Introduction
Influenza A virus (IAV) (family
Orthomyxoviridae) is the causative agent of significant
respiratory infection (1). The
global spread of the 2009 pandemic H1N1 (pH1N1) influenza virus
emphasized that humans are limited in their effective strategies to
control influenza. Vaccines are the best option for prophylaxis of
looming influenza pandemics. However, the time that elapses between
characterization of the new strain and vaccine production may have
devastating consequences (2).
Antiviral therapy is therefore imperative in the control of
influenza pandemics (3). However,
drug-resistant influenza strains to conventional drugs continuously
emerge (4–6). Therefore, the development of
anti-influenza drugs with broad reactivity against all strains and
subtypes is crucial (2,7). As IAVs use host cell machinery to
support their replication and transportation inside the host cell
(8), new antiviral treatments
should focus on overlapping pathways used by host cells and
influenza viruses. The advantage of this approach involves
decreasing the potential of drug resistance (8,9).
Previous studies have shown that influenza
infections lead to uncontrolled elevations of pro-inflammatory
cytokines making this infection a strong risk factor for severe
complications (10,11). Some of the most important
cytokines involved in the above pathway are tumor necrosis factor-α
(TNF-α), interleukin-6 (IL-6) (12) and interferon-γ (IFN-γ) (13). These cytokines induce an innate
immune system to control the infection. However, hypercytokinemia
occasionally occurs which may cause potentially fatal immune
reaction (14). Effective
alternative therapeutics to vaccines and conventional antiviral
drugs can be based on anti-inflammatory and immunomodulatory agents
(7).
Statins, which have the ability to inhibit HMG-CoA
reductase, are considered to be mediators of direct cell effects
beyond their lipid lowering capacity (15–17). Thus, they block downstream
molecules such as isoprenoids and lipid groups, which may be key
factors for the virus infectivity cycle (18,19). Due to the anti-inflammatory
effects statins exert which result in the reduction of inflammatory
reactions, they are regarded as being of clinical significance
(20–22).
Actin filaments are necessary for transportations of
cells. Cytoskeletal changes were reported following interaction
with a number of viruses (23–25). These changes were associated with
the binding of different proteins that regulate the dynamics of the
actin cytoskeleton (26,27). One class of well-known proteins
that regulate actin structures is the Rho family GTPase proteins
(28–30). RhoA, the best studied member of
this family, triggers the progression of different pathogenic
processes (31,32). The effect of statin on disassembly
of the cytoskeleton affecting post-translational modification of
RhoA has also been demonstrated (33,34).
Pathways such as endocytosis and autophagy are also
required for efficient transduction within the cells (35,36). Endocytic pathways reach lysosomes
(37) as the final destination of
autophagosomes for degradation (38). Rab5 and Rab7 are considered
reliable markers for early and late endosomes, respectively.
Protein conversion as the mechanism of cargo transport between
early and late endosomes even of viral particles trafficking has
been previously demonstrated (39,40). By contrast, light chain myosin 3
(LC3) is a reliable marker of autophagosome and autolysosome
formation (41). During autophagy
the level of membrane-associated LC3-II (16 kDa) increases, while
the level of soluble LC3-I (18 kDa) decreases (42,43). Therefore, LC3-II levels correlate
with the number of autophagosomes (44).
Numerous pathogens including viruses hijack the
endocytic and autophagic pathways mediating their internalization
as well as trafficking to the site of replication to avoid being
degraded by the lysosomes (26,38,39).
To the best of our knowledge, limited data are
available on the anti-inflammatory effects and inhibitory role of
statins on RhoA, Rab and LC3 protein functions induced by IAV
infection. The therapeutic role of statins on influenza infection
with regard to their effects on these proteins has not yet been
established. In the present study the effects of simvastatin on IAV
replication were evaluated using MTT cytotoxicity and
hemagglutination (HA) assays. The cytoskeletal effects of
simvastatin in the presence of IAV via its effect on RhoA protein
prenylation were also investigated. This study characterized the
involvement of simvastatin on viral cellular infection via its
effect on Rab proteins during endocytosis and its effect on
lysosomal activity by affecting autophagosomes. The results
suggested that simvastatin should be further investigated in order
to be introduced as a potential anti-influenza compound in the
future.
Materials and methods
Reagents, chemicals and antibodies
Cell culture media and penicillin-streptomycin
solution were purchased from Mediatech Cellgro Co. (Northbrook, IL,
USA). Accutase cell detachment solution was obtained from
Innovative Cell Technologies (San Diego, CA, USA) and fetal bovine
serum (FBS) was purchased from PAA Laboratories (Pasching,
Austria). Plastic wares were obtained from Orange Scientific
(Braine-l’Alleud, Belgium) while 8-well Lab-Tek II Chamber Slides
were purchased from Thermo Scientific (New York, NY, USA).
Simvastatin base, amantadine hydrochloride, oseltamivir phosphate,
RhoA inhibitor Y-27632 dihydrochloride, farnesyl pyrophosphate
(FPP), geranylgeranyl pyrophosphate (GGPP), farnesyl transferase
inhibitor (FTI-276), geranylgeranyl transferase inhibitor
(GGTI-2133), cholesteryl ester, Bafilomycin A1 (BafA1), tosylamide
phenylethyl chloromethyl ketone-treated trypsin (TPCK-Trypsin),
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT) and BCIP-NBT substrate were obtained from Sigma (St. Louis,
MO, USA). Fluorescent rhodamine 110 phalloidin was purchased from
Biotium (Hayward, CA, USA) and ProLong® Gold Antifade
reagent and LysoTracker Red DND-99 were purchased from Invitrogen
(Carlsbad, CA, USA). CytoBuster™ protein extraction reagent was
provided from Novagen (Madison, WI, USA). Protein extraction kit
(ab65400), rabbit primary polyclonal anti-RhoA (ab68826), donkey
secondary polyclonal anti-rabbit (AP-conjugated) (ab97061), rabbit
primary polyclonal anti-Rab5 (ab18211), goat secondary polyclonal
anti-rabbit IgG (AP-conjugated) (ab97048), mouse primary monoclonal
anti-Rab7 (ab50533), goat secondary polyclonal anti-mouse IgG
(AP-conjugated) (ab97020), rabbit primary polyclonal anti-LC3
(ab58610), mouse primary monoclonal anti-GAPDH (ab9484), mouse
primary monoclonal anti-pan-Cadherin (ab6528) and goat secondary
polyclonal anti-mouse (AP-conjugated) (ab97020) were purchased from
Abcam (Cambridge, UK). Simvastatin (10 mg) was dissolved in 1 ml of
DMSO while Y-27632 (10 mg) was dissolved in 1 ml of
dH2O. FTI-276 and GGTI-2133, which served as control
drugs for simvastatin, were also dissolved in DMSO. Amantadine
hydrochloride, which served as the control antiviral drug, was
dissolved in dH2O. The stock solutions were
filter-sterilized using 0.22 μm syringe filter, aliquoted and
stored at −20°C. Control experiments were carried out using
solvents but no effects on these vehicles were observed.
Virus and cell culture
Influenza virus strain A/New Jersey/8/76 (H1N1)
[A/NJ (H1N1)] (ATCC VR-897™) was used in the present study. The
viral stock was propagated in Madin Darby canine kidney (MDCK)
cells in the presence of 1 μg/ml of trypsin-TPCK. The MDCK cell
line was purchased from ATCC (CCL-34™). The cells were grown in
Dulbecco’s modified Eagle’s medium (DMEM) containing 10%
heat-inactivated FBS, 100 U/ml penicillin G and 100 μg/ml
streptomycin. Cultured cells were incubated at 37°C, in a 5%
CO2 atmosphere. During antiviral evaluations, FBS was
washed with phosphate-buffered saline (PBS) and the medium was
supplemented with TPCK-treated trypsin (1 μg/ml). For virus-stock
preparation, the MDCK cells were infected with the virus at a
multiplicity of infection (MOI) of 0.5. Progeny virus was harvested
three days post-infection. Standard HA tests using tissue culture
infectious dose 50 (TCID50) and the Karber formula
(45,46) were conducted to measure virus
infectivity.
Cell viability
MDCK cells sub-cultured in 96-well plates were
exposed to different concentrations of simvastatin at various time
intervals of 24, 48 and 72 h in triplicate. A colorimetric MTT
assay was performed as previously described (47). Color adsorption in the solution
was measured using a microplate reader (BioTek EL 800; BioTek
Instruments Inc., Winooski, VT, USA) at 570 nm. The 50% cytotoxic
concentration (CC50) causing visible morphological
changes in 50% of the cells with respect to cell control, and
effective concentration (EC50), which is the
concentration required to achieve 50% protection against a
virus-induced cytopathic effect and cell viability, were defined by
this method.
In one series of experiments, MDCK cells were
pretreated with FPP, GGPP and cholesterol for a period of 6 h. The
cells were then washed with PBS, followed by treatment with
simvastatin and H1N1 (100 TCID50) for 1 h. The cells
were then washed with PBS and subsequently incubated in the medium
supplemented with simvastatin and TPCK for 48 h at 37°C. FTI, GGTI,
amantadine and oseltamivir treatments were simultaneously
examined.
Hemagglutination assay
MDCK cells were pretreated with FPP, GGPP and
cholesterol as previously mentioned for 6 h. Following washing, the
combined treatment of simvastatin and H1N1 was conducted for 1 h.
The cells were supplemented with simvastatin and TPCK and incubated
for a period of 48 h at 37°C. The effects of FTI, GGTI, amantadine
and oseltamivir were simultaneously examined. The virus titer was
quantified using HA assay from the cell supernatants, as previously
described (48). Briefly, serial
double dilutions of the culture media were added to 96-well
U-shaped microplates in duplicate. HA units were calculated as the
reciprocal of the highest dilution, yielding complete
agglutination. Chicken red blood cells were used at a concentration
of 0.5%.
RNA extraction and cDNA synthesis
MDCK cells were washed with PBS following treatments
with simvastatin (10 μM) and H1N1 (100 TCID50) for 1 h.
In the co-inoculation treatment, the cells were exposed to
simvastatin and H1N1 simultaneously. In the pre-inoculation
treatment the cells were initially treated with simvastatin for 1
h, followed by H1N1 inoculation, whereas in the post-inoculation
treatment the cells were initially inoculated with H1N1 for 1 h
followed by the addition of simvastatin. Following the treatments,
the cells were washed again with PBS and incubated with medium
supplemented with simvastatin and TPCK for 48 h at 37°C.
Viral RNA was extracted using the Viral Nucleic Acid
Extraction kit II (Geneaid, Taipei, Taiwan), as per the
manufacturers’ instructions. The extracted RNAs were
reverse-transcribed using a RevertAid H Minus First Strand cDNA kit
(Fermentas, Burlington, ON, Canada). The incubation cycle was
performed as previously described (49). Virus-inoculated and mock-infected
supernatants were considered as positive and negative controls,
respectively. The gene actin α1 (ACTA1) was targeted as the
housekeeping control.
Quantitative polymerase chain reaction
(qPCR)
qPCR assay was carried out on a 10-fold serially
diluted positive control for each target gene in order to construct
the standard curves. Copy numbers for the standards were calculated
as previously described (50).
The reaction was carried out with Maxima SYBR-Green/Fluorescein
qPCR master mix (Fermentas) using Thermo Cycler Apparatus (Bio-Rad,
Philadelphia, PA, USA) in a total volume of 25 μl. The PCR protocol
was generated using the software program as previously described
(49). Melting curve analysis was
performed to confirm the specificity of the amplified products.
Data acquisition and analysis were performed using CFX Manager
version 2.0. Primer sequences used for M2 amplification were
described in a previous study (49). Amplification for the NP
gene (CY039994) was carried out with NP-A-For primer (CAG ACC AAA
TGA AAA CCC AGC) and NP-A-Rev primer (AAT CTG AAC CCC TCT TGT GG)
at a 147 bp length from position 973 to 1120 of the NP gene.
Amplification for the ACTA1 gene (NM_001195845.1) was
carried out with ACTA1-For primer (TTC CGC TGC CCA GAG GCT CT) and
ACTA1-Rev primer (GCT CAG GGG GTG CGA TGA TCT TG) for the internal
control at a 240 bp length from position 909 to 1249 of
ACTA1 gene.
Cytokine protein quantification
Confluent monolayer of MDCK cells incubated for 24 h
at 37°C, 5% CO2 in 96-well flat-bottom tissue culture
plate was exposed to 100 TCID50 of H1N1 in the presence
or absence of simvastatin. Untreated MDCKs were considered as the
negative control. Cell-free supernatants were treated for 24, 48
and 72 h at different concentrations in duplicate and then stored
at −80°C for the cytokine analysis. Quantitative sandwich ELISA was
performed by Quantikine ELISA kits (R&D Systems, Minneapolis,
MN, USA) as per the manufacturer’ instructions.
Immunofluorescent labeling and confocal
laser scanning microscopy
MDCK cells were cultured and allowed to grow up to
80% confluency in 8-well chamber-slides (8×104
cell/well) for 24 h. The cells were then treated with simvastatin
at a concentration of 10 μM in the presence or absence of H1N1 at a
concentration of 100 TCID50/0.1 ml. At the end of the
incubation period of 24 h, the cells were fixed in 3%
paraformaldehyde in PBS for 10 min at 4°C. The cells were then
washed with PBS and permeabilized at room temperature with 0.2%
Triton-X-100 and blocked with 3% non-fat dry milk in PBS for 1 h at
4°C. The cells were then labeled with rhodamine 110 phalloidin
staining at a concentration of 2 μM (1:50 dilution in blocking
buffer) for 20 min at room temperature. DAPI (1 μg/ml) was used as
the nuclear counterstain. The slide was then mounted with warm
anti-fade reagent and sealed with clear lacquer.
Fluorescent images were obtained by confocal
microscopy (LSM 518F; Zeiss, Jena, Germany) using suitable filters
(excitation at 502 nm and emission at 524 nm for rhodamine 110
phalloidin and excitation at 358 nm and emission at 461 nm for
DAPI) and 100× oil immersion lens, pixel resolution of 0.164 μm,
scan speed of 5, and four-line averaging from an argon-krypton
laser. The micrographs were processed using appropriate software
(LSM5) and the average fluorescence intensities in all the
treatments as a ratio to the untreated control were used to
quantify the intensity results.
Sample preparation for RhoA, Rab and LC3
protein immunoblotting
MDCK cells were cultured in 75 cm2
flasks. To detect prenylated RhoA protein, the cells were
simultaneously treated with simvastatin and Y-27632 (10 μM) in the
presence or absence of H1N1 (100 TCID50/0.1 ml) for 48
h. To detect prenylated Rab proteins, the cells were treated with
simvastatin (10 μM) in the presence or absence of H1N1 (100
TCID50/0.1 ml) for 48 h. The effects of simvastatin for
these detections were disrupted by 6 h pretreatment of exogenous
FPP and GGPP (10 μM). To detect LC3 protein, MDCK cells were
pretreated with BafA1 (20 nM) as a lysosome activity inhibitor for
6 h with and/or without simvastatin (10 μM) in the presence or
absence of H1N1 (100 TCID50/0.1 ml) for 48 h.
For all the treatments, the cells were trypsinized,
scraped, collected in ice-cold PBS and centrifuged to form pellets.
For RhoA and Rabs detection, the cell pellet was re-suspended and
homogenized in 1 ml of homogenizing buffer mixed with protease
inhibitor cocktail. The homogenate was centrifuged at 700 × g for
10 min, at 4°C. The pellet was then discarded and the supernatant
was subjected to high-speed centrifugation at 15,000 × g for 1 h,
at 4°C. The pellet (membrane fraction) was dissolved in 100 μl of
0.5% Triton X-100 in PBS. All the membrane and cytosol fractions
were stored at −80°C. For LC3 detection, the cell pellet was
re-suspended and homogenized in 1 ml of CytoBuster lysis buffer and
protease inhibitor cocktail at room temperature. The homogenate was
centrifuged at high speed at 15,000 × g for 1 h, at 4°C. The pellet
was discarded and the supernatant was stored at −80°C for
subsequent use. The protein content in each sample was quantified
using the Bradford assay.
Detection of RhoA and Rabs translocation
and LC3 lipidation in MDCK cells
A quantity of 0.5 μg protein of each sample was
fractioned using sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) in 15% mini-gels and transferred onto
nitrocellulose membrane (Bio-Rad) by vertical semi-dry
electroblotting. Membranes were blocked for 1 h at room temperature
using milk diluent as blocking buffer (Kirkegaard and Perry
Laboratories, Gaithersburg, MD, USA). Following three washing steps
with Tris-buffered saline (TBS)-Tween, the membranes were incubated
overnight with primary antibody at 4°C. Following incubation with
primary antibody and washing, the membranes were exposed to
alkaline phosphatase-conjugated antibody for 1 h at room
temperature. Following several washing steps, the enzymatic
reaction was visualized using a BCIP-NBT substrate. Antibodies
against Pan-cadherin and GAPDH housekeeping proteins were used for
membrane and cytosolic loading controls, respectively. Blots were
imaged using Odyssey infrared imaging system (Li-COR Biosciences,
Lincoln, NE, USA). Quantification was performed on single channels
using the analysis software provided in the Odyssey Infrared
Imaging System (Li-COR Biosciences, Lincoln, NE, USA). For each
sample, signal intensities were normalized to the appropriate
internal control.
Modulation of the lysosome localization
in MDCK cells
To investigate lysosomal activity and localization,
MDCK cells were cultured in 8-well chamber-slides (8×104
cell/well) for 24 h and further treatment with simvastatin (10 μM)
and H1N1 (100 TCID50/0.1 ml) was conducted in wet
chambers for 24 h. At the end of the incubation period the
lysosomal mass was visualized by staining with LysoTracker Red
DND-99 probe. Briefly, the cells were washed with PBS. Pre-warmed
LysoTracker Red containing media at 50 nM concentration of probe
was added to the coverslip for 25 min under growth conditions
appropriate for the cell culture in the dark. Loading solution was
replaced with fresh media. The cells were fixed with 3%
paraformaldehyde for 10 min at room temperature. The slide was
washed with PBS, mounted with warm mounting buffer
ProLong® Gold anti-fade reagent, sealed with clear
lacquer and stored at 4°C the dark overnight. Images were obtained
by confocal microscopy (Zeiss LSM 518F; Zeiss) with proper filters
(excitation at 577 nm and emission at 592 nm) using 63× objectives
(oil, NA=104), pixel resolution of 0.164 μm, scan speed 5 and
four-line averaging. Images were processed with the appropriate
software (LSM5) and intensity data [mean ± standard deviation (SD)]
were used to quantify the results.
Statistical analysis
Statistical analysis of the data was performed using
SPSS 20.0. Data were presented as mean ± SD. A two-way or one-way
analysis of variance (ANOVA) post-hoc LSD test was used to analyze
the data for cytotoxicity, HA, qPCR and ELISA assays. The Student’s
t-test was used to examine the effect of treatments on actin
filament polymerization, lysosomal mass localization, RhoA and Rab
protein prenylation and LC3 lipidation. For all the tests, P≤0.05
was considered significant.
Results
Cell viability
The viability of MDCK cells following 24, 48 and 72
h treatment with different concentrations of simvastatin as
determined by MTT assay are shown in Table I. The results showed that
simvastatin had no cytotoxic effect on the cell at concentrations
up to 15 μM. The EC50 of simvastatin was calculated from
MTT data using the two-way ANOVA test at a concentration of 10 μM,
which had no significant cytotoxic effect on the cell viability as
compared to the control.
 | Table IMTT cell viability results following
simvastatin treatments on MDCK cells. |
Table I
MTT cell viability results following
simvastatin treatments on MDCK cells.
| Concentration
(μM) | OD (mean ± SD)
Simvastatin |
|---|
| 20 | 0.27±0.19a |
| 15 | 0.40±0.28a |
| 10 | 0.78±0.19 |
| 5 | 0.79±0.12 |
| 2 | 0.83±0.10 |
| 1 | 0.93±0.07 |
| Control | 1.00±0.00 |
Inhibitory effect of statins on the
virus
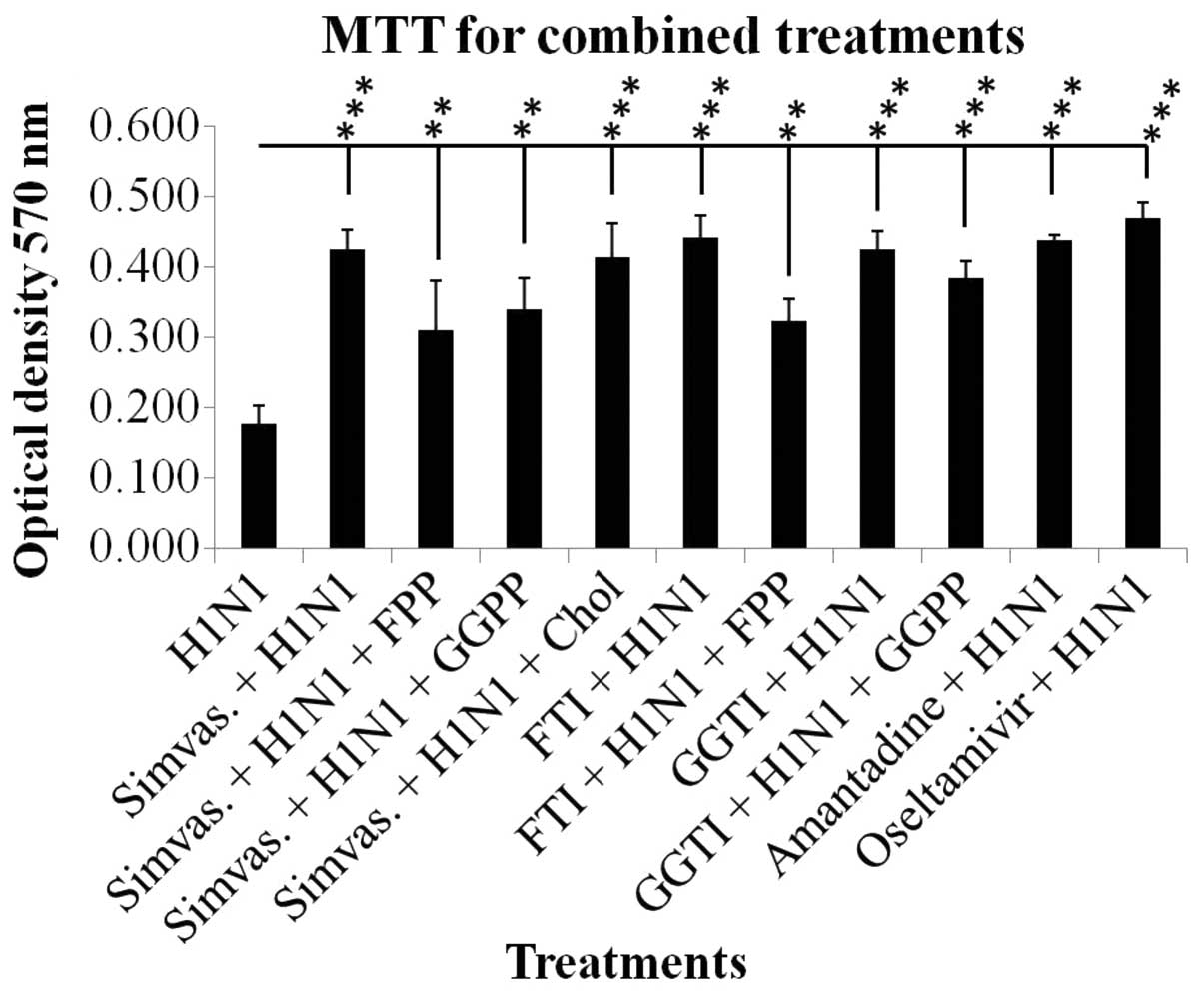
Increased optical density in the combined treatments
of simvastatin and H1N1 as compared to H1N1 alone was markedly
significant (P≤0.001). The effect of simvastatin on cell viability
was affected by exogenous FPP and GGPP although this effect was not
significant; however, cell viability was not affected by
cholesterol (Fig. 1). The
significant increase in cell viabilities as compared to H1N1
infection demonstrated the protective effect of simvastatin on the
cell viability against viral cytopathic effects. The results of
FTI, GGTI, amantadine and oseltamivir treatments are shown in
Fig. 1. Treatment with Y-27632
also showed similar results to simvastatin (data not shown).
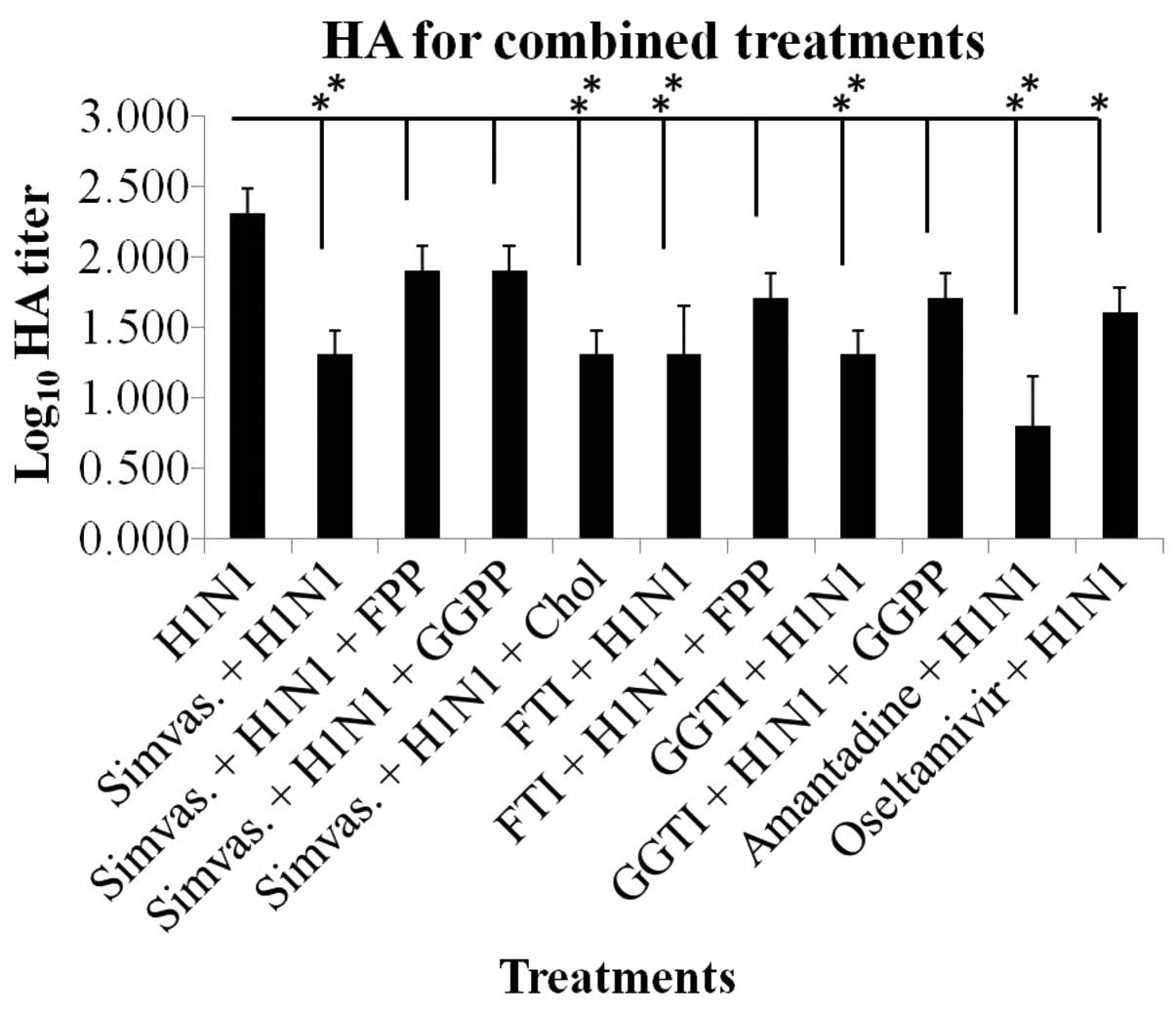
Hemagglutination assay results
Based on HA titration, the inhibitory effect of
simvastatin on viral adsorption to the cell surface was
demonstrated by a significant reduction in the HA titer unit in the
combined treatments (P≤0.01 and P≤0.05) (Fig. 2). The results of FTI, GGTI,
amantadine and oseltamivir treatments are shown in Fig. 2. Y-27632 treatment results were
similar to the simvastatin effects (data not shown). Notably, the
addition of exogenous FPP and GGPP reversed the effects of
simvastatin on HA titration although not significantly, while
cholesterol did not interfere with the effects of statins.
Absolute quantification
Amplification graph and melt curve analysis for each
gene amplification confirmed the specificity of the amplified
products (data not shown). Quantitative analysis of PCR products of
NP and M2 IAV genes at different time points yielded significant
decrements in log10 copy numbers of viral genes compared
to H1N1-inoculated cells (P≤0.01 and P≤0.001). Amantadine treatment
was conducted as the control treatment (Table II).
| Table IINucleoprotein and matrix 2 genes
log10 copy numbers in different treatments. |
Table II
Nucleoprotein and matrix 2 genes
log10 copy numbers in different treatments.
| Log10
copy nos./μl (mean ± SD) |
|---|
|
|
|---|
| Absolute
quantification | Co-inoculation
treatment | Pre-inoculation
treatment | Post-inoculation
treatment |
|---|
| NP gene |
| Infected |
| H1N1 | 11.644±0.008 | | |
| Simvastatin |
11.283±0.002a |
10.990±0.011b |
11.451±0.027b |
| Amantadine |
11.240±0.011a |
11.303±0.053a |
11.036±0.200a |
| M2 gene |
| Infected |
| H1N1 | 11.620±0.039 | | |
| Simvastatin |
10.912±0.013a |
10.548±0.001a |
11.247±0.047a |
| Amantadine |
10.821±0.050a |
10.924±0.187a |
10.902±0.136a |
Quantification of cytokine expression
levels by ELISA assay
TNF-α, IL-6 and IFN-γ proteins were not detectable
in supernatants of MDCK cell culture at 24 and 48 h after exposure
(data not shown). However, at 72 h treatments, H1N1 infection
exhibited highly significant expression levels of these cytokines
compared to the simvastatin-treated supernatants (P≤0.001). Fold
reduction of the protein expression shown in Table III was averaged at 2.5, 1.5 and
2 for TNF-α, IL-6 and IFN-γ, respectively.
| Table IIITNF-α, IL-6 and IFN-γ protein
reduction in MDCK culture supernatants following 72 h exposure time
as compared to the H1N1 treatment. |
Table III
TNF-α, IL-6 and IFN-γ protein
reduction in MDCK culture supernatants following 72 h exposure time
as compared to the H1N1 treatment.
| Treatments | TNF-α fold
reduction compared to virus | IL-6 fold reduction
compared to virus | IFN-γ fold
reduction compared to virus |
|---|
| Negative
control | 2.02 | 1.49 | 2.29 |
| Simvastatin | 3.05 | 1.49 | 1.67 |
| Simvastatin +
H1N1 | 2.44 | 1.61 | 1.57 |
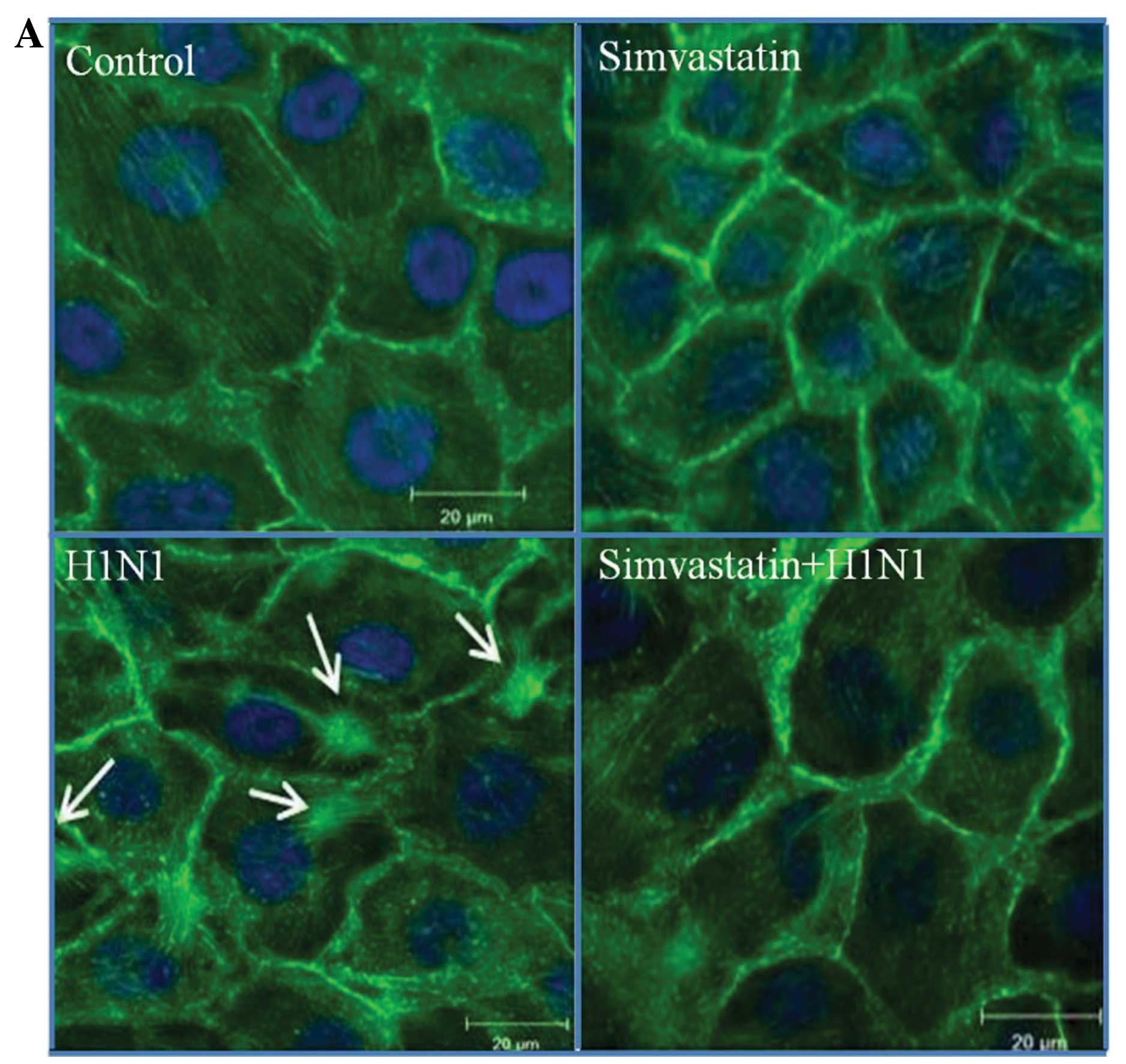
Alterations of actin cytoskeleton
organization
Images of MDCK cells grown under normal conditions
showed spindle-like appearance with numerous distinct and
well-organized actin fibers extending across the cytosol to the
plasma membrane. Following treatment with simvastatin at a
concentration of 10 μM, actin filaments appeared disassembled with
loss of correlation between spanning fibers. Infection of MDCK
cells with H1N1 showed an increased incidence of actin stress
fibers and remodeling of the actin cytoskeleton switching to a
condensed star-shape appearance. Images of the combined treatments
clearly showed the inhibitory effect of simvastatin on actin
filament condensation caused by H1N1 (Fig. 3A). These images were processed
with LSM5 software and the average fluorescence intensities of at
least 10-fold repetitions (mean ± SD) were used to quantify the
results. Data were normalized to the negative control. Statistical
analysis of fluorescent intensity for these treatments showed a
significant decrement in the combined treatment as compared to
H1N1. This difference was highly significant (P≤0.001) (Fig. 3B).
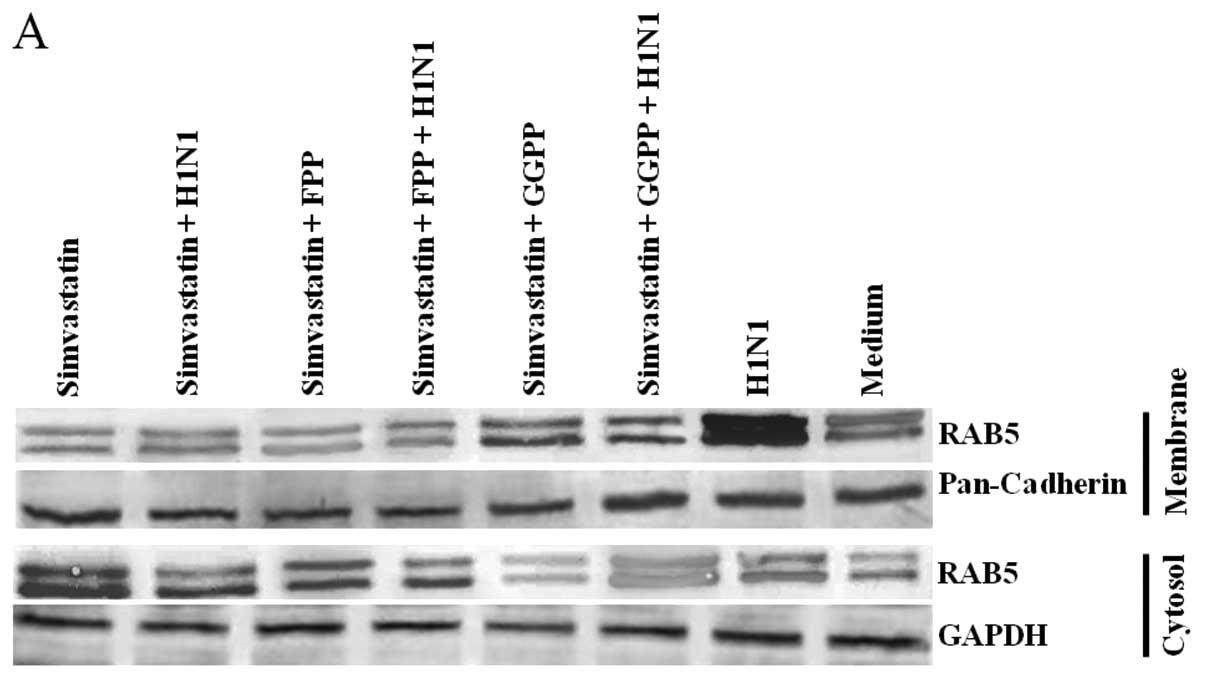
Simvastatin and H1N1 effects on RhoA and
Rabs translocation and LC3 lipidation
Cell distributions of RhoA and Rab proteins in
membrane and cytosol sub-cellular fractions with different
treatments are shown in the western blots of Figs. 4A, 5A and 6A. Simvastatin treatment depleted these
proteins from the membrane fractions, resulting in their enrichment
in the cytosol section. The increase of RhoA and Rabs in membrane
fraction for H1N1 samples and its depletion in cytosol fraction
were evident. Treatment with Y-27632 as a selective inhibitor of
Rho-associated protein kinases also resulted in a decrease in RhoA
protein membrane translocation.
Statistical analysis verified the significant cell
distributions (P≤0.001). Membrane localization of RhoA and Rabs was
significantly decreased in simvastatin- and Y-27632-treated cells
as compared to that of H1N1 (Figs.
4B, 5B and 6B) and their incidence in cytosol showed
a significant increase compared to H1N1 (Figs. 4C, 5C and 6C). The effect of simvastatin was
disrupted by exogenous FPP and GGPP, which increased the protein
membrane translocation and was depleted in the cytosol section.
GAPDH and Pan-cadherin as cytosolic and membranous housekeeping
proteins remained unchanged after the treatments.
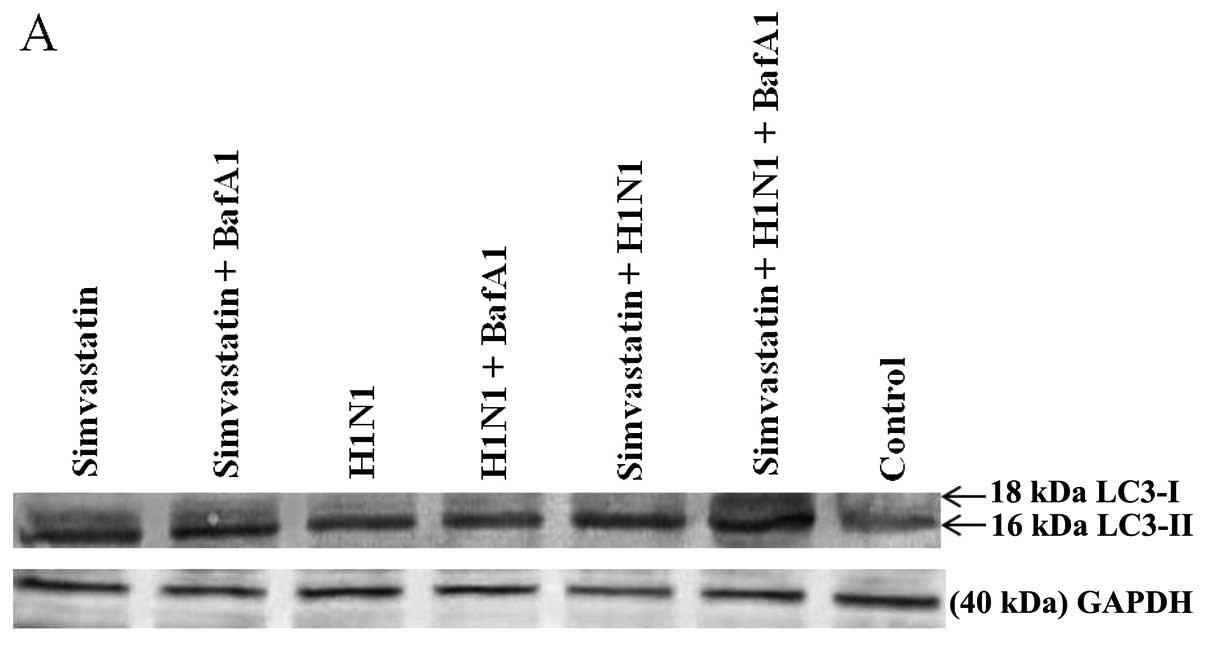
Different distribution of lipidated LC3 (LC3-II) in
different treatments are shown in the western blots in Fig. 7A. Based on statistical analysis
(Fig. 7B), the membrane
localization of LC3 protein in simvastatin and H1N1 samples was
increased as compared to the control sample. Furthermore, LC3-II in
the combined treatments of simvastatin with BafA1 was increased
compared to the simvastatin treatment, but it was not affected in
the combined treatment of H1N1 with BafA1. In addition, BafA1
increased the LC3-II to the highest value in the combined treatment
of simvastatin and H1N1. GAPDH as a housekeeping protein was well
detected and remained intact during the treatments.
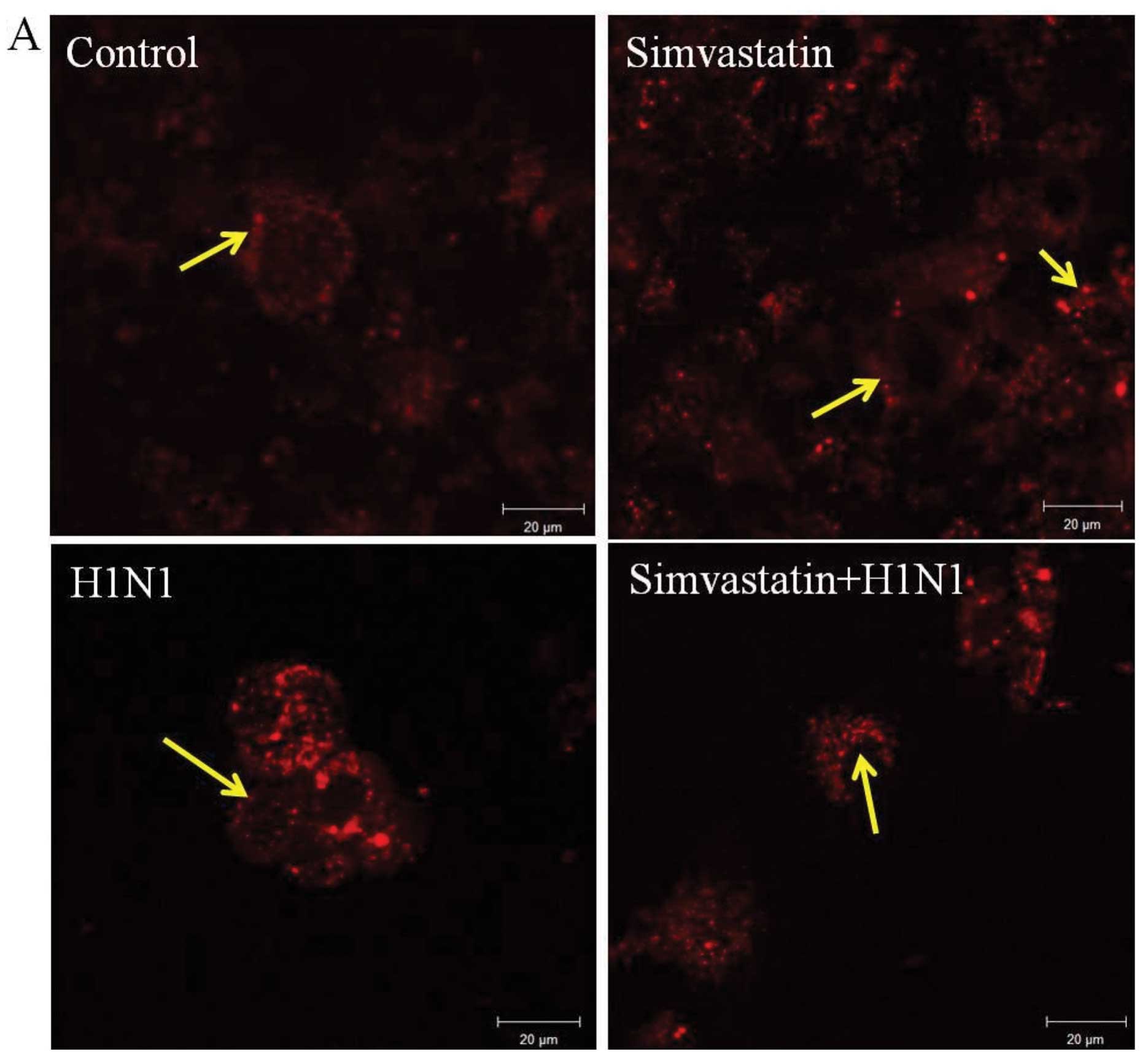
Lysosome localization detection
MDCK cells in the control had a normal appearance in
the punctate staining of lysosomes (Fig. 8A). Following treatment with
simvastatin and H1N1 after 24 h, these localized compartments were
elevated by an increase in spots and dense acidified lysosomes
around the nucleus. The combined treatments of simvastatin and H1N1
showed a similar appearance in density values. These images were
processed with LSM5 software and the average fluorescence
intensities of three repetitions normalized to the control sample
(mean ± SD) were used to quantify the results. The density of the
acidified lysosomes increased similarly in all statins and virus
treatments. Nevertheless, no significant differences were found in
terms of lysosomal volume among mock-infected (control),
simvastatin-treated, H1N1-infected and combined treatments
(Fig. 8B).
Discussion
Influenza A infection affects children, the elderly,
as well as other high-risk groups (51,52). Available studies on the beneficial
effects of statin treatments during the course of IAV infection are
based on retrospective observational studies, which have shown that
statins reduce morbidity during influenza pandemics as well as
hospitalizations and deaths associated with seasonal influenza
outbreaks (53,54). Statins potentially exert
beneficial effects on influenza-infected patients by modifying the
host response to the infection through mechanisms that are not
connected with virus replication. Statins improve outcome in
patients with sepsis and pneumonia (55,56). Furthermore, statin treatment of
patients hospitalized with influenza is associated with a 41%
reduction in a 30-day mortality, a reduction that may be
attributable to previous vaccination or antiviral treatment
(57). Authors of the
abovementioned studies are in agreement that the benefits obtained
from statins are due to their effects on the host response, and not
the pathogen itself. This is of importance as, in any influenza
patient treated with statin, the drug would affect the entire body,
including cells and organs, in which no influenza virus may be
found. However, the exact cell interactions and mechanisms of the
effect of statins on IAV replication remain to be determined.
Previously, we showed significant reductions in the
H1N1 viral titer in combined treatments with atorvastatin (45). The inhibitory effect of statins on
the dysregulation of TNF-α and IL-6 cytokines caused by IAV was
also demonstrated in CRFK cells (58).
The current study used different approaches to
determine the effects of simvastatin treatment in vitro to
reduce the ability of IAV to replicate. The concentration of 10 μM
of simvastatin, which did not cause any toxicity on cell viability
(Table I), was selected as the
effective concentration (EC50) for antiviral
experiments.
MTT and HA assays were conducted to verify the
effect of simvastatin on the cell viability increment and viral
load reduction. The metabolic effects of simvastatin were prevented
by the addition of FPP and GPP (downstream products of the HMG-CoA
reductase reaction) to the culture medium, but not by exogenous
cholesterol, which is a major end-product of this metabolic pathway
(Figs. 1 and 2). No significant effects of FPP and
GGPP on HA and MTT results were found to be associated with the
short-time incubations, which should be tested for longer
incubation time treatments.
The antiviral effects of simvastatin on H1N1 viral
load were examined using qPCR assay. In this test, the quantity of
target genes was evaluated with reasonable accuracy by using a
reliable standard. A standard curve, constructed from standard
concentrations (data not shown) was used to determine the copy
numbers of target genes related to the Ct value. The
log10 copy numbers which were calculated from the
concentrations against mean Ct values confirmed these significant
decrements (Table II). Using
ELISA confirmed that infection by H1N1 causes high expression
levels of target pro-inflammatory cytokines in MDCK, while in all
the combined treatments, the expression of these proteins decreased
significantly and showed notable fold reductions compared to that
of H1N1 (Table III). Therefore,
it is possible to restrict immune system overexpression and lung
inflammation by preventing the cell inflammatory responses to
infection if proper treatment with simvastatin is applied.
The present study hypothesized that inhibition of
RhoA and Rab protein prenylation by simvastatin could lead to
inhibition of H1N1 transfer through the nucleus and cytoplasm by
applying considerable changes in actin filament modulation and
endocytosis.
RhoA regulates the contraction and relaxation of the
actin cytoskeleton by coupling changes in the external environment
leading to alterations in the actin cytoskeleton (59). Moreover, the actin cytoskeleton,
which is imperative to viral translocation (60–62), is correlated with RhoA
prenylation. Thus, the factors that restrict actin contractile
function through inhibition of RhoA isoprenylation may have
anti-influenza beneficial effects. The results of our study
provided sufficient evidence to support the hypothesis that
inhibition of RhoA and Rab protein membrane translocation as an
underlying mechanism of action for simvastatin was responsible for
the suppression of H1N1 replication.
Results of the present study revealed that H1N1
infection caused the activation of RhoA protein prenylation and
induction of actin cytoskeleton remodeling. In addition, it was
demonstrated that simvastatin reduced the replication of H1N1 by
possible blocking of RhoA prenylation and membrane localization. It
was shown that the cell distributions of the prenylated form of
this protein in simvastatin-treated, Y-27632-treated and combined
treatments of H1N1 were significantly different from H1N1 alone
(P≤0.001) (Fig. 4). Furthermore,
the present study demonstrated that simvastatin exerted direct cell
effects on actin filament condensation (Fig. 3). During treatments using
simvastatin, cell spanning actin fibers were reduced resulting in
loosening of the actin cytoskeleton tightness and correlation. In
H1N1-inoculated cells viruses induced actin filament condensation.
This phenomenon suggests that H1N1 is involved in intermediate cell
compartments requiring their movement through endocytosis, which is
the verification of the critical role of simvastatin in regulating
the state of intracellular actin filaments (63). Our findings are in agreement with
the results from previous studies, which showed round and
refractile morphology in lovastatin-treated cells (30 μM), which
was due to the significant reduction in actin cables polymerization
(17,64). These findings were also consistent
with previous reports, which emphasized the role of Rho protein
family in decreasing viral loads in an in vivo model of an
acute HIV-1 infection, where it was suggested that changes in
RhoA-controlled actin cytoskeleton rearrangements inhibit HIV-1
entry and exit from host cells (65).
It was also revealed that isoprenylation of RhoA
facilitated the transport of H1N1 through an actin-dependent
transport system. Inhibition of RhoA isoprenylation is necessary to
suppress H1N1 replication. In addition, simvastatin can affect the
upregulated RhoA pathway induced by H1N1.
Simvastatin treatments also led to a considerable
decrease in Rabs prenylation, which affected H1N1 replication and
promoted delocalization of Rab proteins from the endosomal membrane
to cytosol. Therefore, the transport of viruses through early and
late endosome vesicles may be affected. By contrast, H1N1 promoted
their localization from cytosol to membrane. Density values of the
expression of these proteins in different treatments showed
significant differences in the cellular distribution of these
proteins compared to H1N1 (P≤0.001). This effect was detectable in
early and late endosomes at the level of Rab5 and Rab7 proteins,
however, the expression level in early endosomes (Fig. 5) was more evident as compared to
that of the late endosomes (Fig.
6). This could be deduced from the limited stability of the
late endosomes (66). The
simvastatin effect was significantly disrupted (P≤0.001) by adding
GGPP but not FPP, whereby the GGPP reversed the statin effect and
increased the Rab protein expression level in the membrane
fraction. Inhibiting Rab protein prenylation appears to be another
common mechanism of action for simvastatin as an anti-influenza
agent. Besides H1N1, a previous study has shown the antiviral
effects of simvastatin and lovastatin on HIV production in
HIV-1-infected cells by affecting Rab11 geranylgeranylation
(67). Furthermore, fluvastatin
is also considered clinically useful for the treatment of diabetes
by inhibiting Rab protein prenylation (68).
Other important pathways such as autophagy and
lysis, which are involved in the pathogenesis of IAV (69), were also considered. This study
highlights the effects of simvastatin on autophagy and lysis in the
combined treatment with H1N1.
Modulation of autophagy by affecting LC3 as one of
the most important molecules involved in autophagy may be an
important mechanism to control cell component degradation (70,71). If cells cannot activate autophagy,
protein synthesis predominates over protein degradation. By
contrast, autophagy could be activated to guarantee survival of the
cells (44,72). Previous studies have shown that
during influenza infection, autophagosomes do not fuse with acidic
compartments (70). Thus,
accumulation of autophagosomes in H1N1-infected cells causes an
increase in IAV survival (73).
Therefore, inhibiting autophagy may inhibit virus replication
(74). Viruses such as herpes
simplex virus 1 (HSV-1), kaposi sarcoma-associated herpes virus
(KSHV) and mouse herpes virus 68 (MHV-68) inhibit the autophagosome
formation to escape autophagic degradation (69) while poliovirus, HIV-1 and mouse
hepatitis virus (MHV) can block degradation of autophagosomes and
use them as a platform to assemble their RNA complexes to increase
the viral yield (70).
Therefore, blocking of autophagosome degradation
would lead to gradual accumulation of autophagosomes and LC3-II
(69). However, simvastatin
increased autophagosome formation, leading to a certain degree of
retardation in the maturation process of autophagosomes.
In this study, LC3-II membrane localization by
western blot analysis showed that simvastatin was performed
directly on autophagosome marker LC3 lipidation and its membrane
localization and that the membrane localization of LC3-II was
elevated. Thus it may be hypothesized that simvastatin treatments
increased formation of autophagosomes. Moreover, the combined
treatment of simvastatin with BafA1 as an inhibitor of lysosome
activity increased the LC3-II level significantly (P≤0.01), which
confirmed the results of simvastatin effects on LC3 lipidation. For
H1N1 it was obvious that virus inoculation inhibited autophagosome
degradation by LC3-II accumulation in cell lysate fractions and
inhibited autophagosome maturation as the membrane localization of
LC3-II was the same with or without BafA1 treatment. In the
combined treatments of simvastatin and H1N1, the values showed
increased intensities with the highest significant values amongst
all the treatments (Fig. 7). The
findings from this study were in agreement with those of a previous
study on primary human lung mesenchymal cells, which emphasized
that the relative amount of LC3-II is connected with the dynamic
turnover of LC3-II via the lysosome activity (75). Thus, it is suggested that LC3
protein lipidation and membrane localization was upregulated by
simvastatin and H1N1. However, simvastatin plays a direct role on
LC3 lipidation through cholesterol depletion (42,76), although H1N1 involvement in
autophagy seems to be far more complicated due to the effects of
the upregulation of the expression of several autophagy-related
genes that can increase the autophagic flux (74).
Evaluating lysosomal mass by fluorescent technique
also showed that simvastatin and H1N1 exposure to the cells caused
increments in the lysosomal mass, which, however, were not
significantly different from those of the control (Fig. 8). Simvastatin induced lysosomal
activity by increasing LC3 protein lipidation or promoting its
localization, which is an important factor for autophagosome
maturation. However, simvastatin treatment for H1N1 infection is
potentially a double-edged sword as a decrease of cholesterol
enhances autophagy, whereas if the reduction of cholesterol retards
the maturation of autophagosomes, it may exacerbate the disease
since the infectious agent is alive in the autophagosome.
In conclusion, findings from this study have
demonstrated the ability of simvastatin to function in common cell
pathways in order to block the virus-host interaction system, which
renders it an effective agent against H1N1 infection. This process
is the mechanism underlying the effects of potential anti-influenza
agents such as HMG-CoA reductase inhibitors and other similar
agents that may improve the development of new strategies to
control H1N1 infection in influenza outbreaks. Since simvastatin
exhibits antiviral effects by modulating common cellular machinery
pathways, it is hypothesized that the mechanisms of the antiviral
effects of simvastatin are not limited to its effect on RhoA and
Rab proteins and thus downstream-related pathways in this context
should be investigated. It is also worth considering the timing
and/or duration of statin usage in relation to IAV infection, and
to determine whether the treatment should be administered prior to
infection as a preventative agent, or following onset of
symptoms.
Acknowledgements
This study was supported by grant no. 01-02-04-009
BTK/ER/38 from the Ministry of Science, Technology and Innovation,
Government of Malaysia.
References
|
1
|
Cloutier A, Marois I, Cloutier D,
Verreault C, Cantin AM and Richter MV: The prostanoid
15-deoxy-Δ12,14-prostaglandin-j2 reduces lung inflammation and
protects mice against lethal influenza infection. J Infect Dis.
205:621–630. 2012.
|
|
2
|
Fedson DS: Confronting an influenza
pandemic with inexpensive generic agents: can it be done? Lancet
Infect Dis. 8:571–576. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Calero M, Chen CZ, Zhu W, et al: Dual
prenylation is required for Rab protein localization and function.
Mol Biol Cell. 14:1852–1867. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bright RA, Shay DK, Shu B, Cox NJ and
Klimov AI: Adamantane resistance among influenza A viruses isolated
early during the 2005–2006 influenza season in the United States.
JAMA. 295:891–894. 2006.
|
|
5
|
Hayden FG: Antiviral resistance in
influenza viruses-implications for management and pandemic
response. N Engl J Med. 354:785–788. 2006. View Article : Google Scholar
|
|
6
|
Dharan NJ, Gubareva LV, Meyer JJ, et al:
Infections with oseltamivir-resistant influenza A (H1N1) virus in
the United States. JAMA. 301:1034–1041. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fedson DS: Pandemic influenza: a potential
role for statins in treatment and prophylaxis. Clin Infect Dis.
43:199–205. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haidari M, Zhang W, Ganjehei L, Ali M and
Chen Z: Inhibition of MLC phosphorylation restricts replication of
influenza virus-A mechanism of action for anti-influenza agents.
PLoS One. 6:e214442011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Whittaker GR: Intracellular trafficking of
influenza virus: clinical implications for molecular medicine.
Expert Rev Mol Med. 3:1–13. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rothberg MB and Haessler SD: Complications
of seasonal and pandemic influenza. Crit Care Med. 38(Suppl 4):
e91–e97. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
de Jong MD, Simmons CP, Thanh TT, et al:
Fatal outcome of human influenza A (H5N1) is associated with high
viral load and hypercytokinemia. Nat Med. 12:1203–1207.
2006.PubMed/NCBI
|
|
12
|
Wilkins C and Gale M Jr: Recognition of
viruses by cytoplasmic sensors. Curr Opin Immunol. 22:41–47. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Uetani K, Hiroi M, Meguro T, et al:
Influenza A virus abrogates IFN-gamma response in respiratory
epithelial cells by disruption of the Jak/Stat pathway. Eur J
Immunol. 38:1559–1573. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Osterholm MT: Preparing for the next
pandemic. New Engl J Med. 352:1839–1842. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Darwish I, Mubareka S and Liles WC:
Immunomodulatory therapy for severe influenza. Expert Rev Anti
Infect Ther. 9:807–822. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang CY, Liu PY and Liao JK: Pleiotropic
effects of statin therapy: molecular mechanisms and clinical
results. Trends Mol Med. 14:37–44. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fessler MB, Young SK, Jeyaseelan S, et al:
A role for hydroxy-methylglutaryl coenzyme A reductase in pulmonary
inflammation and host defense. Am J Respir Crit Care Med.
171:606–615. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun X and Whittaker GR: Role for influenza
virus envelope cholesterol in virus entry and infection. J Virol.
77:12543–12551. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng J, Ohsaki Y, Tauchi-Sato K, Fujita A
and Fujimoto T: Cholesterol depletion induces autophagy. Biochem
Biophys Res Commun. 351:246–252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Terblanche M, Almog Y, Rosenson RS, Smith
TS and Hackam DG: Statins and sepsis: multiple modifications at
multiple levels. Lancet Infect Dis. 7:358–368. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Watts KL, Sampson EM, Schultz GS and
Spiteri MA: Simvastatin inhibits growth factor expression and
modulates profibrogenic markers in lung fibroblasts. Am J Respir
Cell Mol Biol. 32:290–300. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alexeeff SE, Litonjua AA, Sparrow D,
Vokonas PS and Schwartz J: Statin use reduces decline in lung
function: VA Normative Aging Study. Am J Respir Crit Care Med.
176:742–747. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bohn W, Rutter G, Hohenberg H, Mannweiler
K and Nobis P: Involvement of actin filaments in budding of measles
virus: studies on cytoskeletons of infected cells. Virology.
149:91–106. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Salas PJI, Misek DE, Vega-Salas DE,
Gundersen D, Cereijido M and Rodriguez-Boulan E: Microtubules and
actin filaments are not critically involved in the biogenesis of
epithelial cell surface polarity. J Cell Biol. 102:1853–1867. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Macejak DG and Luftig RB: Stabilization of
actin filaments at early times after adenovirus infection and in
heat-shocked cells. Virus Res. 19:31–46. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Smythe E and Ayscough RK: Actin regulation
in endocytosis. J Cell Sci. 119:4589–4598. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Winder SJ and Ayscough KR: Actin-binding
proteins. J Cell Sci. 118:651–654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang W, Du L and Gunst SJ: The effects of
the small GTPase RhoA on the muscarinic contraction of airway
smooth muscle result from its role in regulating actin
polymerization. Am J Physiol Cell Physiol. 299:C298–C306. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang J, Nikrad MP, Travanty EA, et al:
Innate immune response of human alveolar macrophages during
influenza A infection. PLoS One. 7:e298792012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hall A: Rho GTPases and the control of
cell behaviour. Biochem Soc Trans. 33:891–895. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen LM, Hobbie S and Galan JE:
Requirement of CDC42 for Salmonella-induced cytoskeletal and
nuclear responses. Science. 274:2115–2118. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ellis S and Mellor H: The novel Rho-family
GTPase rif regulates coordinated actin-based membrane
rearrangements. Curr Biol. 10:1387–1390. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heusinger-Ribeiro J, Fischer B and
Goppelt-Struebe M: Differential effects of simvastatin on mesangial
cells. Kidney Int. 66:187–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schaafsma D, McNeill KD, Mutawe MM, et al:
Simvastatin inhibits TGFβ1-induced fibronectin in human airway
fibroblasts. Respir Res. 12:1132011.
|
|
35
|
Samaj J, Baluska F, Voigt B, Schlicht M,
Volkmann D and Menzel D: Endocytosis, actin cytoskeleton and
signaling. Plant Physiol. 135:1150–1161. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Griffiths G, Hoflack B, Simons K, Mellman
I and Kornfeld S: The mannose 6-phosphate receptor and the
biogenesis of lysosomes. Cell. 52:329–341. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pfeffer S: Membrane domains in the
secretory and endocytic pathways. Cell. 112:507–517. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Matarrese P, Nencioni L, Checconi P, et
al: Pepstatin A alters host cell autophagic machinery and leads to
a decrease in influenza A virus production. J Cell Physiol.
226:3368–3377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sieczkarski SB and Whittaker GR:
Dissecting virus entry via endocytosis. J Gen Virol. 83:1535–1545.
2002.PubMed/NCBI
|
|
40
|
Sieczkarski SB and Whittaker GR:
Differential requirements of Rab5 and Rab7 for endocytosis of
influenza and other enveloped viruses. Traffic. 4:333–343. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mann SS and Hammarback JA: Molecular
characterization of light chain 3. A microtubule binding subunit of
MAP1A and MAP1B. J Biol Chem. 269:11492–11497. 1994.PubMed/NCBI
|
|
42
|
Ghavami S, Eshragi M, Ande SR, et al:
S100A8/A9 induces autophagy and apoptosis via ROS-mediated
cross-talk between mitochondria and lysosomes that involves BNIP3.
Cell Res. 20:314–331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang Q, Yang YJ, Wang H, et al: Autophagy
activation: a novel mechanism of atorvastatin to protect
mesenchymal stem cells from hypoxia and serum deprivation via
AMP-activated protein kinase/mammalian target of rapamycin pathway.
Stem Cells Dev. 21:1321–1332. 2012. View Article : Google Scholar
|
|
44
|
Noda T, Fujita N and Yoshimori T: The late
stages of autophagy: how does the end begin? Cell Death Differ.
16:984–990. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mehrbod P, Ideris A, Omar AR and Hair-Bejo
M: Evaluation of antiviral effect of atorvastatin on H1N1 infection
in MDCK cells. Afr J Mic Res. 6:5715–5719. 2012.
|
|
46
|
Karber G: 50% endpoint calculation. Arch
Exp Pathol Pharmacol. 162:480–483. 1931.
|
|
47
|
Levi R, Beeor-Tzahar T and Arnon R:
Microculture virus titration - a simple colourimetric assay for
influenza virus titration. J Virol Methods. 52:55–64. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hirst GK: The agglutination of red cells
by allantoic fluid of chick embryos infected with influenza virus.
Science. 94:22–23. 1941. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mehrbod P, Ideris A, Omar AR, et al:
Attenuation of influenza virus infectivity with herbal-marine
compound (HESA-A): an in vitro study in MDCK cells. Virol J.
9:442012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Godornes C, Leader BT, Molini BJ,
Centurion-Lara A and Lukehart SA: Quantitation of rabbit cytokine
mRNA by real-time RT-PCR. Cytokine. 38:1–7. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Khandaker G, Dierig A, Rashid H, King C,
Heron L and Booy R: Systematic review of clinical and
epidemiological features of the pandemic influenza A (H1N1) 2009.
Influenza Other Respir Viruses. 5:148–156. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Damak H, Chtara K, Bahloul M, et al:
Clinical features, complications and mortality in critically ill
patients with 2009 influenza A (H1N1) in Sfax, Tunisia. Influenza
Other Respir Viruses. 5:230–240. 2011.
|
|
53
|
Brett SJ, Myles P, Lim WS, et al:
Pre-admission statin use and in-hospital severity of 2009 pandemic
influenza A (H1N1) disease. PLoS One. 6:e181202011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kwong JC, Li P and Redelmeier DA:
Influenza morbidity and mortality in elderly patients receiving
statins: a cohort study. PLoS One. 4:e80872009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kopterides P and Falagas ME: Statins for
sepsis: a critical and updated review. Clin Microbiol Infect.
15:325–334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Falagas ME, Makris GC, Matthaiou DK and
Rafailidis PI: Statins for infection and sepsis: a systematic
review of the clinical evidence. J Antimicrob Chemother.
61:774–785. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Vandermeer ML, Thomas AR, Kamimoto L, et
al: Association between use of statins and mortality among patients
hospitalized with laboratory-confirmed influenza virus infections:
a multistate study. J Infect Dis. 205:13–19. 2012. View Article : Google Scholar
|
|
58
|
Mehrbod P, El Zowalaty M, Omar AR,
Hair-Bejo M and Ideris A: Statins reduce the expression of
proinflammatory cytokines in influenza A virus infected CrFK cells.
Acta Virol. 56:353–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hall A: Rho GTPases and the actin
cytoskeleton. Science. 279:509–514. 1998. View Article : Google Scholar
|
|
60
|
Burridge K and Wennerberg K: Rho and Rac
take center stage. Cell. 116:167–179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Gad AKB and Aspenström P: Rif proteins
take to the RhoD: Rho GTPases at the crossroads of actin dynamics
and membrane trafficking. Cell Signal. 22:183–189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Radtke K, Döhner K and Sodeik B: Viral
interactions with the cytoskeleton: a hitchhiker’s guide to the
cell. Cell Microbiol. 8:387–400. 2006.
|
|
63
|
Kreijtz JH, Fouchier RA and Rimmelzwaan
GF: Immune responses to influenza virus infection. Virus Res.
162:19–30. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Fenton RG, Kung HF, Longo DL and Smith MR:
Regulation of intracellular actin polymerization by prenylated
cellular proteins. J Cell Biol. 117:347–356. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
del Real G, Jiménez-Baranda S, Mira E, et
al: Statins inhibit HIV-1 infection by down-regulating Rho
activity. J Exp Med. 200:541–547. 2004.PubMed/NCBI
|
|
66
|
Simon I, Zerial M and Goody RS: Kinetics
of interaction of Rab5 and Rab7 with nucleotides and magnesium
ions. J Biol Chem. 271:20470–20478. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Amet T, Nonaka M, Dewan MZ, et al:
Statin-induced inhibition of HIV-1 release from latently infected
U1 cells reveals a critical role for protein prenylation in HIV-1
replication. Microbes Infect. 10:471–480. 2008. View Article : Google Scholar
|
|
68
|
Procino G, Barbieri C, Carmosino M, et al:
Fluvastatin modulates renal water reabsorption in vivo through
increased AQP2 availability at the apical plasma membrane of
collecting duct cells. Pflugers Arch. 462:753–766. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Rossman JS and Lamb RA: Autophagy,
apoptosis and the influenza virus M2 protein. Cell Host Microbe.
6:299–300. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Schmid D: Autophagy delivers viral
antigens for MHC class II presentation and is regulated by viral
infection. The Rockefeller University; 205. 2007
|
|
71
|
Tanida I, Minematsu-Ikeguchi N, Ueno T and
Kominami E: Lysosomal turnover, but not a cellular level, of
endogenous LC3 is a marker for autophagy. Autophagy. 1:84–91. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wang S, Li H, Chen Y, et al: Transport of
influenza virus neuraminidase (NA) to host cell surface is
regulated by ARHGAP21 and Cdc42 proteins. J Biol Chem.
287:9804–9816. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Schmid D and Münz C: Innate and adaptive
immunity through autophagy. Immunity. 27:11–21. 2007. View Article : Google Scholar
|
|
74
|
Dai JP, Li WZ, Zhao XF, et al: A drug
screening method based on the autophagy pathway and studies of the
mechanism of evodiamine against influenza A virus. PLoS One.
7:e427062012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ghavami S, Mutawe MM, Hauff K, et al:
Statin-triggered cell death in primary human lung mesenchymal cells
involves p53-PUMA and release of Smac and Omi but not cytochrome c.
Biochim Biophys Acta. 1803.452–467. 2010.PubMed/NCBI
|
|
76
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|