Esquivel JA II, et
al: Autophagy inhibition enhances vorinostat-induced apoptosis via
ubiquitinated protein accumulation. J Cell Mol Med. 14:2448–2459.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar
|