Introduction
Cardiac hypertrophy is characterized by the
augmentation of the ventricular mass against pressure overload in
disease settings, such as hypertension and aortic stenosis
(1,2). In the case of cardiac hypertrophy,
although increased ventricular mass is initially a compensatory
mechanism, sustained hypertrophy can ultimately lead to a decline
in left ventricular function and heart failure. In this regard,
cardiac hypertrophy has been considered as an independent risk
factor for heart failure or sudden death (2,3).
Previous studies have explored the mechanisms involved in the
development of pathological hypertrophy on a molecular and cellular
level (1–3). This has led drug discovery research
to the developement of effective therapies for pathological
hypertrophy. However, current therapeutic agents are limited to
halting the progression of this disease. Therefore, novel
therapeutic strategies are required to inhibit the development of
cardiac hypertrophy before heart failure develops.
In response to pressure overload, cardiomyocytes are
subjected to mechanical stretching and release humoral factors
through autocrine and paracrine signaling, such as through
transforming growth factor-β1 (TGF-β1). TGF-β1 is a pleiotropic and
multifunctional cytokine (4) that
serves as a master switch for cardiac hypertrophy. The activation
of TGF-β1-mediated hypertrophic signaling plays a crucial role in
this process (5). The
mitogen-activated protein kinase (MAPK) superfamily includes p38,
extracellular signal-regulated kinase (ERK)1/2 (also known as
p44/42) and c-Jun N-terminal kinase (JNK), all of which have been
implicated as downstream signaling targets of TGF-β1 and causally
contribute to cardiac hypertrophy (4,6,7).
Nuclear factor-κB (NF-κB) is a well-known pluripotent transcription
factor, which can be activated by MAPK signaling (8). Once activated, NF-κB stimulates gene
expression and the products of inflammatory cytokines, and
ultimately leads to cardiac hypertrophy (8,9).
It is evident that blocking NF-κB binding activity significantly
attenuates cardiac hypertrophy (8). Therefore, the targeting of
TGF-β1-mediated hypertrophic signaling may constitute a suitable
therapeutic intervention for pathological hypertrophy.
Asiatic acid (AA) is a pentacyclic triterpenoid that
has been reported to exhibit a variety of pharmacological effects,
including antioxidant (10),
anti-inflammatory (11) and
anti-apoptotic effects (12). Of
note, a recent study demonstrated that AA inhibited liver fibrosis
in vitro and in vivo, and the anti-hepatofibrotic
effects of AA involved the blocking of the TGF-β1/Smad signaling by
reducing the TGF-β1 expression levels (13). However, to the best of our
knowledge, there is no study available to date on the effects of AA
on cardiac hypertrophy. Thus, we hypothesized that AA may attenuate
cardiac hypertrophy by blocking TGF-β1-mediated hypertrophic
signaling. To examine this hypothesis, in the present study, we
investigated the anti-hypertrophic effects and mechanisms of action
of AA using a TGF-β1-stimulated hypertrophic cardiomyocyte model
in vitro and a pressure overload-induced cardiac hypertrophy
model in vivo.
Materials and methods
Materials
The purified natural product of AA (97%) and
dimethyl sulfoxide (DMSO), were obtained from Sigma-Aldrich (St.
Louis, MO, USA). Recombinant human TGF-β1 was obtained from
PeproTech (Rocky Hill, NJ, USA). Dulbecco’s modified Eagleæs medium
(DMEM) and fetal bovine serum (FBS) were obtained from Gibco BRL
Life Technologies, Inc. (Carlsbad, CA, USA). The cell counting
kit-8 (CCK-8) assay kit was obtained from Dojindo Laboratories
(Kumamoto, Japan). Primary antibodies against total and
phosphorylated (p)-p38, ERK1/2 and JNK1/2 were obtained from Cell
Signaling Technology (Berverly, MA, USA). The electrophoretic
mobility shift assay (EMSA) kit was obtained from Pierce
Biotechnology, Inc. (Rockford, IL, USA). Cytoplasmic and nuclear
protein extraction kits were obtained from KeyGen Biotech Co., Ltd.
(Nanjing, China). Unless otherwise indicated, all other chemicals
and materials were obtained from Sigma-Aldrich.
Ethics statements
Animal handling and use complied with the Guide for
the Care and Use of Laboratory Animals published by the US National
Institutes of Health (NIH Publication no. 85-23, revised 1996) and
were approved by the Animal Care and Use Committee of Nanjing
Medical University (Nanjing, China). They were housed in a room
maintained at 22°C with a 12:12 h light/dark cycle and provided
with standard food and water ad libitum. The experiments
were designed to minimize the pain inflicted and the number of
animals used.
Primary cultures of neonatal rat
ventricular myocytes
Hearts were immediately removed from 1- to 2-day-old
neonatal Sprague-Dawley rats anesthetized by diethyl ether under
aseptic conditions and washed in Ca2+- and
Mg2+-free phosphate-buffered saline (PBS). After the
atria and aorta were discarded, the ventricles were minced and
enzymatically digested with 0.1% collagenase type I (Sigma-Aldrich)
and 0.125% trypsin (Gibco BRL Life Technologies, Inc.). The
liberated cells were collected by centrifugation and incubated in
100-mm culture dishes for 90 min at 37°C in a humidified incubator
with 5% CO2 air. Non-adherent cells were harvested as
cardiomyocytes and seeded at a density of 1×106
cells/well into 6-well culture plates. They were incubated in DMEM
supplemented with 10% FBS, 1% penicillin/streptomycin and
bromodeoxyuridine (BrdU, 100 μM; Sigma-Aldrich). After 48 h, the
culture medium was replaced by DMEM containing 1% FBS. After 24 h
of serum starvation, the cells were incubated with AA for 24 h
prior to treatment with TGF-β1 [4 ng/ml, as previously described in
the study by Lim et al (4)] stimulation, which lasted 24 h.
Untreated cells served as the controls. AA was freshly prepared as
a stock solution in DMSO and diluted with sterile double-distilled
water [0.1% (v/v) DMSO]. TGF-β1 was dissolved in sterile
double-distilled water. There were 5 experimental groups: i)
control, ii) AA, iii) TGF-β1, iv) TGF-β1 + vehicle (DMSO-saline)
and v) TGF-β1 + AA.
Cell viability assay
Cell viability was monitored using a CCK-8 assay
according to the manufacturer’s instructions. In brief, the
cardiomyocytes were initially cultured at a density of
1×104 cells/well in 96-well plates. The cells were then
pre-treated with various concentrations of AA (2.5–30 μM) for 24 h.
CCK-8 solution (10 μl) was then added to each culture well followed
by incubation for 4 h at 37°C. The absorbance at 450 nm was
measured using a microplate reader (Bio-Rad Laboratories, Hercules,
CA, USA). All experiments were performed in triplicate, and cell
viability was calculated as a percentage.
Immunofluorescence analysis of
cardiomyocytes
The cardiomyocytes were cultured on coverslips.
Following TGF-β1 (4 ng/ml) stimulation for 24 h in the presence or
absence of AA for 24 h, the cells were washed with PBS, fixed with
4% paraformaldehyde for 20 min and permeablized with 0.1% Triton
X-100/PBS for 10 min. After blocking with 5% bovine serum for 30
min, the size of the cells was determined by staining the membranes
with specific anti-sarcomeric α-actinin antibody (Sigma-Aldrich)
and visualized under an inverted fluorescence microscope (Nikon,
Tokyo, Japan). The size of the cardiomyocytes was determined using
ImageJ software (NIH, Bethesda, MD, USA).
Animal models of cardiac hypertrophy
An animal model of pressure overload-induced cardiac
hypertrophy was created by transverse aortic constriction (TAC) in
male C57BL/6 mice (8–10 weeks of age, 20–30 g body weight;
Experimental Animal Center of Jiangsu Province, Nanjing, China). To
achieve constriction, a 7-0 suture was snugly tied twice around a
blunt 27-gauge needle, which was positioned adjacent to the aorta
between the right innominate and left carotid arteries and promptly
removed following ligation (14).
This produced a 60–70% constriction with an outer aortic diameter
of approximately 0.4 mm. Acute and chronic mortality from the
ligature procedure was <10%. Sham-operated controls consisted of
age-matched littermates that underwent an identical surgical
procedure, including the isolation of the aorta, only without
placement of the ligature. Twenty-four hours after the operation,
the mice subjected to TAC and the sham-operated mice were orally
gavaged with AA 100 mg/kg/day or the vehicle (DMSO-saline). After 2
weeks of TAC, the hearts were harvested and the ratio of heart
weight to body weight (HW/BW) was calculated. The heart samples
were frozen in liquid nitrogen and stored at −80°C. AA was freshly
prepared as a stock solution in DMSO and diluted with saline to
yield a final AA concentration of 100 mg/kg body weight [0.1% (v/v)
DMSO] (based on our preliminary experiment). The vehicle control
was administered a mixture of DMSO with saline [0.1% (v/v) DMSO].
There were 5 experimental groups: i) sham-operated control (sham),
ii) sham + AA, iii) untreated TAC, iv) TAC + vehicle and v) TAC +
AA.
Transthoracic echocardiography
All mice were anesthetized by a mixture of
isoflurane (1.5%) and oxygen (0.5 l/min). Cardiac dimensions and
functions were evaluated by echocardiography (Vevo 2100 equipped
with a 30-MHz high-resolution phase array transducer; VisualSonics,
Toronto, ON, Canada) after 2 weeks following TAC. The left
ventricle (LV) was assessed in both parasternal long-axis and
short-axis views. End-systolic and end-diastolic volume was defined
as the phase in which the smallest and largest area of the LV were
obtained, respectively. Interventricular septal end-diastolic
dimension (IVSD), left ventricular end diastolic posterior wall
dimension (LVPWD), left ventricular end-systolic diameter (LVESD)
and left ventricular end-diastolic diameter (LVEDD) were measured
from the LV M-mode tracing with a sweep speed of 50 mm/sec at the
papillary muscle level. The percentage of fractional shortening
(%FS) was calculated using a standard formula: %FS =
[(LVEDD−LVESD)/LVEDD] ×100. At each location, for each mouse, 6–10
beats were analyzed.
Nuclear protein extraction and
electrophoretic mobility shift assay (EMSA)
Nuclear proteins were isolated from the LV samples
and cultured cardiomyocytes, as described in a previous study
(9). NF-κB binding activity was
examined using an EMSA kit (Pierce Biotechnology, Inc.), according
to the manufacturer’s instructions. For the competition assay,
specific unlabeled NF-κB competitors (200-fold molar excess) were
employed along with the binding reaction mixture. In brief, a
biotin end-labeled DNA duplex of sequences containing the NF-κB
binding domain (5′-AGTTGAGGGGACTTTCCC AGGC-3′ and
3′-TCAACTCCCCTGAAAGGGTCCG-5′) was incubated with nuclear proteins
at room temperature for 20 min. The reaction mixture was separated
by 6.5% polyacrylamide gel electrophoresis, and transferred onto
nylon membranes. The membranes were subjected to UV light
cross-link for 1 min and were then incubated with blocking buffer
containing stabilized streptavidin-horseradish peroxidase conjugate
(1:2,000) for 15 min. The signals on the membranes were detected
with the Chemiluminescent Nucleic Acid Detection Module (Pierce
Biotechnology, Inc.). The NF-κB binding bands were scanned by
G:BOX-CHEMI-XR5-E (Syngene, Frederick, MD, USA) and the relative
intensities were analyzed using ImageJ software (NIH).
Quantitative RT-PCR (RT-qPCR)
mRNA transcripts were quantified by RT-qPCR.
Briefly, RNA from the LV tissues and cardiomyocytes was isolated
using RNAiso and TRIzol reagent (Invitrogen, Carlsbad, CA, USA),
respectively. cDNA generated from 500 ng of total RNA was reverse
transcribed using the PrimeScript™ RT reagent kit (Takara
Biotechnology, Shiga, Japan). Specific products were determined
using Eppendorf Mastercycler ep realplex analysis software
according to the instructions provided with SYBR® Premix
Ex Taq™ II (Tli RNaseH Plus; Takara Biotechnology). The specific
forward and reverse primers used were as follows: mouse atrial
natriuretic peptide (ANP) forward, 5′-CCAGCATGGGC TCCTTCTCCA-3′ and
reverse, 5′-CCGGAAGCTGTTGCA GCCTAGT-3′; mouse TGF-β1 forward,
5′-GACTCTCCACCT GCAAGACC-3′ and reverse, 5′-ACTGCTTCCCGAATGT
CTGA-3′; mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
forward, 5′-GGCATCGTGGAGGGA-3′ and reverse,
5′-TGAGTTAGACTGAGTGAAGAG-3′; rat ANP forward,
5′-GCTCGAGCAGATCGCAAAAG-3′ and reverse, 5′-CACC
ACCTCTCAGTGGCAAT-3′; and rat GAPDH forward,
5′-ATGGGAAGCTGGTCATCAAC-3′ and reverse, 5′-GTGG
TTCACACCCATCACAA-3′. The expression levels of all transcripts were
normalized to the housekeeping gene, GAPDH, in the same tissue. The
relative mRNA expression was calculated as follows: mRNA expression
= 2−(ΔCT sample − ΔCT control).
Western blot analysis
Total protein was extracted from the LV tissues and
cardiac fibroblasts and assessed by western blot analysis and
enhanced chemiluminescence. The proteins (20–30 μg) were separated
by 10–15% SDS-PAGE and subsequenlty transferred onto polyvinylidene
difluoride (PVDF) membranes using a Mini Trans-Blot electrophoresis
transfer cell (Bio-Rad Laboratories). The membranes were incubated
with appropriate primary antibodies against TGF-β1, p-p38
(Thr180/Tyr182), p38, p-ERK1/2 (Thr202/Tyr204), ERK1/2, p-JNK
(Tyr183/Tyr185), JNK and GAPDH. After extensive washing in TBST (10
mM Tris-HCl, 150 mM NaCl and 0.1% Tween-20, pH 7.6), the membranes
were incubated with the appropriate HRP-conjugated secondary
antibody. The signals were detected using an ECL Western Blot
Detection kit (Thermo Scientific, Rockford, IL, USA), and blot
quantification was performed using densitometry with ImageJ
software (NIH).
Histological analysis
The hearts were fixed in 10% neutral formalin,
dehydrated in 75, 80, 90 and 100% ethanol, transferred to xylene,
embedded in paraffin and sectioned at a thickness of 4–5 μm, then
stained with hematoxylin and eosin (H&E) or with Masson’s
trichrome stain. The diameter of the cardiomyocytes and the
interstitial collagen fraction were measured using NIH ImageJ
software (NIH). At least 3 different hearts with 5 separate fields
of cells (total 50–70 cells for each heart), were quantified for
cellular analysis.
Statistical analysis
Data are expressed as the means ± standard deviation
(SD). The GraphPad Prism 5.01 (GraphPad Software, Inc., La Jolla,
CA, USA) and PASW Statistics 18.0 (SPSS Inc., Fayetteville, NC,
USA) packages were used. Differences among groups were tested by
one-way ANOVA. Comparisons between 2 groups were performed by an
unpaired two-tailed Student’s t-test. If a p-value was <0.05,
the result was considered statistically significant.
Results
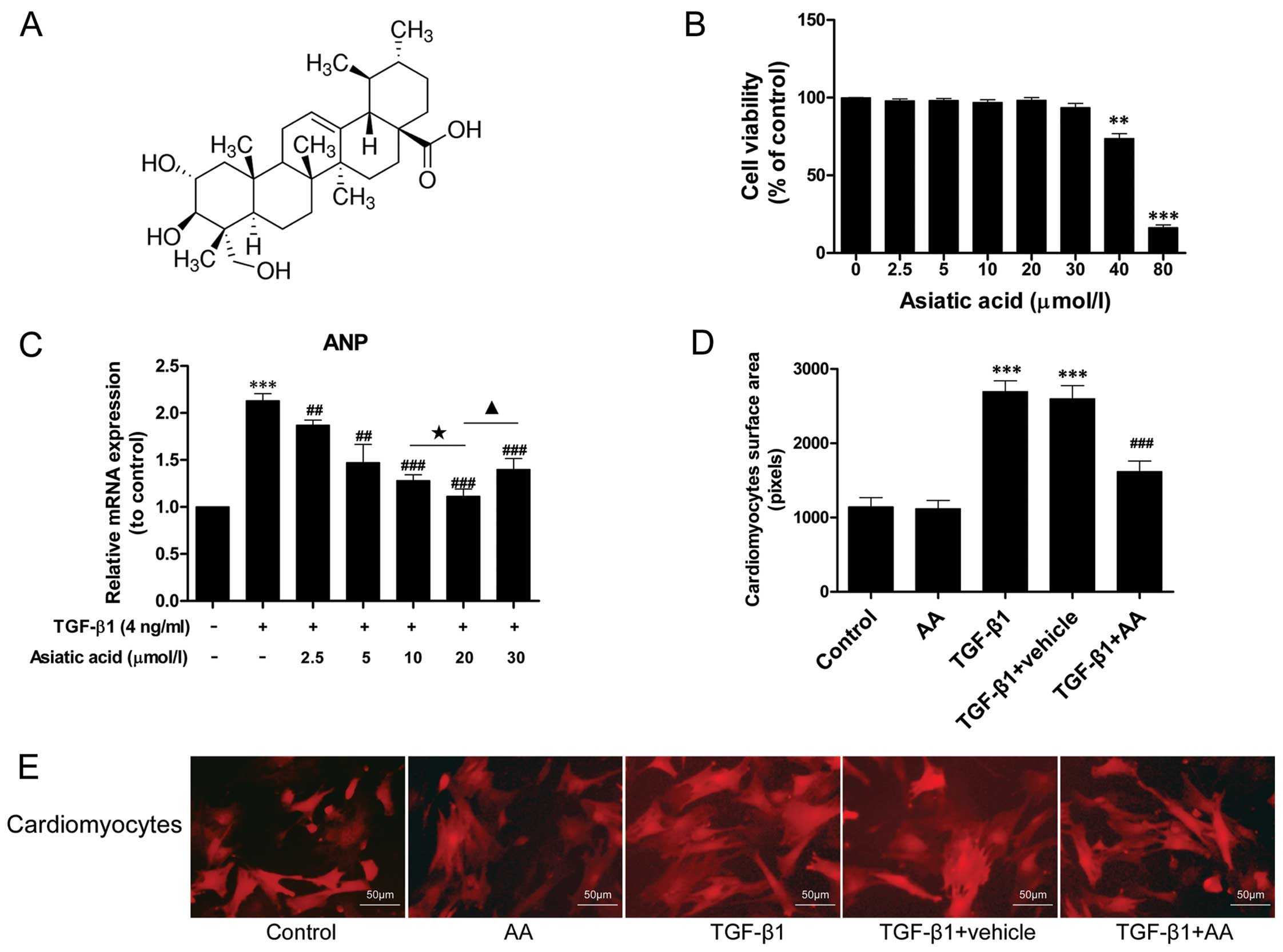
AA inhibits the TGF-β1-induced
hypertrophic response in cardiomyocytes
TGF-β1 stimulation has been demonstrated to induce
hypertrophic effects on cardiomyocytes, promoting the synthesis of
fetal contractile proteins (5).
To determine whether AA inhibits cardiomyocyte hypertrophy induced
by TGF-β1, the cells were treated with TGF-β1 (4 ng/ml) for 24 h in
the presence or absence of AA (2.5–30 μM). The expression of the
fetal gene, ANP, was examined by RT-qPCR. TGF-β1 (4 ng/ml)
stimulation markedly increased the mRNA expression of ANP by
2.13-fold relative to the control group (P<0.001) (Fig. 1C). AA (2.5–30 μM) pre-treatment
significantly decreased ANP mRNA expression, and the level of ANP
mRNA expression in the AA-treated (20 μM) group was 47.89% lower
than that in the TGF-β1-stimulated group (P<0.001).
In addition, the size of the cardiomyocytes was
measured by immunofluorescence staining. TGF-β1 stimulation induced
a noticeable hypertrophic response in the cardiomyocytes that was
not observed in the untreated control cells (Fig. 1D and E). By contrast, AA (20 μM)
pre-treatment attenuated the TGF-β1-induced hypertrophic response.
However, the AA-treated cells were the same size as the untreated
cells.
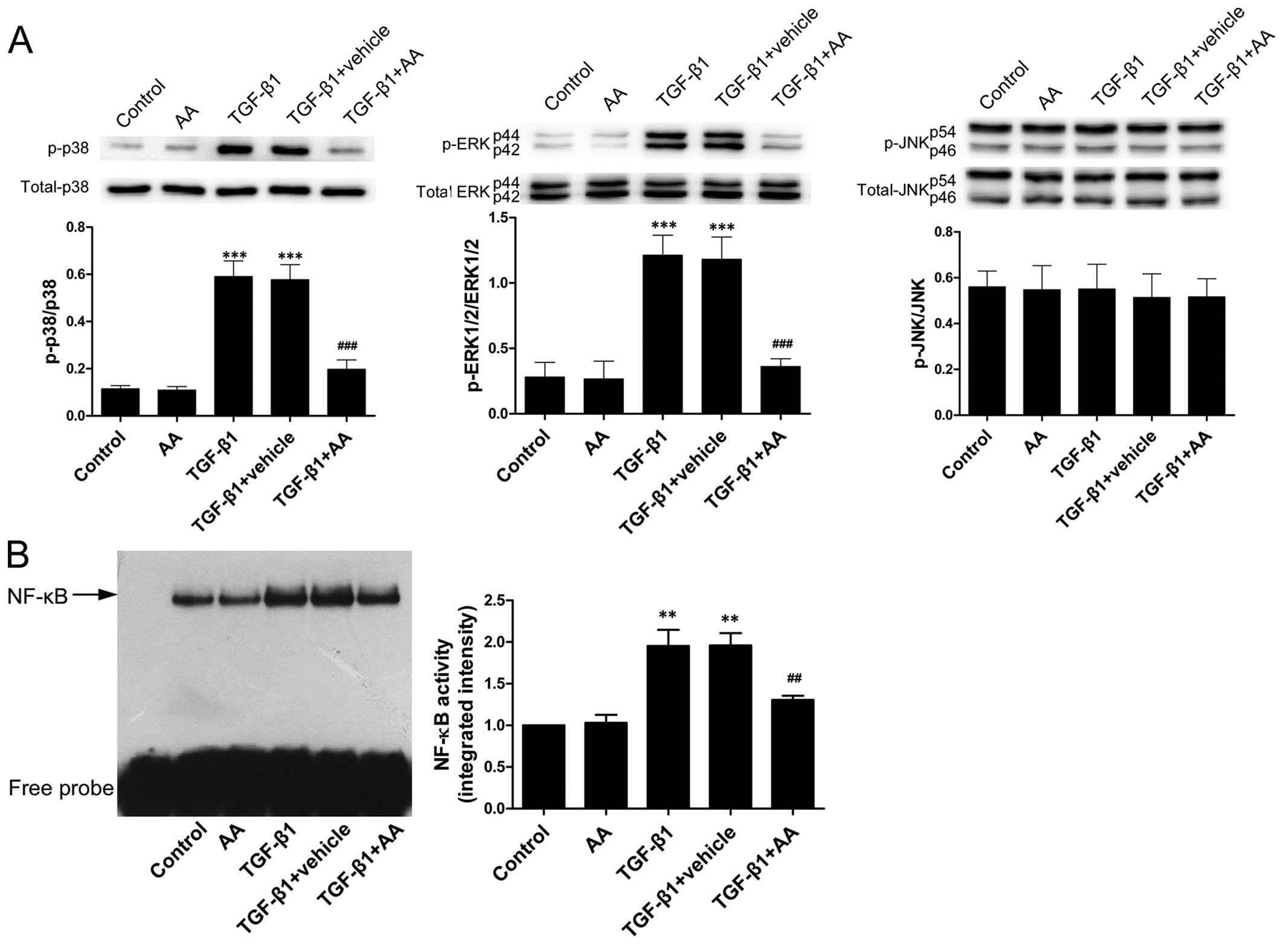
AA prevents the TGF-β1-stimulated
increase in p38 and ERK1/2 phosphorylation and NF-κB binding
activity
The effects of AA on TGF-β1-stimulated NF-κB binding
activity and MAPK phosphorylation were examined. The
phosphorylation levels of p38 and ERK1/2, as well as the NF-κB
binding activity were markedly higher after 24 h of TGF-β1 (4
ng/ml) stimulation compared with the untreated cells (P<0.001,
P<0.001 and P<0.01 for p38, ERK1/2 and NF-κB, respectively)
(Fig. 2). However, TGF-β1 did not
induce the phosphorylation of JNK. AA pre-treatment substantially
reduced the TGF-β1-stimulated increase in the levels of p-p38/p38
and p-ERK1/2/ERK1/2 and NF-κB binding activity (P<0.001,
P<0.001 and P<0.01 for p38, ERK1/2 and NF-κB,
respectively).
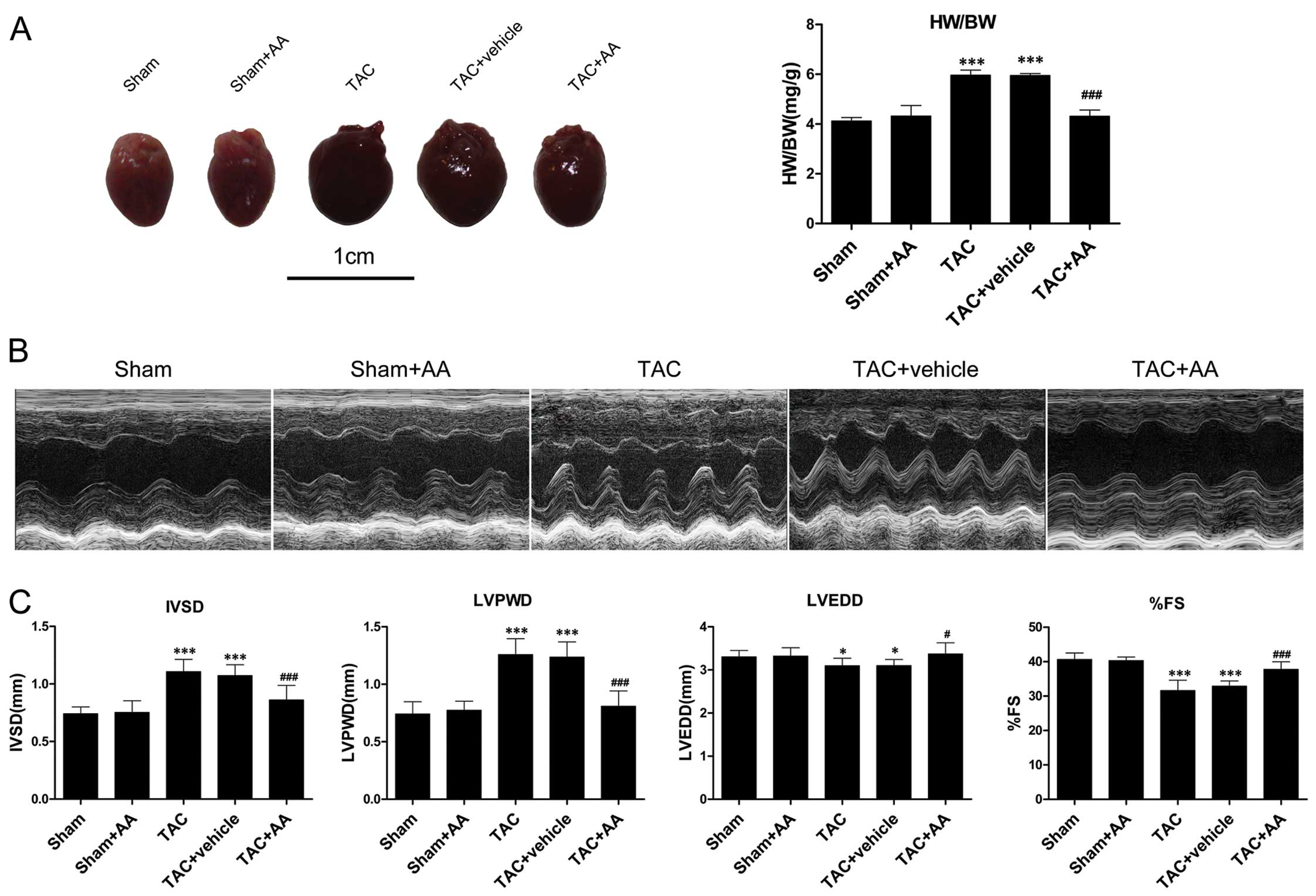
AA administration attenuates TAC-induced
cardiac hypertrophy and cardiac dysfunction
The issue of whether AA can attenuate pressure
overload-induced cardiac hypertrophy was then addressed. The mice
were subjected to TAC and treated with AA (100 mg/kg/daily) or with
the vehicle for 2 weeks. Pressure overload induced by TAC markedly
increased the ratio of HW/BW to 42.06% higher than that in the sham
group (P<0.001) (Fig. 3A). AA
administration reduced the ratio of HW/BW by 28.27% relative to the
TAC group (P<0.001). The mice subjected to pressure overload
displayed markedly higher IVSD and LVPWD compared with the the
sham-operated mice (P<0.001 and P<0.001 for IVSD and LVPWD,
respectively) (Fig. 3B and C). AA
administration reduced the TAC-induced increase in IVSD and LVPWD
by 22.24% (P<0.001) and 33.28% (P<0.001), respectively. LVEDD
decreased after 2 weeks of pressure overload (P<0.05), and AA
administration prevented the TAC-induced decrease in LVEDD
(P<0.05). In addition, %FS significantly decreased by 22.25%
relative to the sham group at 2 weeks after TAC (P<0.001). AA
pre-treatment attenuated the TAC-induced decrease in %FS
(P<0.001).
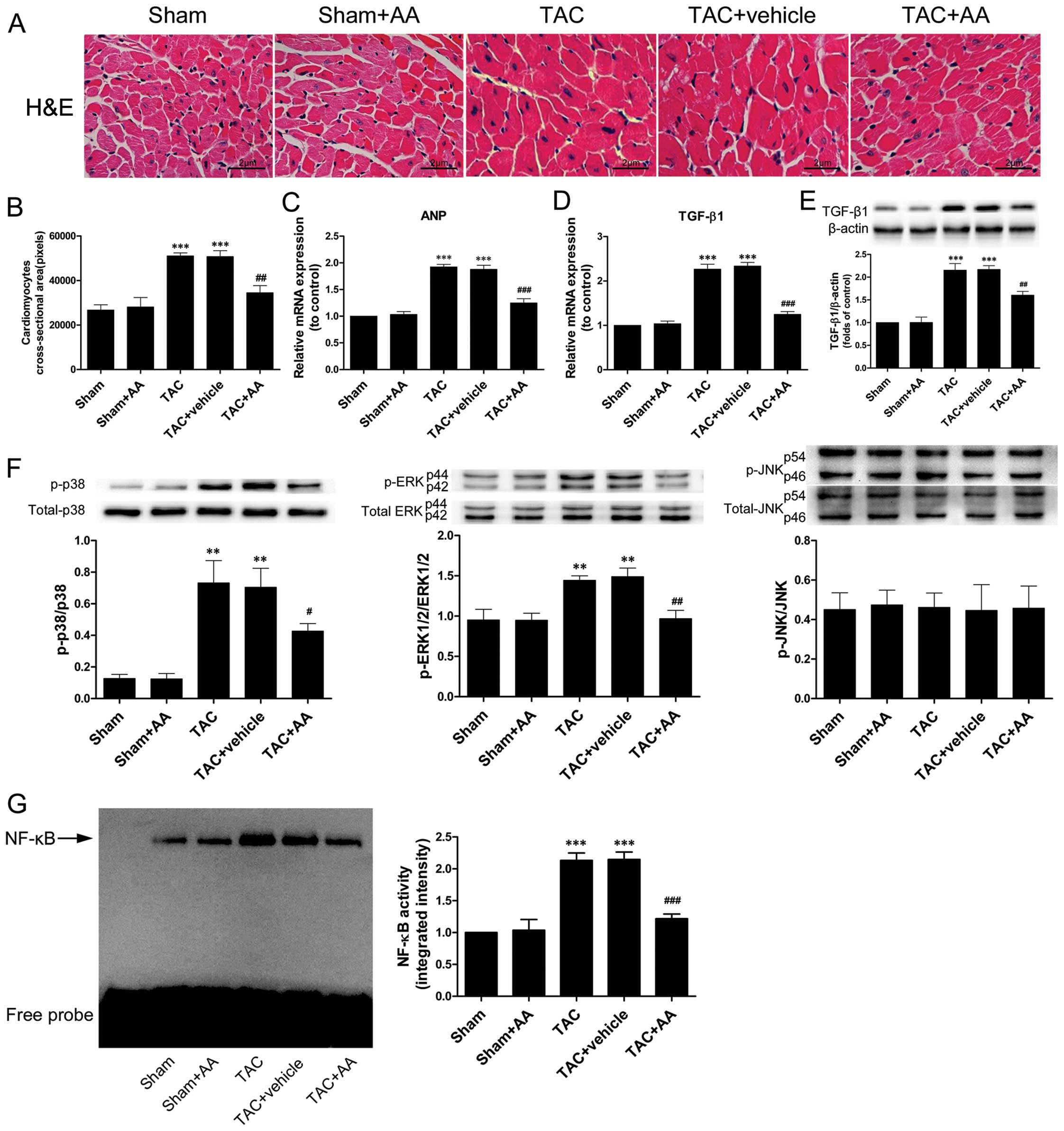
Pressure overload-induced cardiac hypertrophy was
evidenced by H&E staining; the cross-sectional area (CSA) of
the cardiomyocytes in the TAC group was much larger than that of
the cells in the sham group (P<0.001) (Fig. 4A and B). AA reduced the
TAC-induced increase in CSA (P<0.01). ANP mRNA expression is
considered as a predominant hallmark of hypertrophic remodeling
(15). Fig. 4C illustrates that pressure
overload significantly increased the mRNA expression of ANP in the
myocardium by 1.92-fold compared to that of the sham group
(P<0.001). AA prevented the TAC-induced increase in ANP mRNA
expression (P<0.001). In addition, the expression of TGF-β1 mRNA
and protein in the pressure-overloaded myocardium was markedly
higher than in the sham group (Fig.
4D and E) (P<0.001 and P<0.001 for TGF-β1 mRNA and
protein, respectively). By contrast, AA administration
substantially reduced the TAC-induced increase in the expression of
TGF-β1 mRNA and protein (Fig. 4D and
E) (P<0.001 and P<0.01 for TGF-β1 mRNA and protein,
respectively).
AA prevents the TAC-induced increase in
p38 and ERK1/2 phosphorylation and NF-κB binding activity
The activation of MAPKs and NF-κB has been shown to
play an important role in the development of cardiac hypertrophy
induced by TAC (3,9,16).
We thus examined the effect of AA on TAC-induced MAPK
phosphorylation and NF-κB binding activity. The phosphorylation
levels of p38 and ERK1/2 and the NF-κB binding activity were
notably higher in the TAC group compared with the sham group
following pressure overload lasting for 2 weeks (Fig. 4F and G). However, TAC did not
induce the activation of JNK. By contrast, AA administration
prevented the TAC-induced increase in the levels of p-p38/p38 and
p-ERK1/2/ERK1/2 and in the NF-κB binding activity (P<0.05,
P<0.01 and P<0.001 for p38, ERK1/2 and NF-κB,
respectively).
Discussion
The present study is among the first to attempt to
elucidate the biological effects of AA on cardiac hypertrophy. Our
results indicated that AA prevented not only the cardiomyocyte
hypertrophic response induced by TGF-β1 in vitro, but also
the upregulation of TGF-β1 levels and cardiac hypertrophy induced
by TAC in vivo. The anti-hypertrophic effects of AA were
found to be associated with the reduction in TGF-β1 expression, the
deactivation of p38 and ERK1/2 and the inhibition of NF-κB binding
activity. These findings support the conclusion that AA is a
suitable candidate for the prevention and treatment of cardiac
hypertrophy.
TGF-β1 is a pleiotropic and multifunctional
cytokine, which serves as a master switch in the pathogenesis of
cardiac hypertrophy (4,5). It has been previously demonstrated
that TGF-β1 levels substantially increase in the
pressure-overloaded myocardium during hypertrophic growth (17). Moreover, the role of TGF-β1 in
provoking cardiac hypertrophy is evidently supported in transgenic
mice. The overexpression of TGF-β1 in transgenic mice has been
shown to contribute significantly to cardiac hypertrophy, with
other factors being contractile dysfunction and interstitial
fibrosis (18). Conversely,
TGF-β-deficient mice show no marked cardiac hypertrophy in response
to hypertrophic stimuli (4). In
accordance with these reports, the in vitro experiments of
the present study revealed that TGF-β1 stimulation markedly
increased the size of the cardiomyocytes, as well as the mRNA
expression of ANP, which are characteristics of hypertrophic
processes (15). In the present
in vivo pressure overload model, the mice subjected to TAC
displayed a marked upregulation in TGF-β1 mRNA and protein levels
in the myocardium, as well as significant cardiac hypertrophy. AA
treatment was found to prevent the TGF-β1-induced hypertrophic
response of the cardiomyocytes. In vivo experiments revealed
that AA treatment not only reduced the expression of TGF-β1 mRNA
and protein in the pressure-overloaded myocardium, but also
attenuated cardiac hypertrophy and improved cardiac performance by
reducing the dimensions of the left ventricular chamber. This
further demonstrates that TGF-β1 serves as a trigger for cardiac
hypertrophy induced by pressure overload. Taken together, these
data suggest that the inhibition of TGF-β1 signaling may be one of
the mechanisms through which AA attenuates cardiac hypertrophy.
The activation of MAPKs, such as p38, ERK1/2 and
JNK, has been implicated in cardiac hypertrophy (3,19).
p38 and JNK have been reported to significantly contribute to the
induction of specific gene expression and increased protein
synthesis in the hypertrophic myocardium (20,21). ERK1/2 has been implicated in
growth-associated hypertrophic growth (22). More importantly, extensive basic
and clinical studies have demonstrated that MAPKs serve as
downstream signaling targets of TGF-β1 (3,5,6).
For instance, Huang et al (7) reported that TGF-β1 signaling induced
the phosphorylation of p38, ERK1/2 and JNK, and was largely
responsible for cardiac hypertrophy in the pressure-overloaded
myocardium in vivo. In in vitro experiments, Lim
et al (4) observed that
the phosphorylation levels of p38, ERK1/2 and JNK significantly
increased in cardiomyocytes following TGF-β1-induced hypertrophic
growth. In addition, Esposito et al (23) reported that the phosphorylation
levels of p38 and ERK1/2 significantly increased following
treatment with TAC for 1 week. JNK activity also increased, but
decreased over time. In accordance with these studies, our results
showed a significant increase in the phosphorylation levels of p38
and ERK1/2 in cardiomyocytes following TGF-β1-induced hypertrophy
in vitro, but no increase was observed for JNK. The present
in vivo experiments showed that the significant increase in
the p38 and ERK1/2 phosphorylation levels positively correlated
with the marked upregulation of TGF-β1 expression in the
pressure-overloaded myocardium. This indicated that, in the present
study, p38 and ERK1/2 served as downstream signaling targets of
TGF-β1. When AA was administered, the TGF-β1-induced increase in
the p38 and ERK1/2 phosphorylation levels markedly reduced in
vitro and in vivo.
NF-κB, a DNA-binding transcription factor, is known
to play a critical role in controlling the production of
pro-inflammatory cytokines and is required for the development of
cardiac hypertrophy (8,16,24,25). As previously described, MAPK
signaling can phosphorylate and activate their target proteins and
transcription factors, resulting in NF-κB translocation to the
nucleus. This initiates the expression of fetal genes and the
production of inflammatory cytokines, and ultimately provokes the
development of cardiac hypertrophy (6,8,24,26). On the other hand, the inhibition
of NF-κB binding activity may be a means of preventing cardiac
hypertrophy (8,26). For instance, the inactivation of
NF-κB with direct gene transfection of sh-p65 RNA has been shown to
result in the attenuation of cardiac hypertrophy (26). In addition, Li et al
(8) reported that blocking NF-κB
binding activity in the myocardium significantly attenuated the
pressure overload-induced cardiac hypertrophy. In accordance with
these findings, the present study found that the NF-κB binding
activity was significantly increased in cardiomyocytes following
TGF-β1-induced hypertrophy in vitro. In vivo
experiments revealed that the significant increase in NF-κB binding
activity was associated with the marked activation of
TGF-β1-p38/ERK1/2 signaling in the pressure-overloaded myocardium.
AA administration substantially reduced the NF-κB binding activity
in the TGF-β1-stimulated hypertrophic cardiomyocytes in
vitro and in the pressure-overloaded myocardium in
vivo.
In conclusion, to the best of our knowledge, the
present study is the first to demonstrate that AA inhibits the
development of cardiac hypertrophy in vitro and in
vivo. The mechanisms through which AA attenuates this
hypertrophic process involve the downregulation of TGF-β1
expression levels, the inhibition of p38 and ERK1/2
phosphorylation, and the reduction in NF-κB binding activity. These
results suggest that AA may be used as a pharmacological agent for
the prevention and treatment of cardiac hypertrophy.
Acknowledgements
This study was supported in part by grants from the
Natural Science Foundation of Jiangsu Higher Education Institutions
(12KJB320003); the Administration of Traditional Chinese Medicine
of Jiangsu Province (lz13217); the Nanjing Foundation for
Development of Science and Technology (201303036); the National
Natural Science Foundation of China (81300128); Ph.D. Programs
Foundation of Ministry of Education of China (20123234120015); the
Jiangsu Natural Science Foundation (BK20131025); the Project
Sponsored by the Scientific Research Foundation for Returned
Overseas Chinese Scholars, State Education Ministry; and the
Project Funded by the Priority Academic Program Development of
Jiangsu Higher Education Institutions.
References
|
1
|
Frohlich ED and Susic D: Pressure
overload. Heart Fail Clin. 8:21–32. 2012. View Article : Google Scholar
|
|
2
|
Ruwhof C and van der Laarse A: Mechanical
stress-induced cardiac hypertrophy: mechanisms and signal
transduction pathways. Cardiovasc Res. 47:23–37. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heineke J and Molkentin JD: Regulation of
cardiac hypertrophy by intracellular signalling pathways. Nat Rev
Mol Cell Biol. 7:589–600. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lim JY, Park SJ, Hwang HY, Park EJ, Nam
JH, Kim J and Park SI: TGF-beta1 induces cardiac hypertrophic
responses via PKC-dependent ATF-2 activation. J Mol Cell Cardiol.
39:627–636. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dobaczewski M, Chen W and Frangogiannis
NG: Transforming growth factor (TGF)-β signaling in cardiac
remodeling. J Mol Cell Cardiol. 51:600–606. 2011.
|
|
6
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang H, Tang QZ, Wang AB, et al: Tumor
suppressor A20 protects against cardiac hypertrophy and fibrosis by
blocking transforming growth factor-beta-activated kinase
1-dependent signaling. Hypertension. 56:232–239. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Y, Ha T, Gao X, et al: NF-kappaB
activation is required for the development of cardiac hypertrophy
in vivo. Am J Physiol Heart Circ Physiol. 287:H1712–H1720. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li HL, Wang AB, Huang Y, et al:
Isorhapontigenin, a new resveratrol analog, attenuates cardiac
hypertrophy via blocking signaling transduction pathways. Free
Radic Biol Med. 38:243–257. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pittella F, Dutra RC, Junior DD, Lopes MT
and Barbosa NR: Antioxidant and cytotoxic activities of Centella
asiatica (L) Urb. Int J Mol Sci. 10:3713–3721. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yun KJ, Kim JY, Kim JB, et al: Inhibition
of LPS-induced NO and PGE2 production by asiatic acid via NF-kappa
B inactivation in RAW 264.7 macrophages: possible involvement of
the IKK and MAPK pathways. Int Immunopharmacol. 8:431–441. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang X, Wu J, Dou Y, Xia B, Rong W,
Rimbach G and Lou Y: Asiatic acid protects primary neurons against
C2-ceramide-induced apoptosis. Eur J Pharmacol. 679:51–59. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tang LX, He RH, Yang G, et al: Asiatic
acid inhibits liver fibrosis by blocking TGF-beta/Smad signaling in
vivo and in vitro. PLoS One. 7:e313502012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu XH, Xu J, Xue L, Cao HL, Liu X and Chen
YJ: VEGF attenuates development from cardiac hypertrophy to heart
failure after aortic stenosis through mitochondrial mediated
apoptosis and cardiomyocyte proliferation. J Cardiothorac Surg.
6:542011. View Article : Google Scholar
|
|
15
|
Feng JA, Perry G, Mori T, Hayashi T,
Oparil S and Chen YF: Pressure-independent enhancement of cardiac
hypertrophy in atrial natriuretic peptide-deficient mice. Clin Exp
Pharmacol Physiol. 30:343–349. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu Y, Li T, Song J, et al: The
TIR/BB-loop mimetic AS-1 prevents cardiac hypertrophy by inhibiting
IL-1R-mediated MyD88-dependent signaling. Basic Res Cardiol.
106:787–799. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li JM and Brooks G: Differential protein
expression and subcellular distribution of TGFbeta1, beta2 and
beta3 in cardiomyocytes during pressure overload-induced
hypertrophy. J Mol Cell Cardiol. 29:2213–2224. 1997. View Article : Google Scholar
|
|
18
|
Rosenkranz S, Flesch M, Amann K, et al:
Alterations of beta-adrenergic signaling and cardiac hypertrophy in
transgenic mice overexpressing TGF-beta(1). Am J Physiol Heart Circ
Physiol. 283:H1253–H1262. 2002.PubMed/NCBI
|
|
19
|
Wang Y: Mitogen-activated protein kinases
in heart development and diseases. Circulation. 116:1413–1423.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
LaMorte VJ, Thorburn J, Absher D, et al:
Gq- and ras-dependent pathways mediate hypertrophy of neonatal rat
ventricular myocytes following alpha 1-adrenergic stimulation. J
Biol Chem. 269:13490–13496. 1994.PubMed/NCBI
|
|
21
|
Wang Y: Signal transduction in cardiac
hypertrophy-dissecting compensatory versus pathological pathways
utilizing a transgenic approach. Curr Opin Pharmacol. 1:134–140.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Clerk A and Sugden PH: Untangling the Web:
specific signaling from PKC isoforms to MAPK cascades. Circ Res.
89:847–849. 2001.PubMed/NCBI
|
|
23
|
Esposito G, Prasad SV, Rapacciuolo A, Mao
L, Koch WJ and Rockman HA: Cardiac overexpression of a G(q)
inhibitor blocks induction of extracellular signal-regulated kinase
and c-Jun NH(2)-terminal kinase activity in in vivo pressure
overload. Circulation. 103:1453–1458. 2001. View Article : Google Scholar
|
|
24
|
Hall G, Hasday JD and Rogers TB:
Regulating the regulator: NF-kappaB signaling in heart. J Mol Cell
Cardiol. 41:580–591. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gordon JW, Shaw JA and Kirshenbaum LA:
Multiple Facets of NF-κB in the heart: To Be or Not to NF-κB. Circ
Res. 108:1122–1132. 2011.
|
|
26
|
Gupta S, Young D, Maitra RK, et al:
Prevention of cardiac hypertrophy and heart failure by silencing of
NF-kappaB. J Mol Biol. 375:637–649. 2008. View Article : Google Scholar : PubMed/NCBI
|