Introduction
The blood-spinal cord barrier (BSCB) is a highly
specialized structural, transport and biochemical barrier within
the central nervous system (CNS). Similar to the blood-brain
barrier (BBB), the BSCB is primarily formed by endothelial cells
interconnected by tight junctions, which limits passive diffusion
of blood-borne solutes and actively transports nutrients into the
spinal cord (1,2). BSCB dysfunction leading to early
inflammatory response and oxidative stress contributes to secondary
pathogenesis following traumatic spinal cord injury (SCI) (3–10).
BSCB disruption by traumatic SCI also generates harmful substances,
including endothelins (ETs) (11–15), matrix metalloproteinases (MMPs)
(7,9,10),
inflammatory cytokines and reactive oxygen species (ROS) (3–6,8)
that can induce programmed neuronal death and permanently impair
neuron function. This is exemplified by studies showing that the
blockade of ET receptors or ET-converting enzyme (ECE) activity in
brain endothelial cells and glia, which results in endothelial
hyperpermeability and cerebral vasoconstriction, reduces leukocyte
infiltration into the injured spinal cord, which is associated with
significant recovery of motor and neurological functions following
SCI (16–18). Therefore, the brain ET system is
considered to be a therapeutic target of SCI.
The ET system consists of two G-protein-coupled
receptors (ET receptors A and B, ETRs), three peptides (ET-1, ET-2
and ET-3), and two activating peptidases (ECE-1 and ECE-2). It is
the most potent vasoconstrictor and is essential for embryonic
development, vascular remodeling, and wound healing (19,20). Excessive activation of the ET
system can be detrimental, leading to multidimensional pathological
conditions, including BBB or BSCB disruption following ischemic
brain injury and traumatic SCI, as well as inflammation (20,21). For example, the ET system is found
throughout the brain as its components are synthesized in vascular,
neuronal, and glial cells. Expression pattern of ET system
components in many discrete brain areas suggests a variety of
possible functions (19–22). ET-1 is the predominant neural ET
and plays a critical role in abnormal vascular endothelial cell
permeability and inflammation after SCI, while the upregulation of
ET-1 modulates behavior and the metabolism without affecting
cerebral blood flow (23). In
addition, ETs exert their effects through the activation of ET
receptor A (ETAR) and/or ET receptor B (ETBR) (19,22,23). In normal spinal cord, ETAR is
found predominantly in vascular smooth muscle cells and primary
afferent nerve fibers, whereas ETBR is abundantly expressed in
endothelial cells, radial glia, a small population of astrocytes,
and epithelial tissues (23,24). Following SCI, vascular ETAR/ETBR
activation plays a critical role in post-traumatic ischemia, and
astrocyte-only ETBR activation is associated with reactive gliosis
(23–25). However, until recently, there was
a lack of consensus regarding which ETR subtype was the key
determinant of oxidative stress and functional recovery after SCI,
and there has been controversy regarding the exact cellular targets
of ETAR and ETBR in the injured spinal cord.
In the present study, we examined the effects of
ETAR and/or ETBR blockade on early SCI pathogenesis and long-term
neurological recovery in murine models. Our results demonstrated
that ETR blockage markedly reduced inflammatory responses and
oxidative stress, ameliorated MMP-9 activation, and enhanced
long-term neurological function in SCI mice. The results confirm
additive pathogenic roles for ETAR and ETBR in the injured spinal
cord and may aid in the identification of a set of putative
therapeutic targets for neural tissue damage after SCI.
Materials and methods
SCI
All the procedures were conducted in accordance with
the National Institutes of Health Guide for the Care and Use of
Laboratory Animals and with approval from the Animal Subjects
Committee at Zhejiang University. Adult female C57BL/6 (18–22 g)
mice were anesthetized with chloral hydrate (500 mg/kg) and
subjected to a moderate spinal cord contusion injury. A laminectomy
was performed at the T9 level, and a 2-g weight was dropped 5 cm
onto the exposed dura mater. After SCI, the skin was closed with
wound clips. Animal body temperature was maintained at 37°C with a
warming blanket throughout the surgery and during the recovery from
anesthesia (26). For the
sham-operated controls (SHAM), the animals underwent a T9
laminectomy without contusion injury.
Drug treatment
BQ123 and/or BQ788 (both from Sigma, St. Louis, MO,
USA) dissolved in sterile phosphate-buffered saline (PBS) were
administered to corresponding SHAM and SCI mice (SHAM + BQ123, SHAM
+ BQ788, SHAM + BQ123 + BQ788, SCI + BQ123, SCI + BQ788 and SCI +
BQ123 + BQ788) via intraperitoneal injection (10 mg/kg,
respectively) (27–29) at 1 day before SCI and then further
treated once a day for 6 weeks for behavioral testing or for the
indicated time-points (2, 4, 6 and 24 h and 4 days) for other
experiments post-injury. PBS for vehicle control (VEH) was
administered in corresponding SHAM and SCI mice (SHAM + VEH and SCI
+ VEH). Significant side effects resulting from BQ123 and/or BQ788
treatment, such as changes in body weight or an increase in
mortality, were not observed during our experiments.
Leukocyte infiltration assessment
To confirm the depletion of neutrophils, three blood
smears, prepared 1 day post-injury were processed with a Hema
3® stain kit (Fisher Scientific, Pittsburgh, PA, USA).
At least 300 white blood cells were counted, and the percentage of
neutrophils relative to the total number of white blood cells was
determined. To confirm monocyte depletion, blood samples were taken
at 4 days post-injury. A hematology automated white blood cell
analyzer (HemaVet® 850) was used to quantify monocytes.
Data are presented as relative percentages relative to the total
number of white blood cells.
Quantitative polymerase chain reaction
(qPCR)
Total RNA was prepared with the RNeasy kit (Qiagen).
Complementary DNA synthesis and reverse transcriptase PCR were
performed using a previously described method (30). The primers used were: tumor
necrosis factor-α (TNF-α), forward, 5′-CCCAGA CCCTCACACTCAGAT-3′
and reverse, 5′-TTGTCCCTT GAGAGAACCTG-3′; interleukin-1β (IL-1β),
forward, 5′-GCA GCTACCTATGTCTTGCCCGTG-3′ and reverse, 5′-GTCGTT
GCTTGTCTCTCCTTGTA-3′; IL-6, forward, 5′-AAGTTT
CTCTCCGCAAGATACTTCCAGCCA-3′ and reverse,
5′-AGGCAAATTTCCTGGTTATATCCAGTT-3′; inducible nitric oxide synthase
(iNOS), forward, 5′-CTCCATGACTCT CAGCACAGAG-3′ and reverse,
5′-GCACCGAAGATATCC TCATGAT-3′.
Enzyme-linked immunosorbent assay
(ELISA)
To determine cytokine levels, the lesion site was
rapidly dissected and homogenized in PBS at 24 h after SCI. After
centrifugation at 4°C for 15 min at 900 × g, the supernatants were
used to measure the concentrations of TNF-α, IL-1β and IL-6
(R&D Systems, Minneapolis, MN, USA), iNOS, malondialdehyde
(MDA), and superoxide dismutase (SOD) (Cusabio Biotech Co., Ltd.,
Hubei, China) using corresponding ELISA kits.
Western blot analysis
A 0.5-cm length of cord, centered over the site of
impact and representing the epicenter, was lysed on ice for 30 min
with 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 25
mM NaF, 5 mM sodium pyrophosphate, 1 mM
Na3VO4, and protease inhibitors (Roche). Cell
lysates were clarified by centrifugation at 12,000 × g for 15 min
at 4°C, and the supernatants were collected and assayed for the
protein concentration using a BCA protein assay kit (Pierce,
Rockford, IL, USA). Total cell lysates were prepared, and western
blot analysis was performed as described previously (30). The antibodies used were:
anti-hemeoxygenase-1 (anti-HO-1, sc-10789; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), anti-MMP-9 (ab7299;
Abcam), anti-α/β-tubulin (no. 2148; Cell Signaling Technology).
Tubulin served as a loading control.
Gelatin zymography
MMP-9 activity at 24-h post-injury was examined by
gelatin zymography based on a previously described method (26). The epicenter of the injured spinal
cord (0.5 cm in length) was homogenized in lysis buffer containing
50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5% deoxycholate,
and 0.1% SDS. Then, 50-μg protein samples were loaded onto 8%
SDS-polyacrylamide gels and copolymerized with gelatin (1 mg/ml;
Sigma). After electrophoresis, renaturation was achieved by
incubation of the gel in 2.5% Triton X-100 for 30 min and in
substrate buffer (50 mM Tris-HCl at pH 8.5, 5 mM CaCl2)
for 48 h at 37°C. The gel was stained with Coomassie blue solution
for 4 h and then de-stained with 40% methanol/10% acetic acid. For
quantitative analysis, gels were scanned, and the positive band was
measured using NIH ImageJ software.
Behavioral analysis
Three different behavioral tests were performed to
evaluate functional improvements after SCI. The 9-point Basso Mouse
Scale (BMS) was used to examine locomotor recovery in an open field
(53×108×5.5 cm) (31).
This rating scale takes into account limb movement, stepping,
coordination, and trunk stability. Mice were tested at 1 and 3 days
and weekly thereafter until euthanasia at 6 weeks post-injury.
Performance on a rotarod and the ability to traverse a wire grid
were evaluated, in sequence, at 40, 41 and 42 days post-injury.
Three experiments were conducted daily, with a total of nine trials
for each test. In each of these tests, the average score from each
mouse was used to calculate the mean.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism (GraphPad Software, Inc., La Jolla, CA, USA). Statistical
significance was defined at P<0.05. Data were presented as the
mean ± standard error of the mean (SEM) of three independent
experiments.
Results
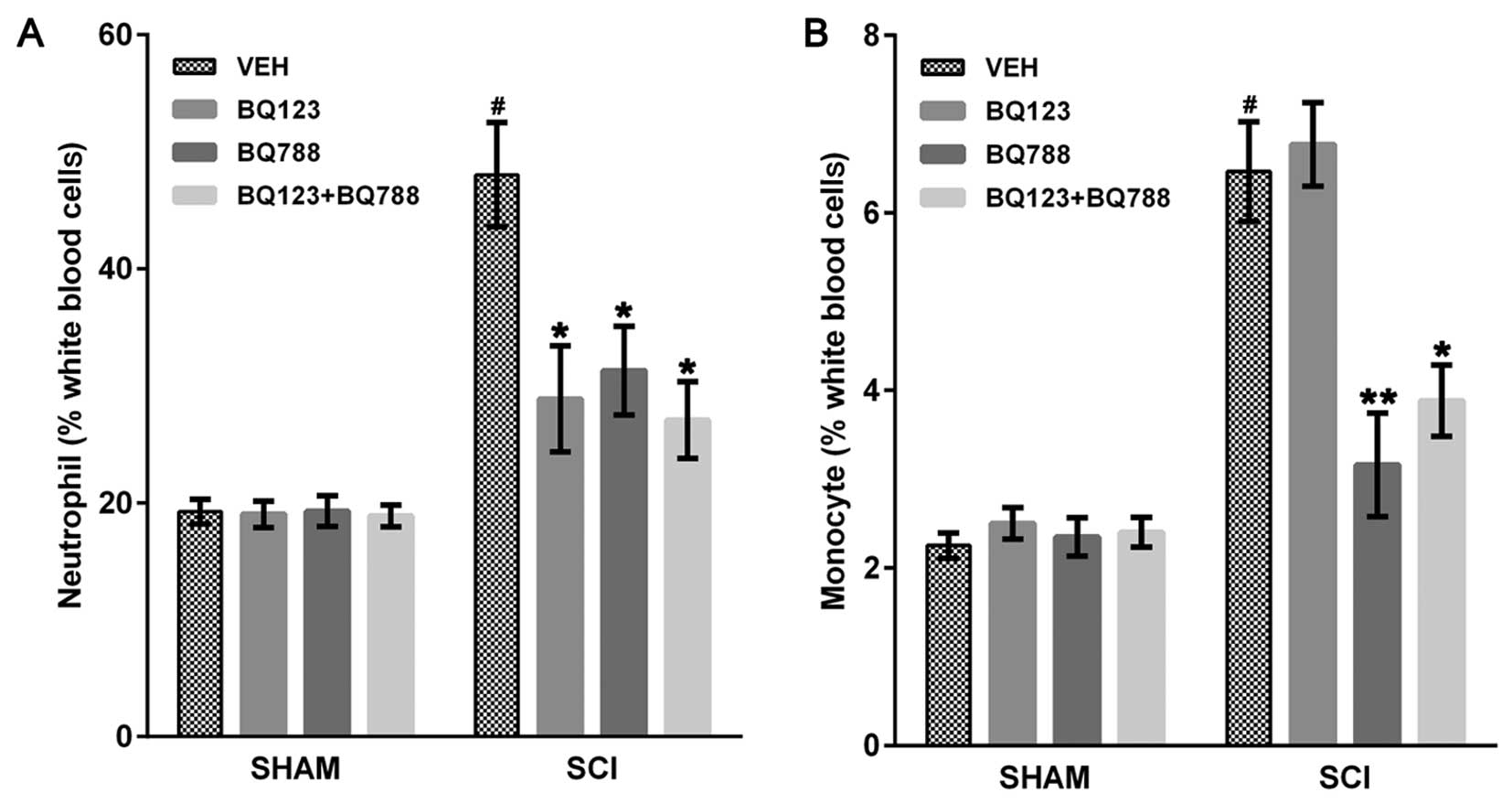
Blockade of ETAR and ETBR reduced
leukocyte infiltration in SCI mice
As previously described (32), the early leukocyte influx
contributes to secondary pathogenesis in the injured spinal cord.
Therefore, we investigated the effects of ETR blockade on leukocyte
influx by performing differential blood cell counts. Compared with
each SHAM mouse, SCI + VEH mice showed an elevated number of
neutrophils and monocytes (Fig.
1). The number of neutrophils in peripheral blood was reduced
in SCI mice at 24 h after blockade of ETAR, ETBR or both (Fig. 1A). Blockade of ETBR or ETAR and
ETBR reduced circulating monocytes 4 days after SCI (Fig. 1B). Notably, blockade of only ETAR
did not affect the number of circulating monocytes in SCI mice
(Fig. 1B).
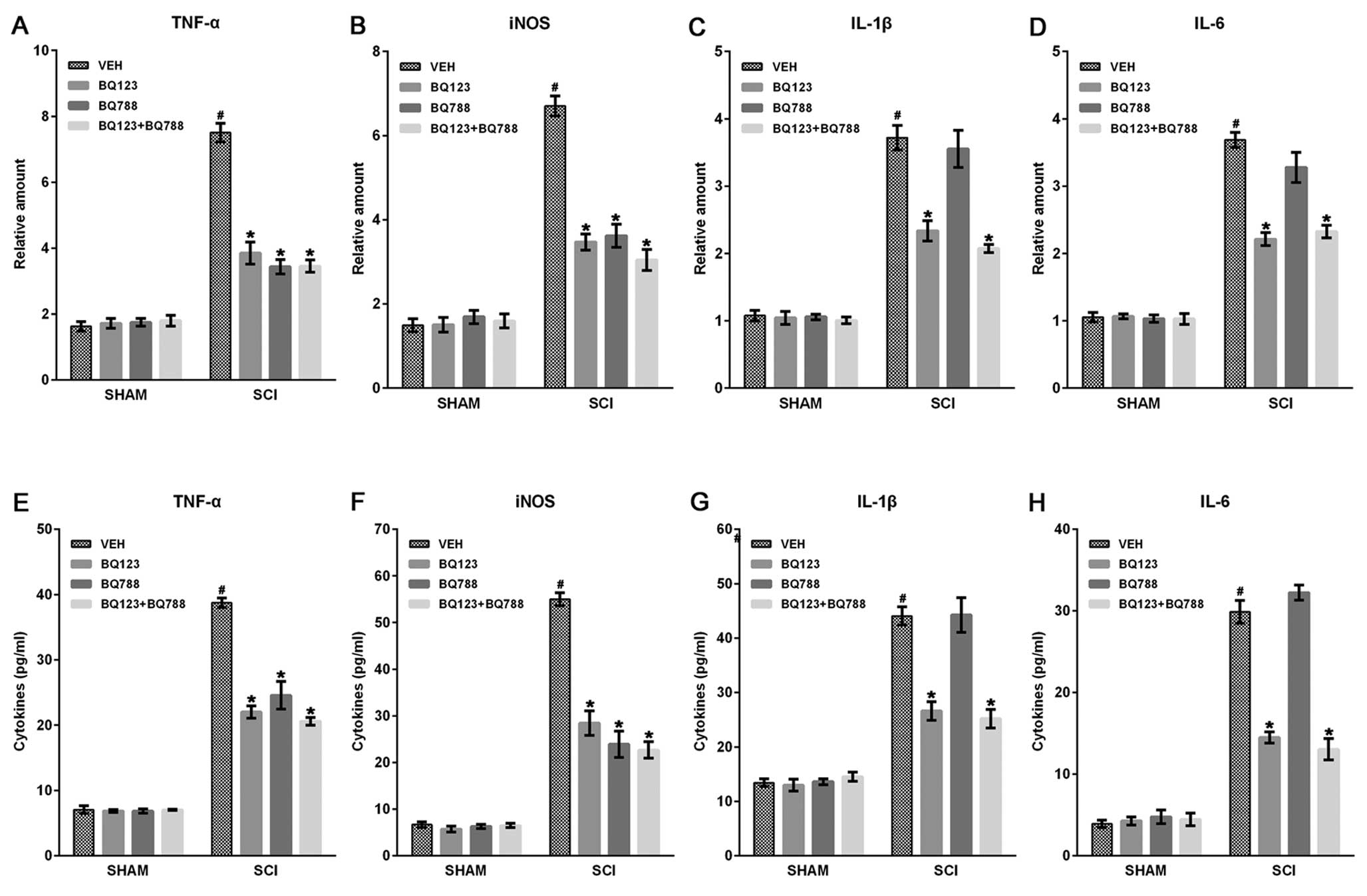
Blockade of ETAR and ETBR inhibited
inflammatory mediator expression in SCI mice
After SCI, leukocyte infiltration following BSCB
disruption initiated inflammatory responses, leading to a secondary
cascade of brain trauma due to the production of inflammatory
mediators, such as TNF-α, IL-1β, IL-6 and iNOS (30). Therefore, we analyzed the effect
of ETR blockade on the expression of inflammatory mediators by
reverse-transcriptase PCR and ELISA assays at indicated time-points
after SCI. Results showed that the mRNA expression levels of TNF-α,
IL-1β (at 2 h), IL-6, and iNOS (at 6 h) were upregulated
post-injury, and blockade of ETAR and ETBR significantly reduced
the levels of these inflammatory mediators compared with VEHs
(Fig. 2A–D). In addition, the
blockade of only ETAR, but not ETBR, decreased IL-1β and IL-6
expression in spinal cords post-injury (Fig. 2B and C). ELISA assays also showed
that the blockade of the two ETRs significantly inhibited the
production of TNF-α, IL-1β, IL-6 and iNOS at 24 h after SCI
(Fig. 2E–H).
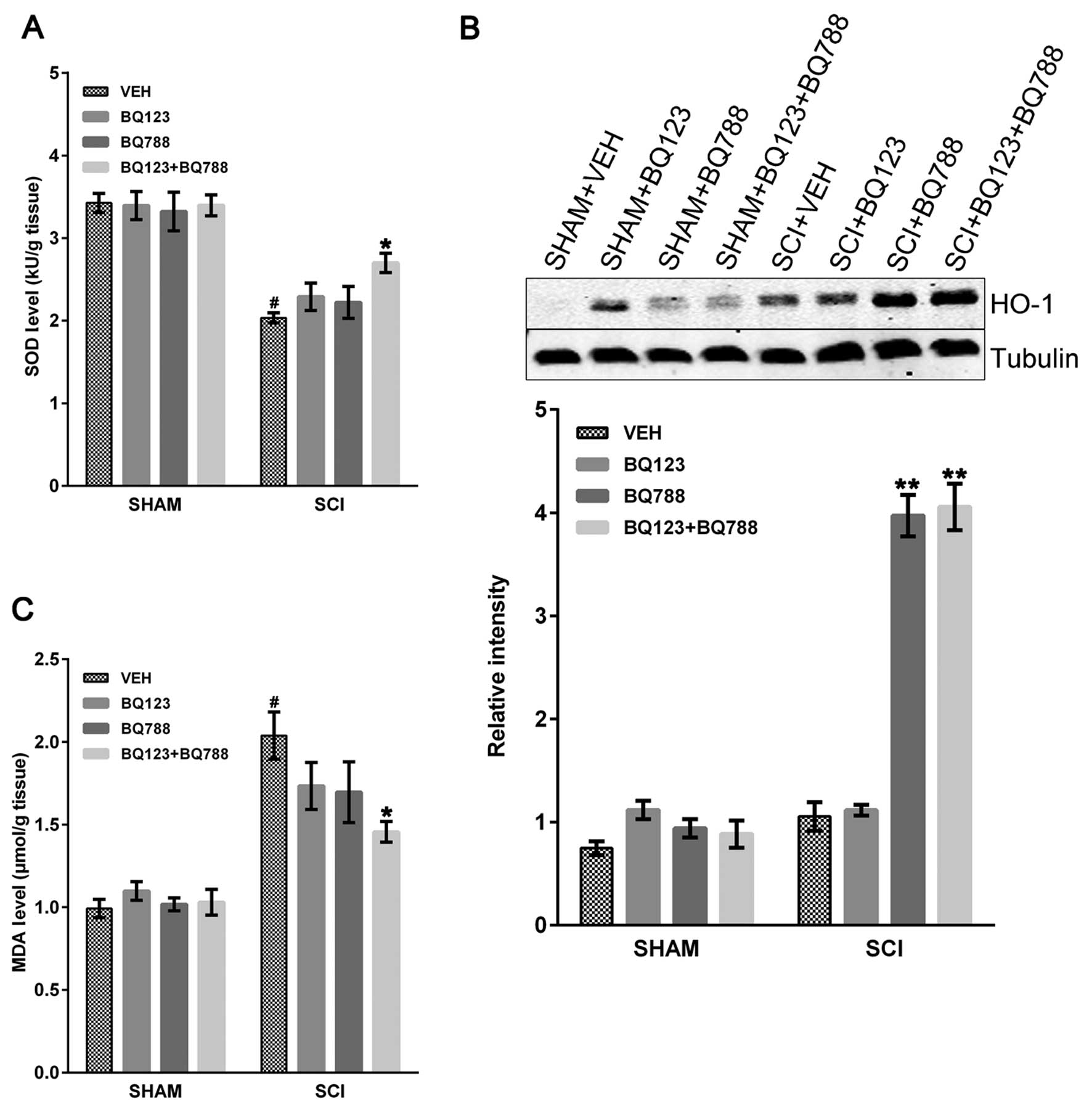
Reduction in oxidative stress in SCI mice
following ETAR and ETBR blockade
Oxidative stress markers (HO-1, MDA and SOD) in
injured mice with or without blockade of ETAR and ETBR were
examined. SOD is an enzyme that neutralizes oxygen-free radicals
and protects cells from being oxidized by superoxide toxicity
(33). SOD is consumed during
oxidative stress in a variety of pathological conditions. SCI
caused a significant decrease in SOD level in SCI + VEH mice
compared to SHAM mice, and the blockade of ETAR and ETBR
significantly rescued the SOD level at 24 h after SCI (Fig. 3A). However, the blockade of only
ETAR or ETBR did not rescue the SOD level in SCI mice (Fig. 3A). Similarly, HO-1, the inducible
form of HO, is an important defense mechanism against early
oxidative stress (5,34). Immunoblot assays revealed a
greater increase in HO-1 levels at 24 h in SCI mice with blockade
of ETBR or both ETAR and ETBR compared to SCI + VEH mice (Fig. 3B). By contrast, no differences
were observed between SCI mice with only ETAR blockade and SCI +
VEH mice (Fig. 3B). Nonetheless,
levels of MDA, the final product of lipid peroxidation (33), were increased at 24 h after SCI
and reduced in SCI mice with ETAR and ETBR blockade, whereas no
statistically significant differences were found in SCI mice with
blockade of only ETAR or ETBR (Fig.
3C).
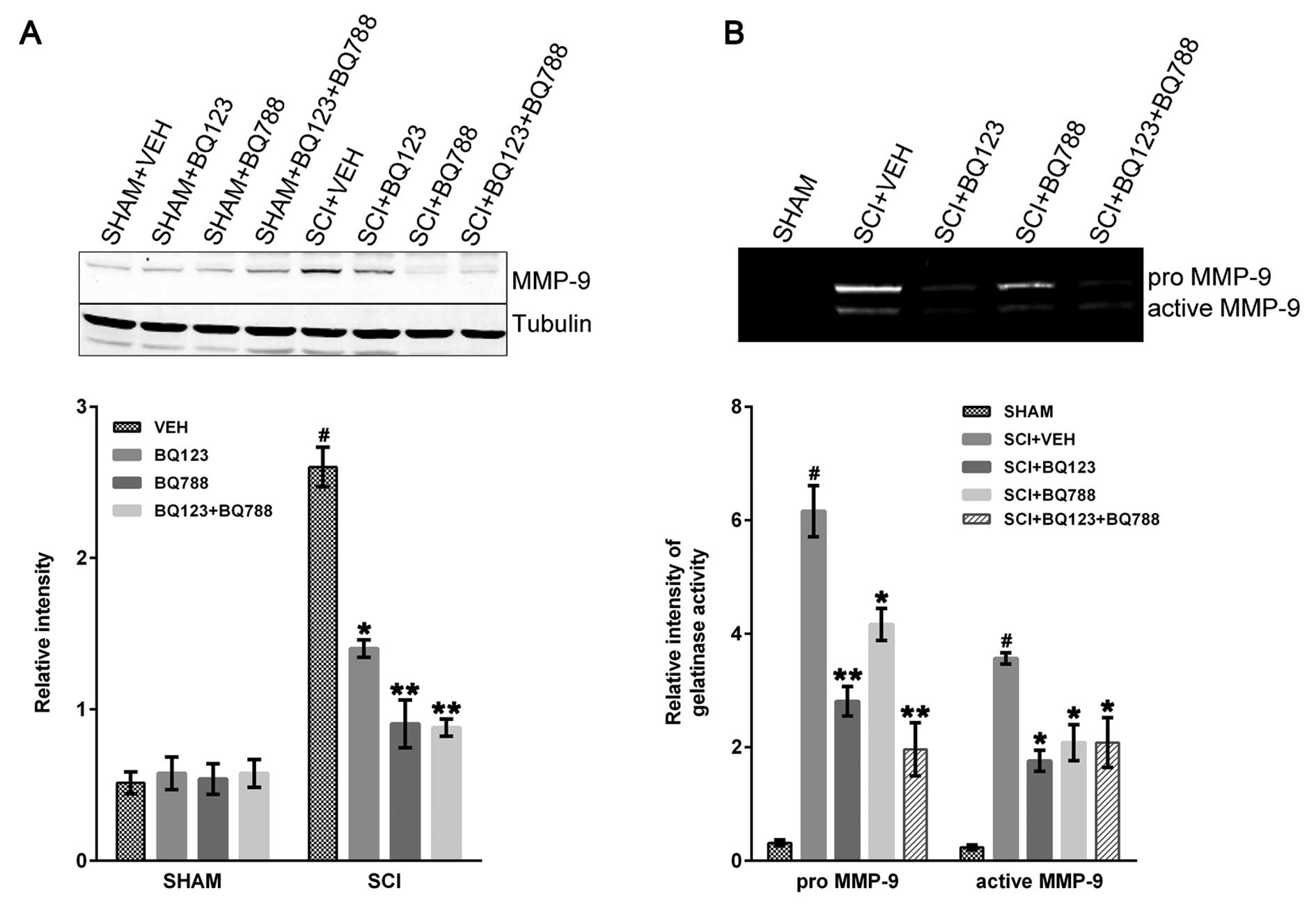
Blockade of ETAR and ETBR reduced MMP-9
expression following SCI
MMP-9 plays a critical role in early neutrophil
infiltration and long-term functional impairment after SCI
(26). The immunoblot results
revealed that MMP-9 was markedly increased at 24 h in SCI + VEH
mice compared to control and was reduced in SCI mice with blockade
of ETAR, ETBR, or a combination thereof (Fig. 4A).
The pro- and active forms of MMP-9 were evaluated in
SHAM and SCI mice by gelatin zymography. The two forms were
significantly reduced in SCI mice that were depleted of ETAR, ETBR,
or both ETRs 24-h post-injury (Fig.
4B).
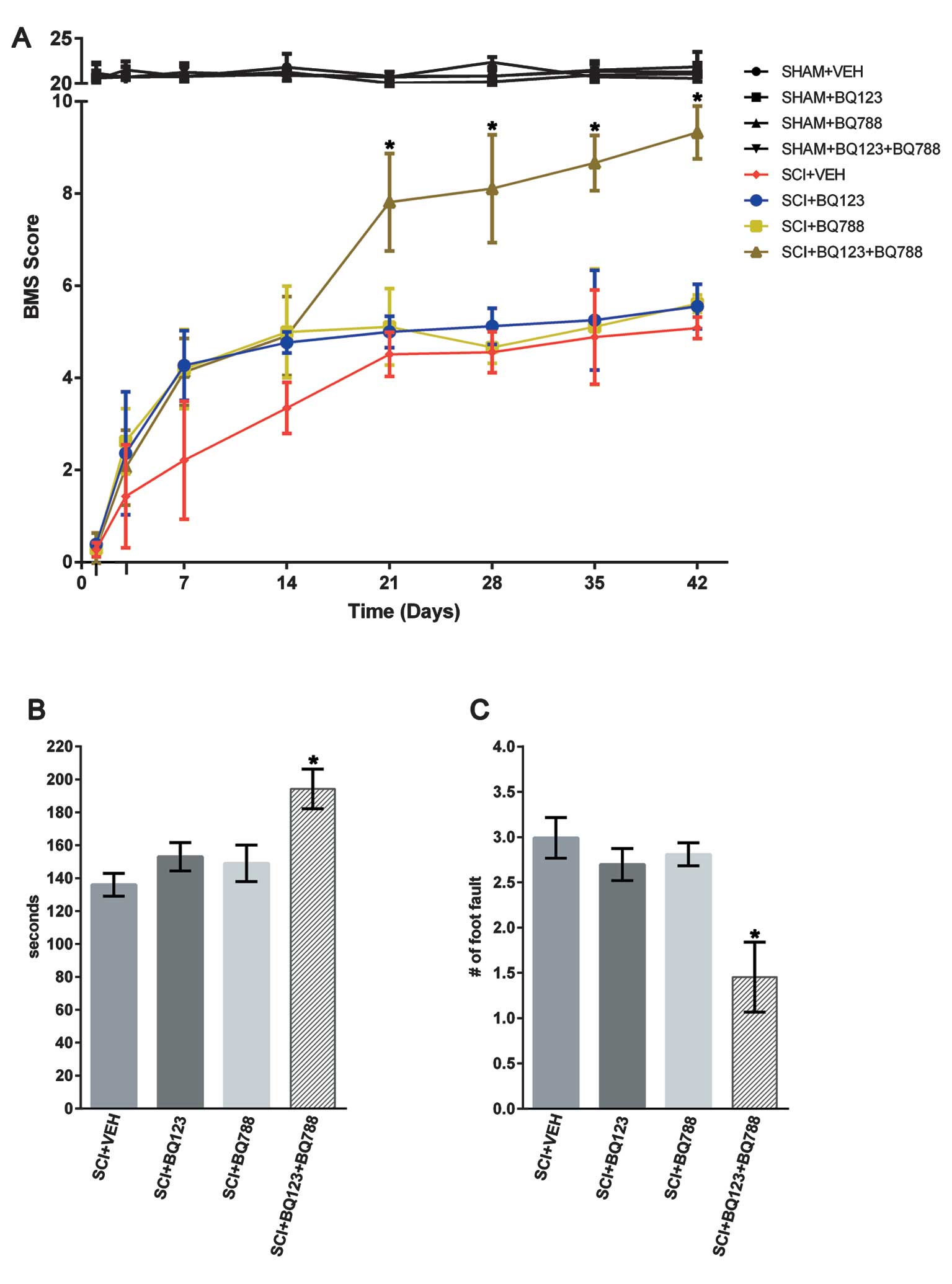
Blockade of ETAR and ETBR improved
functional recovery after SCI
To determine the effects of ETR blockade on the
long-term functional recovery after SCI, locomotor performance was
evaluated in the BMS open field test, on a rotarod, and on a
grid-walking task for 6 weeks. Hind-limb movements were abolished
immediately after SCI as assessed using the BMS scale (31). Although all the groups gradual
improved in locomotor functional recovery after 3 days, this
recovery was markedly higher between 21 and 42 days after SCI in
the group with blockade of ETAR and ETBR (Fig. 5A). Subsequent assessment of the
ability of SCI mice to maintain position on a rotarod and walk
across a grid revealed similar functional improvements in SCI mice
with blockade of ETAR and ETBR, relative to their respective
controls (Fig. 5B–C).
Discussion
ETR activation in the injured spinal cord triggers
several pathologic responses, including leukocyte recruitment,
superoxide generation, and BSCB breakdown, which exacerbate damage
and impair neurological recovery (19–25). Although strategies to block ETRs
in the injured spinal cord can promote locomotor function recovery,
less is known about which ETR subtype influences neurological
recovery processes after SCI (18,35,36). In this study, we have shown that
blockade of ETAR and ETBR in SCI mice resulted in an early
reduction in leukocyte infiltration, oxidative stress, and
expression of inflammatory mediators and MMP-9. Notably, although
all the SCI mice with blockade of ETAR or ETBR showed an early
improvement in locomotor function, blockade of the two receptor
types resulted in significant long-term locomotor function
improvement. Collectively, these results provide evidence for the
additive cooperation between ETAR and ETBR in influencing early
secondary pathogenesis and long-term locomotor recovery after
SCI.
BQ123 is a selective ETAR antagonist that has been
used as a biochemical tool to investigate ETR function (37), whereas BQ788 is a selective ETBR
antagonist used to demonstrate the role of ET-1 and ETBR in
physiological and pathophysiological conditions (29). Results of previous studies have
indicated that BQ123 and BQ788 may exert anti-inflammatory and
anti-oxidative properties in patients with ischemic heart disease
(27–29,38). In our experiments, the mice were
treated with BQ123 and BQ788 pre- and post-SCI, which reduced ETAR
and ETBR activity in the injured tissue, respectively. Thus, we
assessed the specific roles of ETAR and ETBR in the injured spinal
cord by using BQ123 and BQ788 in SCI mice.
Inflammatory neutrophils and monocytes are the first
leukocytes to infiltrate the CNS after SCI (39–41). The number of circulating
neutrophils increased 12–24 h post-injury (32,40), whereas the monocytes infiltrated
into the CNS as early as day 1, reaching peak levels between 4 and
7 days after SCI (32). In our
experiments, an increased number of circulating neutrophils and
monocytes was detected in the peripheral blood in SCI mice. We also
found that blockade of ETAR or ETBR pre- and post-SCI reduced
circulating neutrophil numbers. Nevertheless, the blockade of ETBR
or a combination of the two ETR subtypes decreased circulating
monocytes in SCI mice. However, blockade of only ETAR had no effect
on the number of monocytes after SCI. In parallel with this result,
the expression levels of inflammatory mediators, such as TNF-α,
IL-1β, IL-6 and iNOS, were upregulated after SCI, while the
expression levels were significantly reduced following blockade of
ETAR and ETBR. In addition, blockade of only ETBR did not decrease
IL-1β and IL-6 expression levels in SCI mice. These results were
consistent with previous findings (42–47) suggesting that the relationship
between ETAR and ETBR in SCI mice is cooperative, and not
antagonistic. The specific blockade of the two ETRs may inhibit
inflammatory responses by suppressing the expression of
inflammatory chemokines and reducing leukocyte infiltration
following SCI-induced inflammatory mediator production.
Oxidative stress plays a critical role in secondary
pathogenesis following SCI as it can lead to inflammation and
apoptotic cell death. Increased oxidative stress reflects an
imbalance between ROS and anti-oxidant levels (17,32). We assessed three oxidative stress
parameters in SCI mice: HO-1, MDA and SOD. MDA is a reactive
aldehyde resulting from the degradation of polyunsaturated lipids
that causes toxic stress in cells (7). However, endogenous anti-oxidative
enzymes SOD and HO-1 catalyze superoxide into oxygen and hydrogen
peroxide (7,8,33,48). Consistent with previous
observations (7,8,33,48), SCI induced an increase in MDA and
reduced HO-1 and SOD expression. To the best of our knowledge, the
results have shown for the first time that blockade of ETAR and
ETBR in SCI mice significantly reversed reduction in MDA and SOD
levels, thereby restoring the oxidative stress balance.
HO-1 is regarded as a sensitive and reliable
indicator of cellular oxidative stress (7,8).
Increased HO-1 expression in SCI rodent models can reduce neural
tissue damage and improve locomotor function (32,49). HO-1 induction is regulated by the
stress-responsive element and the Maf recognition element.
Furthermore, HO-1 is rapidly induced by the specific regulation of
oxidant-responsive transcription factors AP-1, NF-κB, and
Nrf2/Keap1-Bach1 in oxidative stress (49,50). We found that blockade of ETBR or
the two ETR subtypes resulted in a significant elevation of HO-1
expression in SCI mice, whereas the blockade of only ETAR had no
effect. These results may be due to the critical role of ETBR on
the ERK pathway signaling, which may affect the Nrf2/Keap1-Bach1
equilibrium, resulting in a decreased HO-1 expression (50,51).
Pro- and active MMP-9 is markedly accumulated in
acute SCI, and it is able to degrade components of the
extracellular matrix, such as microvasculature basal lamina and
myelin basic protein (26,32).
MMP-9 is derived primarily from neutrophils, although it can also
originate from monocytes. It is likely that MMP-9 is released in
response to activation of these cells following SCI. MMP-9 has been
shown to contribute to early BSCB disruption, leukocyte
infiltration, and white matter damage in SCI rodents (26,32). Furthermore, MMP-9-deficient mice
show functional improvement in locomotion following cerebral focal
ischemia (52). Similar to
previous studies (30), we
detected an increased MMP-9 expression and activity in SCI mice. We
found that blockade of ETAR, ETBR, or a combination thereof
significantly reduced levels of pro- and active forms of MMP-9.
These results are highly dependent on the blockade of ETRs, which
reduces leukocyte infiltration after SCI.
It was previously reported that use of
pharmacological or genetic strategies to block ET signaling
enhanced the recovery of locomotor function in SCI rodents
(23,25). In our experiments, although
blockade of only ETAR or ETBR in SCI mice gradually enhanced
locomotor function recovery, improved long-term locomotor recovery
was limited to SCI mice with blockade of ETAR and ETBR. These
beneficial effects may be partially due to improvement of the SCI
molecular environment, including reductions in inflammation,
oxidative stress, and MMP-9 activation, all of which are dependent
on cooperativity between the two ETR subtypes (26,32,48). It is also possible that ETAR or
ETBR exert distinct effects on pro-inflammatory and oxidative
stress states in SCI, which may subsequently influence neurological
functional recovery. For example, blockade of vascular ETAR and
ETBR and astrocyte ETBR after SCI may reduce ischemia and
astrogliosis and facilitate neuronal survival, regeneration, and
neurological function recovery (23). Further studies are needed to
address the involvement of specific ETR subtypes in SCI.
Taken together, to the best of our knowledge,
findings of this study provide the first evidence for collaboration
between ETAR and ETBR in creating an environment that is hostile to
neurological recovery in SCI. These results may serve as a
foundation for developing combination ET-based therapy against
multiple targets that alleviate early inflammatory response and
oxidative stress and promote long-term functional recovery
following SCI. Although the mechanisms involved in ETR blockade
protection against SCI in mice requires further investigation, our
data suggest that ETR blockade is an effective treatment for SCI
and may be used to improve SCI recovery in humans in the
future.
Acknowledgements
This study was supported by the Science and
Technology Bureau Foundation of Ningbo (no. 2012A610230).
References
|
1
|
Hawkins B and Davis T: The blood-brain
barrier/neurovascular unit in health and disease. Pharmacol Rev.
57:173–185. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abbott N, Rönnbäck L and Hansson E:
Astrocyte-endothelial interactions at the blood-brain barrier. Nat
Rev Neurosci. 7:41–53. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bao F, Chen Y, Schneider K and Weaver L:
An integrin inhibiting molecule decreases oxidative damage and
improves neurological function after spinal cord injury. Exp
Neurol. 214:160–167. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goldbaum O and Richter-Landsberg C: Stress
proteins in oligodendrocytes: differential effects of heat shock
and oxidative stress. J Neurochem. 78:1233–1242. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin Q, Weis S, Yang G, et al: Heme
oxygenase-1 protein localizes to the nucleus and activates
transcription factors important in oxidative stress. J Biol Chem.
282:20621–20633. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lindsey M, Wedin K, Brown M, et al:
Matrix-dependent mechanism of neutrophil-mediated release and
activation of matrix metalloproteinase 9 in myocardial
ischernia/reperfusion. Circulation. 103:2181–2187. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu D, Li L and Augustus L: Prostaglandin
release by spinal cord injury mediates production of hydroxyl
radical, malondialdehyde and cell death: a site of the
neuroprotective action of methylprednisolone. J Neurochem.
77:1036–1047. 2001. View Article : Google Scholar
|
|
8
|
Liu Y, Tachibana T, Dai Y, et al: Heme
oxygenase-1 expression after spinal cord injury: the induction in
activated neutrophils. J Neurotrauma. 19:479–490. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nguyen H, O’Barr T and Anderson A:
Polymorphonuclear leukocytes promote neurotoxicity through release
of matrix metalloproteinases, reactive oxygen species, and
TNF-alpha. J Neurochem. 102:900–912. 2007. View Article : Google Scholar
|
|
10
|
Dang A, Tay B, Kim H, Nauth A,
Alfonso-Jaume M and Lovett D: Inhibition of MMP2/MMP9 after spinal
cord trauma reduces apoptosis. Spine (Phila Pa 1976). 33:E576–E579.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dehouck M, Vigne P, Torpier G, Breittmayer
J, Cecchelli R and Frelin C: Endothelin-1 as a mediator of
endothelial cell-pericyte interactions in bovine brain capillaries.
J Cereb Blood Flow Metab. 17:464–469. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hama H, Kasuya Y, Sakurai T, et al: Role
of endothelin-1 in astrocyte responses after acute brain damage. J
Neurosci Res. 47:590–602. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Siren A, Knerlich F, Schilling L,
Kamrowski-Kruck H, Hahn A and Ehrenreich H: Differential glial and
vascular expression of endothelins and their receptors in rat brain
after neurotrauma. Neurochem Res. 25:957–969. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Martin D, Schoenen J, Delrée P, et al:
Experimental acute traumatic injury of the adult rat spinal cord by
a subdural inflatable balloon: methodology, behavioral analysis,
and histopathology. J Neurosci Res. 32:539–550. 1992. View Article : Google Scholar
|
|
15
|
Lampl Y, Fleminger G, Gilad R, Galron R,
Sarova-Pinhas I and Sokolovsky M: Endothelin in cerebrospinal fluid
and plasma of patients in the early stage of ischemic stroke.
Stroke. 28:1951–1955. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reijerkerk A, Lakeman K, Drexhage J, et
al: Brain endothelial barrier passage by monocytes is controlled by
the endothelin system. J Neurochem. 121:730–737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Leonard M, Briyal S and Gulati A:
Endothelin B receptor agonist, IRL-1620, reduces neurological
damage following permanent middle cerebral artery occlusion in
rats. Brain Res. 1420:48–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chou A, Chen T, Winardi W, et al:
Functional neuroprotective effect of CGS 26303, a dual ECE
inhibitor, on ischemic-reperfusion spinal cord injury in rats. Exp
Biol Med (Maywood). 232:214–218. 2007.PubMed/NCBI
|
|
19
|
Barnes K and Turner A: The endothelin
system and endothelin-converting enzyme in the brain: molecular and
cellular studies. Neurochem Res. 22:1033–1040. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kallakuri S, Kreipke C, Schafer P, Schafer
S and Rafols J: Brain cellular localization of endothelin receptors
A and B in a rodent model of diffuse traumatic brain injury.
Neuroscience. 168:820–830. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McKenzie A, Hall J, Aihara N, Fukuda K and
Noble L: Immunolocalization of endothelin in the traumatized spinal
cord: relationship to blood-spinal cord barrier breakdown. J
Neurotrauma. 12:257–268. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
MacCumber M, Ross C and Snyder S:
Endothelin in brain: receptors, mitogenesis, and biosynthesis in
glial cells. Proc Natl Acad Sci USA. 87:2359–2363. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peters C, Rogers S, Pomonis J, et al:
Endothelin receptor expression in the normal and injured spinal
cord: potential involvement in injury-induced ischemia and gliosis.
Exp Neurol. 180:1–13. 2003. View Article : Google Scholar
|
|
24
|
Huggins J, Pelton J and Miller R: The
structure and specificity of endothelin receptors: their importance
in physiology and medicine. Pharmacol Ther. 59:55–123. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Koyama Y, Takemura M, Fujiki K, Ishikawa
N, Shigenaga Y and Baba A: BQ788, an endothelin ET(B) receptor
antagonist, attenuates stab wound injury-induced reactive
astrocytes in rat brain. Glia. 26:268–271. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Noble L, Donovan F, Igarashi T, Goussev S
and Werb Z: Matrix metalloproteinases limit functional recovery
after spinal cord injury by modulation of early vascular events. J
Neurosci. 22:7526–7535. 2002.PubMed/NCBI
|
|
27
|
Fu L, Guo Z and Longhurst J: Endogenous
endothelin stimulates cardiac sympathetic afferents during
ischaemia. J Physiol. 588:2473–2486. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Douglas SA, Vickery-Clark LM, Louden C and
Ohlstein EH: Selective ETA receptor antagonism with BQ-123 is
insufficient to inhibit angioplasty induced neointima formation in
the rat. Cardiovasc Res. 29:641–646. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Okada M and Nishikibe M: BQ-788, a
selective endothelin ET(B) receptor antagonist. Cardiovasc Drug
Rev. 20:53–66. 2002. View Article : Google Scholar
|
|
30
|
Lee J, Kim H, Choi H, Oh T and Yune T:
Fluoxetine inhibits matrix metalloprotease activation and prevents
disruption of blood-spinal cord barrier after spinal cord injury.
Brain. 135:2375–2389. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Basso D, Fisher L, Anderson A, Jakeman L,
McTigue D and Popovich P: Basso mouse scale for locomotion detects
differences in recovery after spinal cord injury in five common
mouse strains. J Neurotrauma. 23:635–659. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee S, Rosen S, Weinstein P, van Rooijen N
and Noble-Haeusslein L: Prevention of both neutrophil and monocyte
recruitment promotes recovery after spinal cord injury. J
Neurotrauma. 28:1893–1907. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu W, Chi L, Xu R, et al: Increased
production of reactive oxygen species contributes to motor neuron
death in a compression mouse model of spinal cord injury. Spinal
Cord. 43:204–213. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cuadrado A and Rojo A: Heme oxygenase-1 as
a therapeutic target in neurodegenerative diseases and brain
infections. Curr Pharm Des. 14:429–442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Salzman S, Acosta R, Beck G, Madden J,
Boxer B and Ohlstein E: Spinal endothelin content is elevated after
moderate local trauma in the rat to levels associated with
locomotor dysfunction after intrathecal injection. J Neurotrauma.
13:93–101. 1996. View Article : Google Scholar
|
|
36
|
Nagasaka J, Tsuji M, Takeda H and
Matsumiya T: Role of endothelin receptor subtypes in the behavioral
effects of the intracerebroventricular administration of
endothelin-1 in conscious rats. Pharmacol Biochem Behav.
64:171–176. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ishikawa K, Fukami T, Nagase T, et al:
Cyclic pentapeptide endothelin antagonists with high ETA
selectivity. Potency- and solubility-enhancing modifications. J Med
Chem. 35:2139–2142. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Khimji A and Rockey D: Endothelin -
biology and disease. Cell Signal. 22:1615–1625. 2010. View Article : Google Scholar
|
|
39
|
Beck K, Nguyen H, Galvan M, Salazar D,
Woodruff T and Anderson A: Quantitative analysis of cellular
inflammation after traumatic spinal cord injury: evidence for a
multiphasic inflammatory response in the acute to chronic
environment. Brain. 133:433–447. 2010. View Article : Google Scholar
|
|
40
|
Letellier E, Kumar S, Sancho-Martinez I,
et al: CD95-ligand on peripheral myeloid cells activates Syk kinase
to trigger their recruitment to the inflammatory site. Immunity.
32:240–252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Stirling D and Yong V: Dynamics of the
inflammatory response after murine spinal cord injury revealed by
flow cytometry. J Neurosci Res. 86:1944–1958. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ahnstedt H, Stenman E, Cao L, Henriksson M
and Edvinsson L: Cytokines and growth factors modify the
upregulation of contractile endothelin ET(A) and ET(B) receptors in
rat cerebral arteries after organ culture. Acta Physiol (Oxf).
205:266–278. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zarpelon A, Pinto L, Cunha T, et al:
Endothelin-1 induces neutrophil recruitment in adaptive
inflammation via TNFα and CXCL1/CXCR2 in mice. Canadian Can J
Physiol Pharmacol. 90:187–199. 2012.PubMed/NCBI
|
|
44
|
Tonari M, Kurimoto T, Horie T, Sugiyama T,
Ikeda T and Oku H: Blocking endothelin-B receptors rescues retinal
ganglion cells from optic nerve injury through suppression of
neuroinflammation. Invest Ophthalmol Vis Sci. 53:3490–3500. 2012.
View Article : Google Scholar
|
|
45
|
Wang H, Hsieh H and Yang C: Nitric oxide
production by endothelin-1 enhances astrocytic migration via the
tyrosine nitration of matrix metalloproteinase-9. J Cell Physiol.
226:2244–2256. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gallelli L, Pelaia G, D’Agostino B, et al:
Endothelin-1 induces proliferation of human lung fibroblasts and
IL-11 secretion through an ET(A) receptor-dependent activation of
MAP kinases. J Cell Biochem. 96:858–868. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Solini A, Santini E, Madec S, Cuccato S
and Ferrannini E: Effects of endothelin-1 on fibroblasts from type
2 diabetic patients: possible role in wound healing and tissue
repair. Growth Factors. 25:392–399. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yin X, Yin Y, Cao F, et al: Tanshinone IIA
attenuates the inflammatory response and apoptosis after traumatic
injury of the spinal cord in adult rats. PLoS One. 7:e383812012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kanno H, Ozawa H, Dohi Y, Sekiguchi A,
Igarashi K and Itoi E: Genetic ablation of transcription repressor
Bach1 reduces neural tissue damage and improves locomotor function
after spinal cord injury in mice. J Neurotrauma. 26:31–39. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Goven D, Boutten A, Lecon-Malas V,
Boczkowski J and Bonay M: Prolonged cigarette smoke exposure
decreases heme oxygenase-1 and alters Nrf2 and Bach1 expression in
human macrophages: roles of the MAP kinases ERK(1/2) and JNK. FEBS
Lett. 583:3508–3518. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sallum CO, Wilson JL, Rupasinghe C, et al:
Enhancing and limiting endothelin-1 signaling with a
cell-penetrating peptide mimicking the third intracellular loop of
the ETB receptor. Chem Biol Drug Des. 80:374–381. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Romanic AM 1, White RF, Arleth AJ,
Ohlstein EH and Barone FC: Matrix metalloproteinase expression
increases after cerebral focal ischemia in rats: inhibition of
matrix metalloproteinase-9 reduces infarct size. Stroke.
29:1020–1030. 1998. View Article : Google Scholar
|