Introduction
Neurofibromatosis type 1 (NF1) (OMIM 162200) is a
progressive autosomal dominant inherited disease and is one of the
most widespread genetic disorders worldwide with a prevalence of 1
in 2500- to -3000 live births (1). The clinical characteristics in the
NF1 diagnostic criteria include café-au-lait spots, neurofibromas,
Lisch nodules, intertriginous freckling, typical osseous lesions
and optic pathway gliomas (2). At
least 78% of patients who fulfill the NIH diagnostic criteria for
NF1 have NF1 gene mutations (3). Moreover, 5–10% of the cases are
caused by a deletion in the NF1 gene (4). However, the positive rates of the
NF1 mutation findings in clinical diagnostic laboratories
vary considerably according to the proportions of the samples from
clinically definite or suspected patients. NF1 is located on
17q11.2 and spans 28,2751 bp in length. This gene contains 60 exons
and encodes neurofibromin, a key component in the RAS-MAPK
signaling pathway. The RAS-MAPK pathway regulates the proliferation
and differentiation of neuronal cells and myocytes (5). Neurofibromin functions as an
inhibitor of RAS activation and as a tumor suppressor with a
central region that is homologous to RAS-GTPase activation proteins
(GAPs) (6). Mutations in the
NF1 gene cause a loss in neurofibromin function, resulting
in downstream cell growth activation (7–9).
Previous studies have reported over 1,400 different mutations due
to the high mutation rate of the NF1 gene. A high number of
these mutations arise as novel mutations; however, there is no hot
spot for the pathogenic variations of NF1 (3,10,11).
In this study, we performed a retrospective review
of 378 cases and compiled the mutations identified at both the
genomic and mRNA level. We present 127 different mutations of the
NF1 gene; 54 of which are novel mutations. In addition, the
deletion of the NF1 gene was detected in 5 cases using
fluorescence in situ hybridization (FISH) or the comparative
genomic hybridization (CGH) array method. With the advent of the
genomic DNA and cDNA sequencing approach, splicing abnormalities
caused by exonic variants were captured and presented in our data.
In addition, 7 of these 13 exonic mutations were novel. Of note,
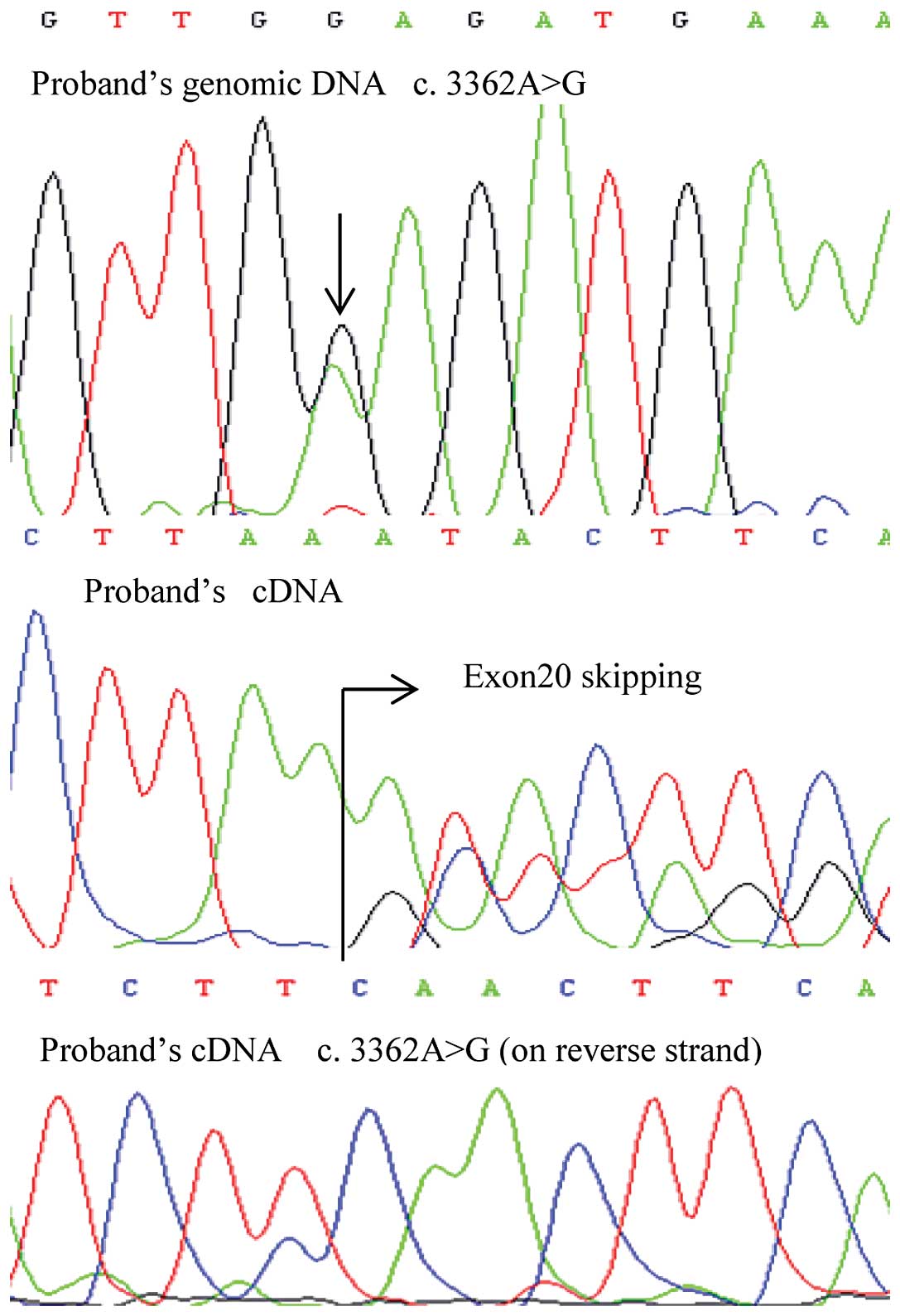
one of these mutations, c.3362A>G, produced mosaicism of a point
mutation and mutant exon skipping at the mRNA level.
Accurate splicing of pre-mRNA is not only controlled
by the 5′/3′ splice sites (ss), but also by other cis-acting
elements, as well as trans-acting factors, i.e., SR proteins and
heterogeneous nuclear ribonucleoproteins (hnRNPs). These cis-acting
elements generally include the splicing enhancers related to
exon-inclusion enhancement, splicing silencers related to
exon-inclusion inhibition, the intronic branch point and the
polypyrimidine tract (12,13).
Although the NF1 mutation spectrum continues to expand,
studies investigating these splicing aberrations are limited
(14–18). Thus, integrated analyses using the
bioinformatics tools were further applied to provide insight into
the mechanisms of these splicing defects caused by exonic variants,
as well as other intronic variants at non-consensus splice
sites.
Patients and methods
Patients
A total of 378 cases were referred for NF1
gene testing in our laboratory from January, 2006 to May, 2013 and
were recruited in this study. The subjects consisted of 338
unrelated probands with clinically definite or suspected NF1
diagnosis and 40 family members. Consent forms were signed by the
patients or authorized representatives. All cases underwent a
NF1 gene-sequencing test developed in our laboratory, which
was approved by the Ethics Committees at the University of Oklahoma
Health Sciences Center, Oklahoma City, OK, USA.
Mutation screening by Sanger
sequencing
Genomic DNA was isolated from peripheral blood
samples of the patients using the QIAamp DNA Mini kit (Qiagen,
Valencia, CA, USA). mRNA was isolated from the peripheral blood
samples using the QIAamp RNA Blood Mini kit (Qiagen). First-strand
cDNA was reverse-transcribed using the SuperScript III Reverse
Transcriptase kit and random primers (both from Invitrogen,
Carlsbad, CA, USA). PCR was performed using specific primers
targeting the mRNA coding region of the NF1 gene. For
confirmation, exon-specific genomic DNA sequencing was also
performed using specific primers. Primer information will be
provided upon request. Sanger sequencing was performed using the
BigDye Terminator v3.1 Cycle Sequencing kit (Life Technologies,
Foster City, CA, USA) and an ABI 3130xl genetic analyzer (Life
Technologies). Sequences were analyzed using Mutation Surveyor
software (SoftGenetics, State College, PA, USA).
In silico analysis
Splice Site Prediction by Neural Network (SSPNN;
www.fruitfly.org/seq_tools/splice.html) and the Human
Splicing Finder (HSF; www.umd.be/HSF/) were used to
investigate the mechanisms of the splicing abnormalities caused by
the mutations at the non-consensus splice sites. HSF contains its
own programs and other prediction platforms, including the exonic
splicing enhancer (ESE) finder (http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home),
RESCUE-ESE (http://genes.mit.edu/burgelab/rescue-ese), FAS-ESS
(http://genes.mit.edu/fas-ess), Putative
Exonic Splicing Enhancers/Silencers (PESX) designed by Zhang and
Chasin (30) and splicing
silencer motifs designed in the study by Sironi et al
(19). This assessment system
contains 2 sets (HSF and SSPNN) to examine the potential splice
sites, where 1 set (HSF) was used for the potential branch points,
4 sets (PESx, RESCUE-ESE, ESE finder and HSF) for the ESE and 3
sets (PESx, FAS-ESS and splicing silencer motifs) for the exonic
splicing silencer (ESS). The query sequences were obtained from the
normal and mutated sequences. The SSPNN, HSF and ESE finder
provided the scores to value the strength of the splicing-relative
sequence motifs using corresponding weight matrices. PolyPhen2
(http://genetics.bwh.harvard.edu/pph2)
and SIFT (http://sift.bii.a-star.edu.sg) were applied to predict
the potential effect of an amino acid substitution on the structure
and function of the NF1 protein. BLASTP was used to align the NF1
protein sequences from the multiple species, including human,
chimpanzee, gorilla, cat, dog, mouse, rat, cattle, chicken and
zebrafish.
Real-time PCR
The alternative post-transcriptional profiles in one
specific case were produced using the SYBR Master Mix (Life
Technologies) and ABI PRISM 7000 Sequencing Detection System (Life
Technologies). The ACTB gene encoding β-actin was used for
normalization. For ACTB, the forward primer was
5′-AGCTCCTCCCTGGAGAAGAG-3′ and the reverse primer was
5′-AGCACTGTGTTGGCGTACA-3′. For NF1, the forward primer was
5′-GATGTAAAATGTCTTACAAG-3′ and the reverse primer was
5′-CTGCCACCTGTTTGCGCACT-3′. Amplicons targeting on the NF1
and ACTB genes were confirmed using Sanger sequencing.
Real-time PCR was performed in triplicate using 5, 2.5, 1.25 and
0.625 ng cDNA.
Results
Mutation spectrum
Mutational screening of the NF1 gene was
performed on samples obtained from 378 clinically diagnosed or
individuals suspected of having NF1. The mutation nomenclature was
based on the NCBI reference NM_000267.3. The exon number was given
according to the conventional rule used in the NF1 testing
community and previous literatures (10,11,17,18). The mutations were confirmed using
the Biobase (HGMD professional version database) and Leiden Open
Variation Database (LOVD) to determine the recurrence. In addition,
the missense mutations were searched in 1000 Genomes project, dbSNP
and Exome Variant Server to rule out normal variants. NF1
mutations were identified in 169 out of 378 cases; 127 different
mutations were observed (Tables I
and II). Of these mutations, 54
mutations were novel, of which, 23 were frameshift mutations, 10
were splicing defects, 7 were nonsense mutations and 14 were
missense mutations (Table III).
The mutations affected almost all exons apart from exon 4c, 14,
23.1, 35, 38 and 49 in the mutation spectrum of the patients, which
was consistent with the finding of no hot spot mutations in
previous studies (3,10,11). The nonsense mutations were the
most common molecular defects found in this study (33/127),
followed by the splice-site mutations (32/127) and missense
mutations (27/127). In the group of frameshift mutations, deletion
(n=23) was prone to occurring compared to insertion/duplication
(n=8) and indels (n=4), but most of the insertion/duplication and
all of the indels were novel mutations.
 | Table IThe 127-mutation spectrum apart from
the splicing abnormalities. |
Table I
The 127-mutation spectrum apart from
the splicing abnormalities.
| cDNA | Protein | Exon |
|---|
| Missense
mutations |
| 1A>G | Met1Val | 1 |
| 410C>G | Ser137Cys | 4a |
| 1241T>G | Leu414Arg | 9 |
| 1646T>C | Leu549Pro | 11 |
| 2350 T>C | Trp784Arg | 15 |
|
2975T>A | Met992Lys | 17 |
|
3104T>C |
Met1035Thr | 18 |
| 3142T>G | Trp1048Gly | 19a |
|
3211G>C |
Ala1071Pro | 19b |
| 3295A>G | Lys1099Glu | 19b |
|
3296A>C |
Lys1099Thr | 19b |
|
3362A>G |
Glu1121Gly | 20 |
| 3494T>C | Ile1165Thr | 20 |
|
3752A>C |
His1251Pro | 22 |
| 3827G>A | Arg1276Gln | 22 |
|
4172G>C |
Arg1391Thr | 24 |
| 4288A>T | Asn1430Tyr | 25 |
| 4306A>G | Lys1436Glu | 25 |
| 4306A>C | Lys1436Gln | 25 |
|
4318A>G |
Met1440Val | 25 |
| 4493G>A | Gly1498Glu | 26 |
|
5131G>C |
Ala1711Pro | 28 |
| 5425C>T | Arg1809Cys | 29 |
|
5489C>T |
Pro1830Leu | 29 |
|
5498T>C |
Leu1833Pro | 29 |
|
5855C>T |
Ala1952Val | 31 |
|
7106T>C |
Leu2369Ser | 39 |
| Nonsense
mutations |
|
569T>G |
Leu190* | 4b |
| 574C>T |
Arg192* | 4b |
|
586G>T |
Glu196* | 4b |
| 668G>A |
Trp223* | 5 |
| 1238C>G |
Ser413* | 9 |
| 1246C>T |
Arg416* | 9 |
| 1275G>A |
Trp425* | 10a |
| 1318C>T |
Arg440* | 10a |
| 1381C>T |
Arg461* | 10a |
| 1754T>A |
Leu585* | 12a |
| 2041C>T |
Arg681* | 13 |
| 2352G>A |
Trp784* | 15 |
| 2446C>T |
Arg816* | 16 |
| 3049C>T |
Gln1017* | 18 |
| 3826C>T |
Arg1276* | 22 |
| 4006C>T |
Gln1336* | 23.2 |
|
4066G>T |
Glu1356* | 23.2 |
| 4084C>T |
Arg1362* | 23.2 |
| 4107C>G |
Tyr1369* | 23.2 |
|
4267A>T |
Lys1423* | 24 |
| 4537C>T |
Arg1513* | 27a |
| 5264C>G |
Ser1755* | 29 |
| 5401C>T |
Gln1801* | 29 |
|
5708T>G |
Leu1903* | 30 |
| 5839C>T |
Arg1947* | 31 |
| 5941C>T |
Gln1981* | 31 |
|
6243C>G |
Tyr2081* | 33 |
| 6709C>T | Arg2237
* | 36 |
| 7285C>T |
Arg2429* | 41 |
| 7486C>T |
Arg2496* | 42 |
| 7843C>T |
Gln2615* | 45 |
| 7993C>T |
Gln2665* | 46 |
|
8072G>A |
Trp2691* | 47 |
| Frameshift
mutations |
| 118delA |
Lys40Argfs*4 | 2 |
| 154delT |
Ser52euLfs*4 | 2 |
| 499_502delTGTT |
Cys167Glnfs*12 | 4b |
|
802_803delCCinsG |
Pro268Aspfs*13 | 6 |
|
1010_1021del12 |
Glu337_Ser340del | 7 |
| 1541_1542delAG |
Gln514Argfs*43 | 10c |
|
1664_1667delTAGA |
556Aspdelfs*13 | 11 |
|
1756_1759delACTA |
Phe586delfs*19 | 12a |
| 1882delT |
Tyr628Thrfs*3 | 12b |
| 1908delT |
Ser636Valfs*52 | 12b |
|
2032_2033delCCinsA |
Pro678Lysfs*10 | 13 |
|
2342dupA |
His781Glnfs*13 | 15 |
|
2984_2988delTGGTC |
Leu995Glnfs*24 | 17 |
|
3054delT |
Leu1018* | 18 |
|
3108_3109ins23 |
Lys1036Thrfs*8 | 18 |
|
3852_3854delAAT |
Lle1284fs | 22 |
|
4312_4314delGAA | Glu1438del | 25 |
| 4418_4419delAT |
His1473Glnfs*7 | 26 |
|
4498_4505delTATCTTTC |
Tyr1500_Ile1501delfs*6 | 26 |
|
4688_4689insAA |
Phe1563Leufs*5 | 27b |
|
4810dupT |
Tyr1604Leufs*16 | 28 |
|
4906dupA |
Asp1636Argfs*5 | 28 |
|
4930_4937indels |
Val1644Serfs*3 | 28 |
| 5010delG |
Lys1670Asnfs*7 | 28 |
|
5040_5043delAGGTinsTA |
Lys1680Asnfs*16 | 28 |
|
5909dupC |
Ile1971Tyrfs*17 | 31 |
| 6791dupA |
Tyr2264* | 37 |
|
7096_7101delAACTTT |
Asn2366_Phe2367del | 39 |
| 7125delA |
Tyr2375Thrfs*20 | 39 |
|
7160delG |
Arg2387Lysfs*10 | 40 |
| 7267dupA |
Thr2423Asnfs*4 | 41 |
|
7388_7384+12del19 | Unknown | 41 |
|
7510delG |
Asp2504Thrfs*23 | 42 |
|
7581_7582delAT |
Ser2528Glnfs*21 | 43 |
|
7726delT |
Ser2576Glnfs*27 | 44 |
| Table IIThe mutation spectrum of the splicing
abnormalities. |
Table II
The mutation spectrum of the splicing
abnormalities.
| Splicing mutations
at the consensus splice sites |
|---|
|
|---|
| Mutation | IVS | cDNA effect | Effect on splice
site |
|---|
|
60+1delG |
IVS1+1delG | Unknown | Inactive 5′ss |
| 204+1G>A | IVS2+1G>A | 100_204del105 | Cryptic 5′ss |
| 205-2A>C | IVS2-2A>C | ΔE3 | Inactive 3′ss |
| 889-2A>G | IVS6-2A>G | ΔE7 | Inactive 3′ss |
| 889-1G>A | IVS6-1G>A | ΔE7 | Inactive 3′ss |
| 1642-1G>A | IVS10c-1G>A | ΔE11 | Inactive 3′ss |
| 2409+1G>A | IVS15+1G>A | ΔE15 | Inactive 5′ss |
| 2850+1G>A | IVS16+1G>A | ΔE16 | Inactive 5′ss |
| 3114-2A>G | IVS18-2A>G | ΔE19a | Inactive 3′ss |
| 3708+2T>A | IVS21+2T>A | ΔE21 | Inactive 5′ss |
| 3709-2A>G | IVS21-2A>G |
3709_3718delGATGAACTAG | Cryptic 3′ss |
| 4367+1G>A | IVS25+1G>A | ΔE25 | Inactive 5′ss |
| 6579+1G>A | IVS34+1G>A | ΔE34 | Inactive 5′ss |
| 6858+1G>A | IVS37+1G>A | ΔE37 | Inactive 5′ss |
| 7676-2A>G | IVS43-2A>G | ΔE44 | Inactive 3′ss |
| 8098-1G>A | IVS47-1G>A | ΔE48 | Inactive 3′ss |
|
| Intronic mutations
at non-consensus splice sites |
|
| Mutation | Related IVS | cDNA effect | Cryptic splice
site |
|
|
1260+3A>T |
IVS9+3A>T |
1261_1262insGTTAGTCCAAAAG | Cryptic 5′ss |
|
2410-18C>G |
IVS15-18C>G |
2410_2411ins17 | Cryptic 3′ss |
| 5944-5A>G | IVS31-5A>G |
5943_5944insCTAG | Cryptic 3′ss |
|
| Exonic
mutations |
|
| Mutation | Related exon | cDNA effect | Cryptic splice
site |
|
| 910C>T | In E7 | ΔE7 | No |
|
972T>A | In E7 | ΔE7 | No |
|
1185G>A | Last NT of E8 | ΔE8 | No |
| 1466A>G | In E10b | 1466_1572del62 | Cryptic 5′ss |
|
1720A>G | Last second NT of
E11 | ΔE11 | No |
| 1885G>A | In E12b | 1846_1886del | Cryptic 3 ′ss |
|
3362A>G | In E20 | ΔE20 | No |
|
3467A>G | In E20 | ΔE20 | No |
|
3496G>A | Last NT of E20 | ΔE20 | No |
| 5546G>A | Last NT of E29 | ΔE29 | No |
| 6792C>A | In E37 | ΔE37 | No |
| 6792C>G | In E37 | ΔE37 | No |
|
7694delC | In E44 | ΔE44 | No |
| Table IIIThe number of each type of
abnormality in 127 mutations and the frequency of the novel
mutations. |
Table III
The number of each type of
abnormality in 127 mutations and the frequency of the novel
mutations.
| Missense | Nonsense | Deletion | Ins/Dup | Indels | Splicing
defect |
|---|
| Total no. | 27 | 33 | 23 | 8 | 4 | 32 |
| Novel
mutations | 14 | 7 | 13 | 6 | 4 | 10 |
Only 50% of the splicing defects disrupted the
conserved GT/AG or AT/AC dinucleotides of the splice sites in this
study (Table II). By contrast, 3
intronic mutations at non-consensus splice sites (Table IV, subgroup I) generated cryptic
5′ss or 3′ss, resulting in the insertion into the mRNA. Thirteen
exonic variants, that were 40.6% of the splicing defects, induced
exon skipping or aberrant exons instead of a point mutation based
on the genomic DNA and cDNA sequencing methods. Among these exonic
mutations, c.1466A>G and c.1885G>A (Table IV, subgroup II) induced aberrant
exons with a deletion by generating cryptic 5′ss or 3′ss,
respectively. In particular, the 1185G>A mutation was a silent
mutation at the genomic DNA level. The other 4 exonic sequence
alterations, which resulted in exon skipping, were identified as
substitutions at the last nucleotide position of exon 8, 20 and 29,
and the last second nucleotide of exon 11 (Table IV, subgroup III). The remaining 7
exonic mutations, which caused exon skipping, did not perturb the
natural 3′/5′ss or create cryptic splice sites (Table IV, subgroup IV).
| Table IVThe scores and results assessed using
the computational tools on the normal and mutant sequences. |
Table IV
The scores and results assessed using
the computational tools on the normal and mutant sequences.
| Predicted scores by
HSF | Predicted scores by
SSPNN | Prediction |
|---|
|
|
|
|
|---|
| Germline
mutations | Authentic ss on
normal sequences | Authentic ss on
mutant sequences | New ss
generated | Authentic ss on
normal sequences | Authentic ss on
mutant sequences | ESE, ESS and other
motifs on mutant sequences |
|---|
| Subgroup I:
Intronic mutation |
| 1260+3A>T | Donor site
(88.18) | Donor site
(83.15) | Donor site
(79.6) | Donor site
(0.99) | Donor site
(0.80) | Putative ESS |
| 2410-18C>G | Acceptor site
(84.37) | Acceptor site
(84.37) | Acceptor site
(86.14) | Acceptor site
(0.46) | Acceptor site
(none) | Putative ESE,
abolished BP |
| 5944-5A>G | Acceptor site
(80.89) | Acceptor site
(80.83) | Acceptor site
(92.26) | Acceptor site
(0.78) | Acceptor site
(0.74) | Abolished ESS |
| Subgroup II: Exonic
mutation with cryptic ss |
| 1466A>G | Donor site
(82.29) | Donor site
(82.29) | Donor site
(82.62) | Donor site
(none) | Donor site
(0.97) | Abolished ESE |
| 1885G>A | Acceptor site
(82.35) | Acceptor site
(82.35) | Acceptor site
(94.06) | Acceptor site
(none) | Acceptor site
(0.98) | ESE and ESS
strength decreased |
| Subgroup III:
Substitution at last NT or last 2nd NT of an exon |
| 1185G>A | Donor site
(95.55) | Donor site
(84.97) | No | Donor site
(0.99) | Donor site
(0.51) | Abolished ESE |
| 1720A>G | Donor site
(77.05) | Donor site
(72.19) | No | Donor site
(0.79) | Donor site
(none) | Abolished ESE |
| 3496G>A | Donor site
(94.52) | Donor site
(71.5) | No | Donor site
(0.79) | Donor site
(none) | Abolished ESE |
| 5546G>A | Donor site
(85.96) | Donor site
(75.38) | No | Donor site
(0.97) | Donor site
(none) | No original
ESE |
| Subgroup IV: Exonic
mutation without cryptic ss |
| 910C>T | - | - | No | - | - | Abolished ESS |
| 972T>A | - | - | No | - | - | ESE strength
decreased |
| 3362A>G | - | - | No | - | - | ESE/ESS ratio
decreased |
| 3467A>G | - | - | No | - | - | ESS strength
decreased |
| 6792C>A/G | - | - | No | - | - | Abolished ESS |
| 7694delC | - | - | No | - | - | ESE/ESS ratio
decreased |
Of note, one exonic mutation, c.3362A>G, produced
mosaicism of E1121G and exon 20 skipping at the mRNA level
(Fig. 1). This germline mutation
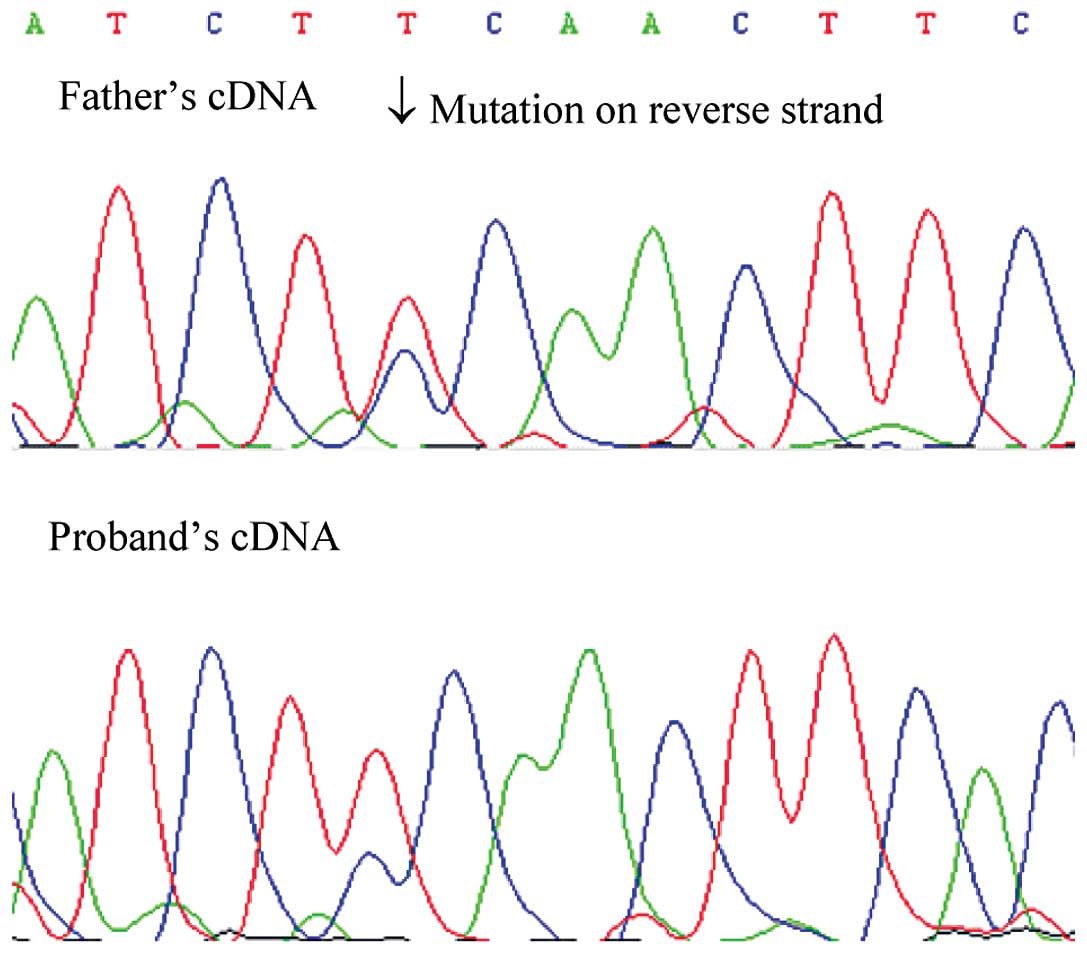
in the proband was inherited from his father, and the
post-transcriptional mosaicism was presented in the mRNA of both
patients. However, the missense mutation of the son showed a lower
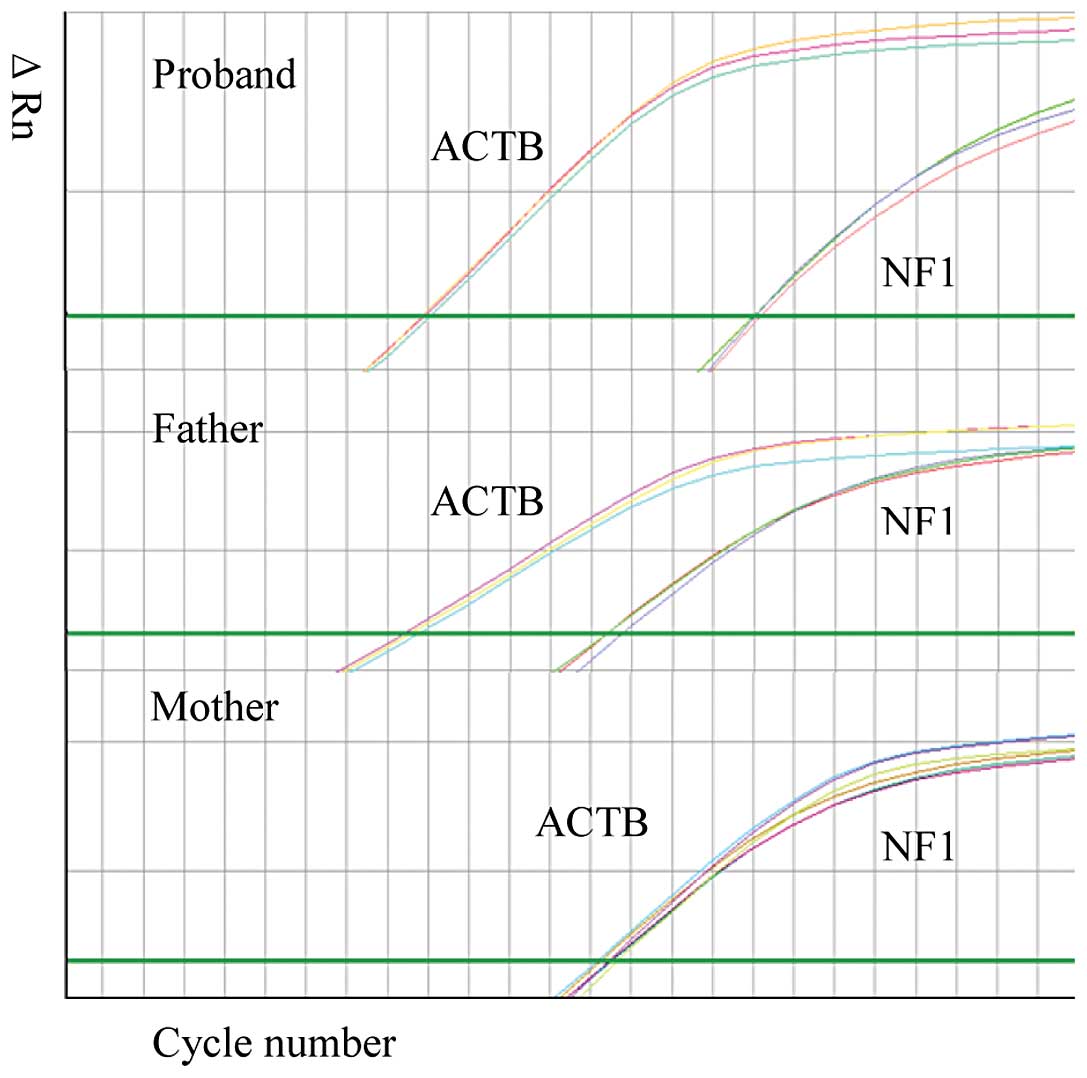
signal intensity using Sanger sequencing (Fig. 2). The mosaicism of the
post-transcriptional profile was further examined using real-time
PCR with cDNA obtained from this family (Fig. 3). The sample from the mother was
used as the wild-type sample in this assay. The skipping rate of
the mutant exon in the samplefrom the son was higher compared to
the rate in the sample from the father.
In silico analysis
To better understand the underlying mechanisms of
these 16 unusual splicing errors, 7 computational tools were
employed to examine how these mutations affected the
splicing-relative sequence motifs. The comparison was made between
the results of the prediction on the normal and mutated sequences
(Table IV). SSPNN predicted a
deletion of the authentic acceptor site (3′ss) caused by
2410-18C>G, and a decrease in the strength of the authentic
splice sites caused by 1260+3A>G and 5944-5A>G in the
subgroup I. By contrast, the cryptic splice sites generated by
these 3 mutations and the subgroup II mutations were given high
scores by HSF or SSPNN. Substitutions in the subgroup III all
abolished the authentic donor sites (5′ss) in the SSPNN evaluation,
compared to the prediction of the strength reduction by HSF. No
cryptic splice sites were predicted in these substitution sequences
and the subgroup IV sequences.
Further investigation on other splicing-relative
sequence motifs revealed that some cis-acting factors were altered
in these mutated sequences. In the subgroup I sequences, the
branch-point motif was deleted and a putative ESE was generated by
2410-18C>G; a putative ESS or an abolished ESS was predicted to
accompany the strength-decreased donor site or acceptor site in the
mutated sequences of 1260+3A>G and 5944-5A>G, respectively.
In the subgroup II sequences, the ESE was deleted or the strength
of the ESE was decreased simultaneously with the strong new splice
site generated by 1466A>G and 1885G>A, respectively. The
original ESE was deleted or no ESE was embedded in the subgroup III
sequences. The architectural alterations of the splicing-regulatory
elements were more complex in the subgroup IV sequences. In
general, the decreased strength of the ESE or decreased ratio of
the ESE/ESS was presented. An abolished ESS was also predicted in
mutated exon 7 and 37, but no abolished ESE was predicted.
Discussion
NF1 is a multisystem genetic disorder with extreme
diversity of clinical expression (3,10,11,20,21,22). A clear correlation of genotype and
phenotype has been previously demonstrated in only two types of
mutations (23). Patients with an
NF1 microdeletion have more severe clinical characteristics
(4,23). We found 5 cases with an NF1
gene deletion in patients with various severe conditions, such as
developmental delay, seizures, and early onset skin/subcutaneous
tissue disorders. One 4 year-old patient harboring a novel splice
site mutation (60+1delG) also exhibited the developmental delay.
This alternation, which induced skipping in exon 1 may cause the
same effect as an NF1 deletion at the protein level. Another
mutation, c.2970-2972delAAT, in exon 17, which was not shown in our
study, has been reported to be associated with the absence of
cutaneous neurofibromas (11,24). However, due to the lack of
detailed clinical information, our investigation of the correlation
between genotype and phenotype was limited. The present study
focused largely on the mutation spectrum, particularly the splicing
errors.
The mutation rate of the NF1 gene is one of
the highest reported in the human genome. We presented a mutation
spectrum of the NF1 gene with 127 different variants in this
study. In the summarized data, exon 19b, 25 and 29 harbored more
missense mutations, exon 4b, 10a and 23.2 appeared to have more
nonsense mutations, and the highest rate of frameshift mutations
was found in exon 28. In addition, exon 7, 20 and 37 were prone to
splicing errors. A total of 54 of the 129 different mutations were
novel mutations that were categorized as either frameshift
mutations, splicing defects, nonsense mutations or missense
mutations. With the exception of missense mutations, the other 3
types of mutations are considered highly likely to be deleterious.
Patients with NF1 with a missense mutation have a lower incidence
of multiple neurofibromas and plexiform neurofibromas compared to
patients with a different type mutation; In addition, it is also
true that no evidently milder NF1 phenotype was concluded to be
distinctly associated with a missense mutation (23). The in silico analysis of
the functional consequence of these novel missense mutations was
performed using the Polyphen2 and SIFT program. A total of 12 of
these 14 novel missense mutations were predicted to be potentially
damaging. Although P1830L and A1952V were determined to be benign,
these mutations and other novel missense mutations altered amino
acids that were conserved across the different species according to
the results obtained from an orthologous alignment using BLASTP. It
is widely believed that mutations in a highly conserved region may
result in a functional change. Moreover, we found 2 second missense
variants, M645V and I1658V, with nonsense mutations. These 2
variants can be treated as neutral polymorphism after the parental
study.
In the present study, 25.2% of the different
mutations induced aberrant splicing and 50% of these splicing
errors were caused by exonic mutations and intronic mutations at
non-consensus splice sites. These results are consistent with those
of previous findings, showing that the NF1 gene is
susceptible to having splicing errors in post-transcription
(14,15). Accurate splice site recognition is
critical in pre-mRNA splicing (25). This process is a coordinated
program involving the strong splice sites, correct splicing
regulatory elements (SRE) embedded in the genome and the associated
proteins. Bioinformatics assessments focused on revealing how the
authentic splice sites and the hidden sequence motifs of these SREs
were interfered by these exonic and intronic variants at
non-consensus splice sites in this study. We found that the change
in these splice sites acted by tethering the alteration of the ESE,
ESS and other cis-acting elements, which resulted in aberrant
splicing. The error-prone splicing occurred in the subgroups I and
II in which the high-score cryptic 5′/3′ss or the next AG/GT with a
higher score, as the case of 1260+3A>G, replaced the
strength-decreased or lower-scored authentic ones in the new
microenvironment of splicing regulatory elements. For example, the
2410-18C>G mutation generated a higher score cryptic splice site
while forming a putative ESE and abolishing the original branch
point, and then 17 base pairs were inserted into the mRNA as the
consequences of a strong cryptic splice site in coordination with
the gain and/or loss of SREs. Importantly, the 2410-16A>G,
2410-15A>G, 2410-12T>G mutations have been previously
documented in the Biobase HGMD database. Given 2410-18C>G, it is
an obvious sign to alert that this cluster near the intron 15/exon
16 boundary is a splicing-aberration harbor.
In some cases, the disruption of ESE elements was
the principal cause of the splicing error due to the consequences
of the reduced splicing enhancement activity (16,26,27). As is known, not all of the
substitutions at the last nucleotide position of an exon will cause
exon skipping, although the mutants are also predicted to delete
the authentic splice site or decrease its strength. The loss of ESE
motifs may explain why the exon skipping was caused by the exonic
mutations in the subgroup III. An exception in this case was exon
29 that had no ESE motif. Exon 29 skipping resulted from the
weakened donor site caused by the mutation 5546G>A and the lack
of the ESE to support the splice-site recognition.
There was no cryptic splice site generated and no
strength reduction of the authentic splice sites in the subgroup IV
sequences. However, the acceptor site strength was characterized to
be an important and sensitive parameter in splice-site recognition
(28). The low-score exons, such
as exon 7 and 37 were observed to have more splicing defects in
this study. In addition, our in silico analysis also showed
more complex changes in the ESE and ESS. Not only was the ESE
demonstrating a decrease in strength, but it also showed that
weaker ESS, decreased ratio of ESE/ESS and abolished ESS were
predicted to be the architectural weakness in the exon definition,
resulting in exon exclusion in the subgroup IV sequences. The ratio
of ESE/ESS was important for exon recognition and intron
identification in the complexity of splicing. Several studies have
observed that a higher density of ESEs was in the exons compared to
the introns and vice versa for ESSs (12,29,30). The ratio decreasing of ESE/ESS
will break the delicate balance of SREs, and will be prone to exon
exclusions. Although enhancers and silencers have apparently
opposite effects as suggested by their terms, the Composite Exonic
Regulatory Elements of Splicing (CERES) has already been proposed
when accumulating evidence has suggested ESE and ESS shared
additional properties (31,32). The findings of the weaker or
abolished ESS caused by the mutations in our analysis reflected the
overlapping function of these 2 elements. The SREs became more
critical in determining the splice-site recognition in these
cases.
When the trans-acting factors navigated the new
landscape in which the mutation plays a make-or-break role, the
consequences of the competition and coordination in the splicing
process is more evident in the case with the 3362A>G mutation,
in which the germline mutation decreased the ratio of the ESE/ESS.
The splicing in the mutated microenvironment resulted in the
missense mutation and exon skipping coexisting in the mRNA of an
11-year-old boy and his father with different rates. Furthermore,
real-time PCR clearly confirmed this mosaicism. Both patients have
multiple café-au-lait spots but no other profoundly different and
NF1-related clinical features. The higher skipping rate found in
the sample of the son may be caused by individual’s genetic
variability.
Taken together, this study presents 54 novel
NF1 mutations and reveals the high frequency of the unusual
splicing defects in the pathogenicity of NF1. Integrated analyses
using the bioinformatics tools provided insight in order to better
understand the underlying mechanisms of these splicing errors. In
particular, as the NF1 gene was susceptible to having
aberrant splicing caused by exonic variants, including the silent
mutation, such as 1185G>A, this result underscored the large
consequences of NF1 gene testing at both the genomic and
mRNA levels. In addition, the mutation data may contribute
information to the ongoing antisense therapeutics for NF1 caused by
intronic mutations (33), thus
shedding light on a targeted treatment to restore NF1 gene
function.
Acknowledgements
We would like to thank all of the patients and their
families for their support. We also acknowledge the referring
physicians for providing clinical information. In addition, we
appreciate that Andrea Sternenberger worked closely on editing the
manuscript.
References
|
1
|
Williams VC, Lucas J, Babcock MA, Gutmann
DH, Korf B and Maria BL: Neurofibromatosis type 1 revisited.
Pediatrics. 123:124–133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
No authors listed. Neurofibromatosis.
Conference statement. National Institutes of Health Consensus
Development Conference. Arch Neurol. 45:575–578. 1988.PubMed/NCBI
|
|
3
|
Griffiths S, Thompson P, Frayling I and
Upadhyaya M: Molecular diagnosis of neurofibromatosis type 1: 2
years experience. Fam Cancer. 6:21–34. 2007.PubMed/NCBI
|
|
4
|
Pasmant E, Sabbagh A, Spurlock G,
Laurendeau I, Grillo E, Hamel MJ, Martin L, Barbarot S, Leheup B,
Rodriguez D, Lacombe D, Dollfus H, Pasquier L, Isidor B, Ferkal S,
Soulier J, Sanson M, Dieux-Coeslier A, Bièche I, Parfait B, Vidaud
M, Wolkenstein P, Upadhyaya M and Vidaud D; members of the NF
France Network. NF1 microdeletions in neurofibromatosis type 1:
from genotype to phenotype. Hum Mutat. 31:1506–1518. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aoki Y, Niihori T, Narumi Y, Kure S and
Matsubara Y: The RAS/MAPK Syndromes: Novel roles of the RAS pathway
in human genetic disorders. Hum Mutat. 29:992–1006. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cichowski K and Jacks T: NF1 tumor
suppressor gene function: narrowing the GAP. Cell. 104:593–604.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yunoue S, Tokuo H, Fukunaga K, Feng L,
Ozawa T, Nishi T, Kikuchi A, Hattori S, Kuratsu J, Saya H and Araki
N: Neurofibromatosis type I tumor suppressor neurofibromin
regulates neuronal differentiation via its GTPase-activating
protein function toward Ras. J Biol Chem. 278:26958–26969. 2003.
View Article : Google Scholar
|
|
8
|
Schubbert S, Shannon K and Bollag G:
Hyperactive Ras in developmental disorders and cancer. Nat Rev
Cancer. 7:295–308. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Trovó-Marqui AB and Tajara EH:
Neurofibromin: a general outlook. Clin Genet. 70:1–13. 2006.
|
|
10
|
Nemethova M, Bolcekova A, Ilencikova D,
Durovcikova D, Hlinkova K, Hlavata A, Kovacs L, Kadasi L and
Zatkova A: Thirty-nine novel neurofibromatosis 1 (NF1) gene
mutations identified in Slovak patients. Ann Hum Genet. 77:364–379.
2013.PubMed/NCBI
|
|
11
|
Ko JM, Sohn YB, Jeong SY, Kim HJ and
Messiaen LM: Mutation spectrum of NF1 and clinical characteristics
in 78 Korean patients with neurofibromatosis type 1. Pediatr
Neurol. 48:447–453. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang C, Li WH, Krainer AR and Zhang MQ:
RNA landscape of evolution for optimal exon and intron
discrimination. Proc Natl Acad Sci USA. 105:5797–5802. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kolovos P, Knoch TA, Grosveld FG, Cook PR
and Papantonis A: Enhancers and silencers: an integrated and simple
model for their function. Epigenetics Chromatin. 5:12012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Messiaen LM, Callens T, Mortier G, Beysen
D, Vandenbroucke I, Van Roy N, Speleman F and Paepe AD: Exhaustive
mutation analysis of the NF1gene allows identification of 95% of
mutations and reveals a high frequency of unusual splicing defects.
Hum Mutat. 15:541–555. 2000.PubMed/NCBI
|
|
15
|
Ars E, Serra E, Garcia J, Kruyer H, Gaona
A, Lázaro C and Estivill X: Mutations affecting mRNA splicing are
the most common molecular defects in patients with
neurofibromatosis type 1. Hum Mol Genet. 9:237–247. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zatkova A, Messiaen L, Vandenbroucke I,
Wieser R, Fonatsch C, Krainer AR and Wimmer K: Disruption of exonic
splicing enhancer elements is the principal cause of exon skipping
associated with seven nonsense or missense alleles of NF1. Hum
Mutat. 24:491–501. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wimmer K, Roca X, Beiglböck H, Callens T,
Etzler J, Rao A, Krainer A, Fonatsch C and Messiaen L: Extensive in
silico analysis of NF1 splicing defects uncovers determinants for
splicing outcome upon 5′ splice-site disruption. Hum Mutat.
28:599–612. 2007.PubMed/NCBI
|
|
18
|
Pros E, Gómez C, Martín T, Fábregas P,
Serra E and Lázaro C: Nature and mRNA effect of 282 different NF1
point mutations: focus on splicing alterations. Hum Mutat.
29:E173–E193. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sironi M, Menozzi G, Riva L, Cagliani R,
Comi GP, Bresolin N, Giorda R and Pozzoli U: Silencer elements as
possible inhibitors of pseudoexon splicing. Nucleic Acids Res.
32:1783–1791. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ars E, Kruyer H, Morell M, Pros E, Serra
E, Ravella A, Estivill X and Lázaro C: Recurrent mutations in the
NF1 gene are common among neurofibromatosis type 1 patients. J Med
Genet. 40:e822003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Castle B, Baser ME, Huson SM, Cooper DN
and Upadhyaya M: Evaluation of genotype-phenotype correlations in
neurofibromatosis type 1. J Med Genet. 40:e1092003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kluwe L, Friedrich R, Korf B, Fahsold R
and Mautner V: NF1 mutations in neurofibromatosis 1 patients with
plexiform neurofibromas. Hum Mutat. 19:3092002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
van Minkelen R, van Bever Y, Kromosoeto J,
Withagen-Hermans C, Nieuwlaat A, Halley D and van den Ouweland A: A
clinical and genetic overview of 18 years neurofibromatosis type 1
molecular diagnosis in the Netherlands. Clin Genet. May
18–2013.(Epub ahead of print).
|
|
24
|
Upadhyaya M, Huson SM, Davies M, Thomas N,
Chuzhanova N, Giovannini S, Evans DG, Howard E, Kerr B, Griffiths
S, Consoli C, Side L, Adams D, Pierpont M, Hachen R, Barnicoat A,
Li H, Wallace P, Van Biervliet JP, Stevenson D, Viskochil D,
Baralle D, Haan E, Riccardi V, Turnpenny P, Lazaro C and Messiaen
L: An absence of cutaneous neurofibromas associated with a 3-bp
inframe deletion in exon 17 of the NF1 gene (c.2970–2972 delAAT):
evidence of a clinically significant NF1 genotype-phenotype
correlation. Am J Hum Genet. 80:140–151. 2007.PubMed/NCBI
|
|
25
|
Nelson KK and Green MR: Mechanism for
cryptic splice site activation during pre-mRNA splicing. Proc Natl
Acad Sci USA. 87:6253–6257. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Blencowe BJ: Exonic splicing enhancers:
mechanism of action, diversity and role in human genetic diseases.
Trends Biochem Sci. 25:106–110. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Colapietro P, Gervasini C, Natacci F,
Rossi L, Riva P and Larizza L: NF1 exon 7 skipping and sequence
alterations in exonic splice enhancers(ESEs) in a
neurofibromatiosis 1 patient. Hum Genet. 113:551–554. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vandenbroucke I, Callens T, De Paepe A and
Messiaen L: Complex splicing pattern generates great diversity in
human NF1 transcripts. BMC Genomics. 3:132002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Z, Rolish ME, Yeo G, Tung V, Mawson M
and Burge CB: Systematic identification and analysis of exonic
spicing silencers. Cell. 119:831–845. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang XH and Chasin LA: Computational
definition of sequence motifs governing constitutive exon splicing.
Genes Dev. 18:1241–1250. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Baralle D and Baralle M: Splicing in
action: assessing disease causing sequence changes. J Med Genet.
42:737–748. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Haque A, Buratti E and Baralle FE:
Functional properties and evolutionary splicing constraints on a
composite exonic regulatory element of splicing in CFTR exon 12.
Nucleic Acids Res. 38:647–659. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gottfried O, Viskochil D and Couldwell W:
Neurofibromatosis type 1 and tumorigenesis: molecular mechanisms
andtherapeutic implications. Neurosurg Focus. 28:E82010. View Article : Google Scholar : PubMed/NCBI
|