Introduction
As one of the largest produced and used pesticides,
chlorpyrifos (CPF) is widely distributed in various environmental
media, and in fruits and vegetables (1–3).
Numerous studies have suggested that CPF may elicit adverse effects
on human health (4–6). Thus far, CFP is known as an
acetylcholinesterase (AChE) inhibitor, and induces significant
damage to the central nervous system (7). In addition to neurological
toxicities, CFP has been found to induce reactive oxygen species
(ROS), and thus cause DNA damage in several organs of animal models
(8,9). Other studies have shown that CPF
exposure is associated with developmental disorders, autoimmune
disorders (10,11), and even cancers, such as lung and
breast cancer (12,13). Moreover, histological examinations
also show that CPF administration leads to substantial injury to
liver, resulting in infiltration of macrophages in the portal area
in an animal study (9).
Liver is the central organ governing systemic iron
homeostasis, where hepcidin is primarily secreted by hepatocytes
and regulates iron flux by interacting with its receptor
ferroportin, which is associated with the internalization and
degradation of ferroportin, thereby reducing iron egress from
macrophages and iron absorption from the small intestine (14–16). Iron, an essential element, is
important for a variety of biological processes (17,18). Systemic iron homeostasis is
deliberately modulated by the hepcidin-ferroportin regulatory axis
(14,19–21). Ferroportin is the only known
mammalian iron exporter, mainly expressed in epithelial cells in
duodenum and macrophages, and is critical for controlling iron
egress (15,16). Through the hepcidin-ferroportin
axis, downregulated hepatic hepcidin increases the serum iron level
by promoting iron absorption from enterocytes in duodenum and iron
release from macrophages (22).
Increased ferroportin concentration enhances iron egress from
cells, thereby decreasing intracellular iron (23).
However, studies available on CPF-conducted
impairments to liver-centered iron homeostasis are currently
limited, and the present literature even remains contradictory.
Furthermore, little insight has been gained into potential effects
of CPF on the hepcidin-ferroportin axis, and the molecular
mechanisms responsible for CPF-mediated actions on iron metabolism
remains to be clarified. To the best of our knowledge, in the
present study, we demonstrated for the first time that CPF was able
to disturb iron metabolism by enhancing ferroportin expression in
macrophages and inhibiting hepcidin expression in hepatocytes at
sublethal concentrations.
Materials and methods
Chemicals, reagents and cell culture
CPF was purchased from Shuangma Fine Chemical Co.,
Ltd. (Nantong, China) with the purity of >99.99%. The THP-1
human macrophage, HepG2 human hepatic carcinoma and HEK293T human
embryonic kidney cell lines were obtained from the Shanghai Cell
Bank of Type Culture Collection of Chinese Academy of Sciences. The
cells were cultured in RPMI-1640 medium (Gibco) supplemented with
10% fetal bovine serum (FBS) and 100 U/ml penicillin/streptomycin
(both from HyClone, Logan, UT, USA) in a humidified incubator
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) with 5%
CO2 at 37°C. Differentiation of THP-1 cells into
macrophages was induced with 1 μg/ml PMA (Promega, Madison, WI,
USA) in complete medium for 18 h.
Alamar blue assay
Cell viability was determined through the Alamar
blue assay according to the manufacturer’s instructions (Thermo
Fisher Scientific, Inc.). Briefly, THP-1 and HepG2 cells were
seeded in 96-well plates at a concentration of 5.0×103
cells/well. The cells were then treated with CPF for 24 h, followed
by emission detection at 590 nm with excitation at 530 nm.
Western blotting
Cells were harvested and washed twice with cold
phosphate-buffered saline (PBS). Total proteins were then extracted
with RIPA lysis buffer (Solarbio, Beijing, China) supplemented with
protease inhibitor cocktail (Roche Applied Science, San Francisco,
CA, USA). Equal amounts of protein lysates (30–50 μg/sample) were
subjected to 10% SDS-PAGE and western blot analysis was performed
as described previously (24).
Antibodies included anti-ferroprotin (1:500; Sigma-Aldrich, St.
Louis, MO, USA) and anti-glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) (1:1,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA). Intensity of bands was assessed with the ImageJ software
(http://rsb.info.nih.gov/ij/). GAPDH was
used as an internal control for normalization.
RNA extraction and RT-PCR analysis
Total RNAs were isolated from cells with TRIzol
according to the manufacturer’s instructions (Invitrogen Life
Technologies, Carlsbad, CA, USA). The first strand of cDNA was
synthesized from 2 μg total RNAs using oligo(dT) primer. Primer
sequences used for the PCR reaction were: hepcidin,
5′-CCTGACCAGTGGCTCTGTTT-3′ (forward) and 5′-CACATCCCACACTTTGATCG-3′
(reverse); GAPDH, 5′-GAAGGTGAAGGTCGGAGT-3′ (forward) and
5′-GAAGATGGTGATGGGATTTC-3′ (reverse). GAPDH was used as an internal
control.
Luciferase reporter assays
A DNA fragment with ferroportin promoter region was
subcloned into the pGL3-Promoter luciferase reporter vector
(Promega) to replace the SV40 promoter. The final construct was
validated by DNA sequencing (Invitrogen). Then, 0.8 μg target
construct plus 80 ng Renilla luciferase construct were
co-transfected into HEK293T cells in 24-well plates using
Lipofectamine™ 2000 (Invitrogen). After 24 h, the cells were washed
with PBS, and assayed for luciferase activity using a
dual-luciferase reporter assay system according to the
manufacturer’s instructions (Promega). Relative firefly luciferase
activities were subsequently normalized to those of Renilla
luciferase.
Labile iron pool (LIP) assay
Intracellular LIP was measured as previously
described (25). Briefly, the
cells were collected and washed following treatments, and
subsequently incubated with 0.5 μM calcein acetoxymethyl ester
(CA-AM) (Sigma-Aldrich) for 15 min at 37°C. After washing with PBS,
the cells were equally divided into two groups. One group was
treated with 100 μM desferoxamine (DFO; Sigma-Aldrich) for 1 h at
37°C, and the second group was left untreated. The cells were
subjected to FACS analysis (FACSCalibur; BD Biosciences, San Jose,
CA, USA), where calcein was excited at 488 nm and measured at 525
nm. Intracellular LIP was obtained through deduction of the
cellular fluorescence of DFO-treated cells from that of the
untreated cells.
Statistical analysis
Significance of data between two groups was
determined by the two-tailed independent t-test. One-way analysis
of variance (ANOVA) was employed to assess the mean differences
among groups relative to the control. Experimental data were shown
as mean ± standard error (SE). P<0.05 was considered to indicate
a statistically significant result.
Results
Cytotoxicity following CPF treatment in
macrophages and hepatocytes
To examine the potential effects of CPF exposure on
iron metabolism, two cardinal types of cells were employed to
determine systemic iron flux, macrophages and hepatocytes (16,26,27). The THP-1 human macrophage and
HepG2 human hepatic carcinoma cell lines were used. In order to
dissect the non-toxicity biological effects from cytotoxicity, in
other words, to focus on the biological effects of CPF without
exerting significant injuries on cells, concentrations that did not
induce significant toxicity to cells were initially obtained. THP-1
and HepG2 cells were treated with different concentrations of CPF
for 24 h, and cell viabilities were measured using the Alamar blue
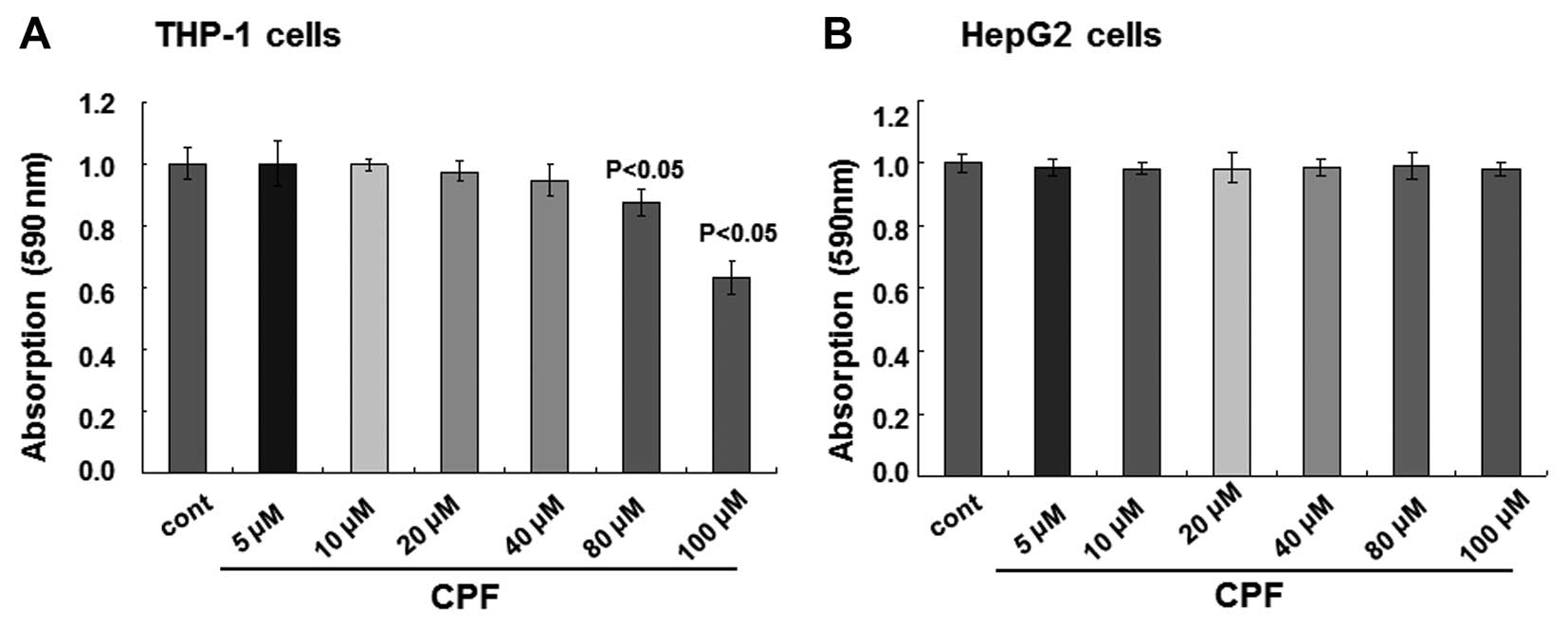
assay. No alterations of cell viability were observed when the
concentrations of CPF were <80 μM for THP-1 cells, while cell
viability was reduced >10% at 80 μM (P<0.05) and subsequently
reduced by >30% at 100 μM (P<0.05) (Fig. 1A). With respect to HepG2 cells, no
detectable cytotoxicity occurred when the cells were treated with
CPF up to 100 μM CPF (P>0.05) (Fig. 1B). Therefore, in the present
study, a non-toxic (i.e., sublethal) concentration, 20 μM, was
selected for the subsequent experiments. Notably, the medium of 20
μM CPF contained 0.001% DMSO only. Medium with 0.001% DMSO caused
no toxicity to cells compared to the complete blank control, and it
served as a control in the present study (designated as control or
cont in the text and figures).
CFP elevates ferroportin expression
through transcriptional regulation
Ferroportin is the only known iron exporter in
mammal animals, and its dysfunction is involved in various iron
disorders, such as thalassemias and hemochromatosis (16,27,28). We therefore investigated the
effect of CPF treatment on ferroportin concentration in THP-1
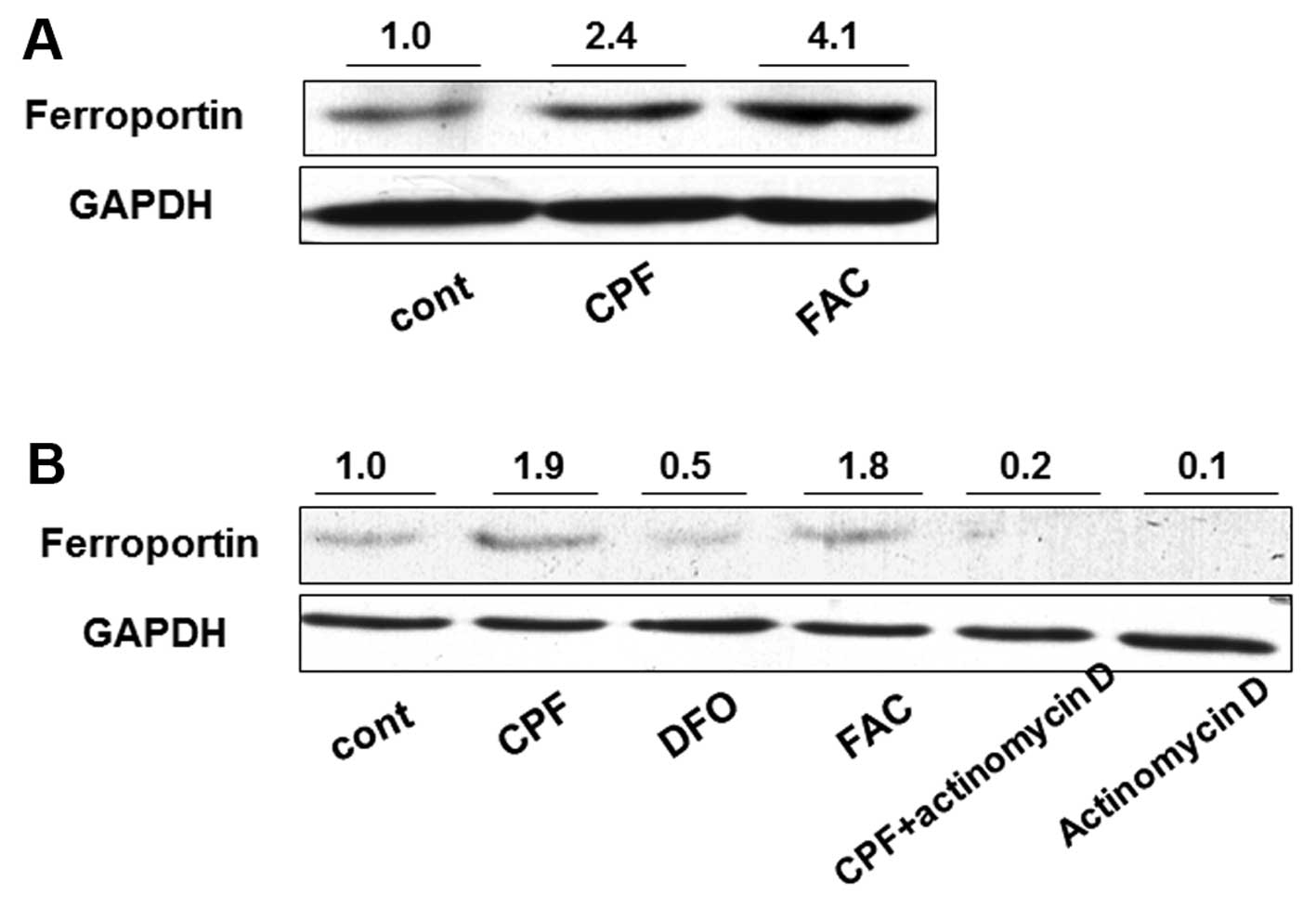
macrophages. Following treatment with CPF at 20 μM for 24 h, the
ferroportin concentration was significantly induced by >2-fold
in CPF-treated cells compared to the untreated cells (Fig. 2A). Ferric ammonium citrate (FAC)
(100 μM), as a positive control, was used to increase ferroportin
concentration (29) (Fig. 2A). To investigate the mechanism
underlying the increase of ferroportin concentration, a
transcription inhibitor, actinomycin D, was used to attenuate
global mRNA transcription. Actinomycin D treatment alone greatly
reduced ferroportin concentration in THP-1 cells, suggesting marked
transcriptional inhibition by actinomycin D (Fig. 2B). The increase of ferroportin
concentration by CPF was markedly reduced when cells were
simultaneously treated with actinomycin D compared to that in cells
treated with CPF only (Fig. 2B).
The ferroportin concentration in the cells treated with CPF +
actinomycin D was greater than that in the cells treated with
actinomycin D only (Fig. 2B).
These results suggested that CPF promoted the ferroportin level
through a transcriptional regulatory mechanism. FAC was used as a
positive control to elevate ferroportin, and DFO (100 μM), a
negative control (30), was used
to impede the ferroportin increase (Fig. 2B).
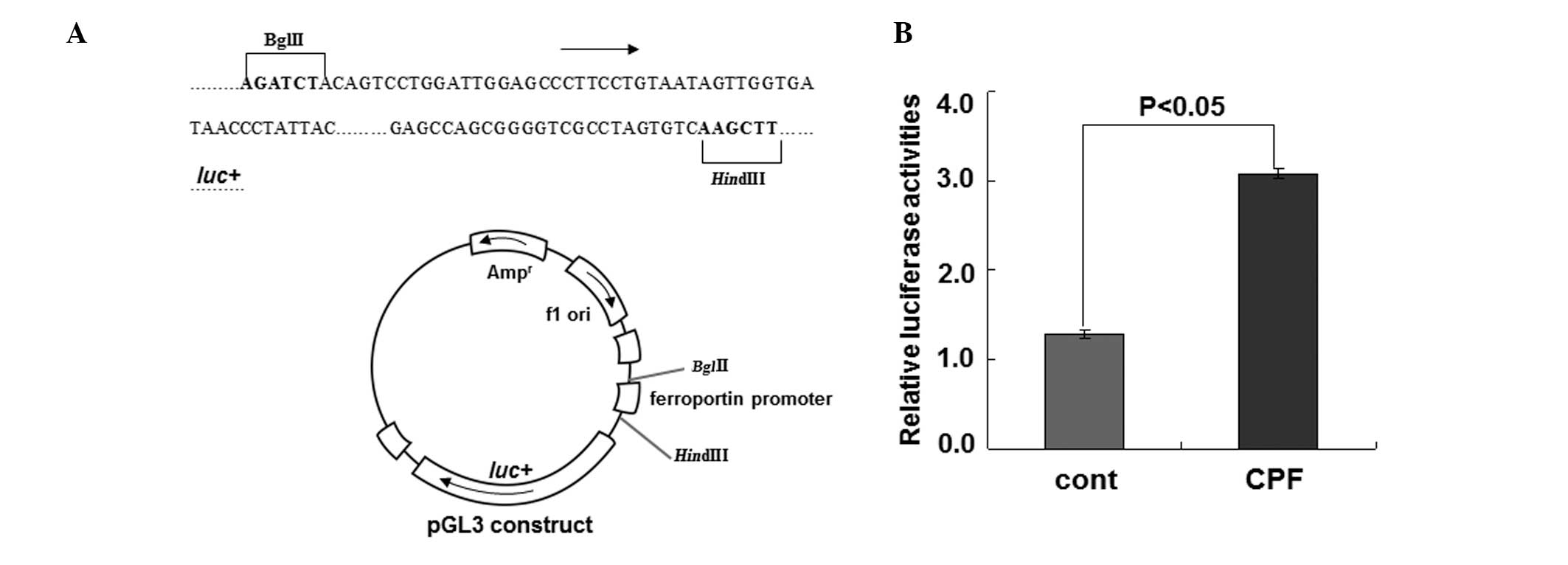
To substantiate the regulation of ferroportin by CPF
at the transcriptional level, we performed the luciferase reporter
assay to examine ferroportin expression driven by its own promoter
upon CPF (Fig. 3). Following
exposure to CFP at 20 μM for 24 h in HEK293T cells, luciferase
activity was measured. Compared to the control, CPF treatment
increased luciferase activity by ~2.5-fold (P<0.05) (Fig. 3), supporting the transcriptional
regulation of ferroportin by CPF. These data together identified a
novel effect of CPF on promoting ferroportin expression in
macrophages.
CPF inhibits hepatic hepcidin
expression
To understand the biological effects of CPF on iron
metabolism, hepatic hepcidin expression was investigated. As the
master regulator of systemic iron homeostasis, hepcidin is
predominantly secreted by hepatocytes (31). Additionally, hepcidin directs iron
flow by suppressing ferroportin-conducted iron release (32). Hepcidin reduction is associated
with increased iron egress and intracellular iron depletion. HepG2
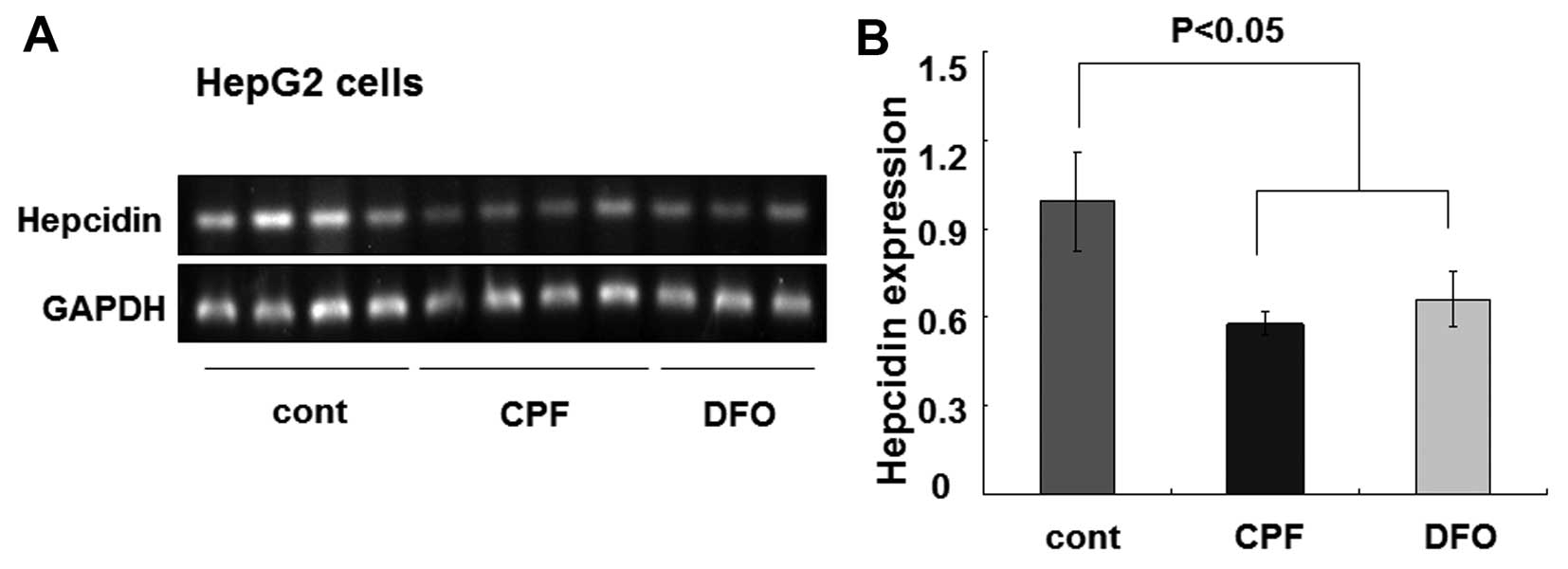
cells were similarly treated with CPF at 20 μM. After 24 h, we
found that the hepcidin mRNA level was greatly reduced by CPF
(Fig. 4A). The quantified data
showed an ~50% reduction of the hepcidin mRNA level in CPF-treated
cells relative to the untreated cells (P<0.05) (Fig. 4B). Consistent with previous
studies (33), DFO was used as a
control to inhibit hepcidin expression (P<0.05) (Fig. 4). These results demonstrated a
considerable inhibitory effect of CPF on hepatic hepcidin
expression.
Decreased LIP following CFP treatment in
macrophages and hepatocytes
The available section of intercellular iron occurred
in loosely-bound divalent iron form (i.e., LIP). It was readily and
rapidly involved in the synthesis of iron-sulfur clusters, heme and
other iron-containing proteins (34,35). LIP was largely determined by the
concentrations of iron exporter ferroportin and iron-storage
protein ferritin (35). Thus, the
intracellular iron concentration was sensitively reflected by the
availability and abundance of cellular LIP. To elucidate the
consequential effects on iron storage following CPF exposure in
THP-1 and HepG2 cells, we assayed LIP in the two types of cells
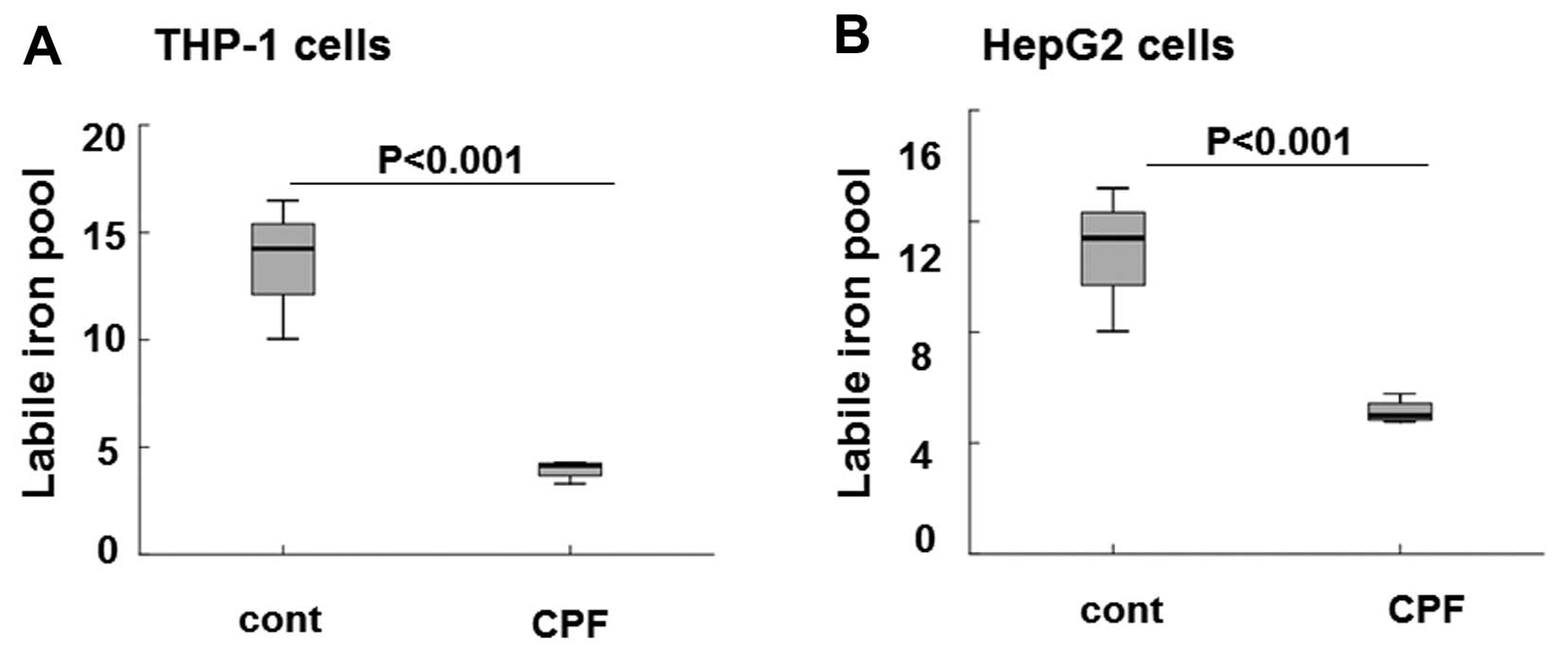
following CPF treatment at 20 μM for 24 h. As shown in Fig. 5A the level of intracellular LIP
was markedly reduced by ~3-fold in THP-1 cells in CPF-treated cells
relative to the untreated cells (P<0.001), consistent with the
significant induction of ferroportin subsequent to CPF treatment
(Fig. 2). The level of
intracellular LIP was greatly reduced by >2-fold in HepG2 cells
treated with CPF compared to the untreated cells (Fig. 5B) (P<0.001), concomitant to
hepcidin reduction in these cells following CPF (Fig. 4). These data signified the
biological consequences of CPF on iron flow in macrophages and
hepatocytes, by modulating ferroportin and hepcidin expression,
respectively.
Discussion
When CPF is uptaken, it may accumulate in various
organs, such as liver, kidney, ovary and uterus (36–38), with liver being one of the
preferential targets (39).
Long-term exposure to CPF results in significant histopathological
alterations to liver, including hepatocytic vacuolation, sinusoidal
dilation and focal necrosis (40). The potential non-toxic biological
effects of CPF on liver, i.e., changes of biological functions
without significant cytotoxicity, has been been recognized.
Hepatocytes and macrophages, synergistically play a pivotal role in
governing systemic iron homeostasis (41). In the present study, we identified
a novel effect of CFP treatment on iron flow by altering the iron
gene expression in hepatocytes and macrophages. Hepcidin expression
was inhibited in HepG2 hepatocytes following CPF treatment,
accompanied by reduced LIP, while ferroportin expression was
elevated in THP-1 macrophages following CPF treatment, which was
associated with LIP reduction.
Reticuloendothelial macrophages are the most
important cells for iron storage, as they are involved in aborbing
senescent red blood cells to recycle iron for erythropoiesis
(42), where ferroportin, as the
only known iron exporter, controls iron egress out of macrophages
(43). An increased ferroportin
concentration enhances iron release and reduces intracellular iron
storage, as evidenced by LIP reduction (44,45). In the present study, we
demonstrate that CPF exposure increased ferroportin concentration
in THP-1 macrophages, and this increase was mainly due to the
upregulation of ferroportin transcription. As a result of the
ferroportin increase, the available iron content as reflected by
LIP was reduced in THP-1 macrophages. By contrast, we identified a
reduced hepatic hepcidin expression in HepG2 hepatocytes following
CPF treatment, which led to a reduced intracellular iron level as
evidenced by LIP reduction presumably due to the hepcidin-induced
degradation of ferroportin in hepatocytes. These results suggest
that CPF exposure elicited significant changes to cellular iron
homeostasis by regulating hepcidin and ferroportin expression in
macrophages and hepatocytes. The detailed mechanisms underlying
CPF-promoted activity in transcribing ferroportin and CPF-mediated
hepcidin repression should be further investigated.
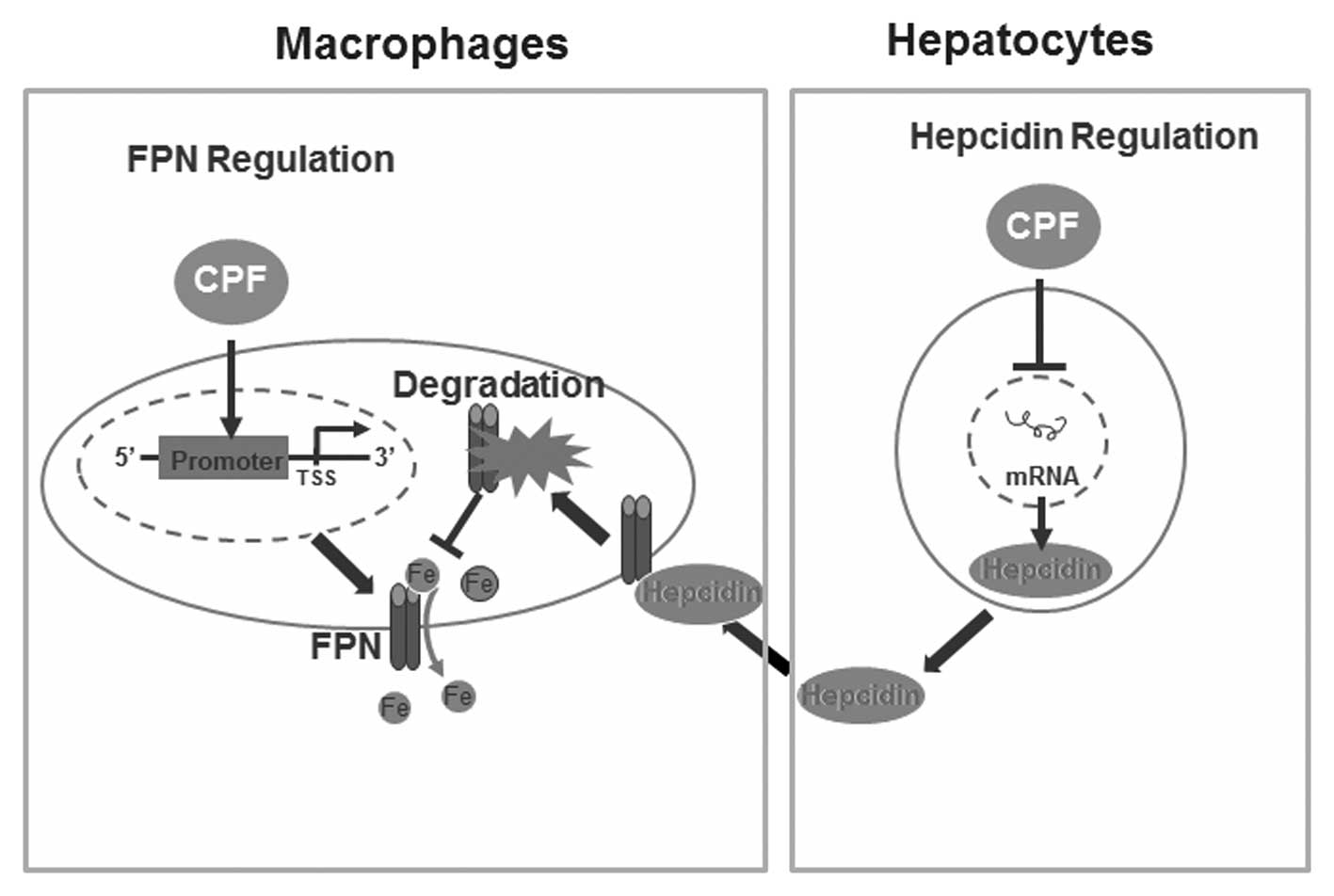
In conclusion, results of the present study have
identified a novel finding of CPF in altering cellular iron
homeostasis without imparing cell viability. We have demonstrated
that CPF exposure significantly altered the expression of central
iron genes, i.e., CPF elevates ferroportin expression in
macrophages and represses hepcidin expression in hepatocytes,
coupled with LIP reduction in macrophages and hepatocytes. A
proposed schematic diagram deciphering the mechanisms responsible
for CPF-induced alterations to cellular iron homeostasis is
presented in Fig. 6.
Acknowledgements
This study was supported by a grant under the
national ‘973’ program (grant no. 2014CB932000) and the National
Natural Science Foundation of China (grant no. 21377159). We would
like to thank the laboratory members for their great assistance
with the experiments and reagents.
References
|
1
|
Zhao L, Wang F and Zhao J: Identification
and functional characteristics of chlorpyrifos-degrading and plant
growth promoting bacterium Acinetobacter calcoaceticus. J
Basic Microbiol. May 26–2013.(Epub ahead of print).
|
|
2
|
Kumar Singh B, Walker A and Wright DJ:
Persistence of chlorpyrifos, fenamiphos, chlorothalonil, and
pendimethalin in soil and their effects on soil microbial
characteristics. Bull Environ Contam Toxicol. 69:181–188.
2002.PubMed/NCBI
|
|
3
|
Gebremariam SY, Beutel MW, Yonge DR, Flury
M and Harsh JB: Adsorption and desorption of chlorpyrifos to soils
and sediments. Rev Environ Contam Toxicol. 215:123–175.
2012.PubMed/NCBI
|
|
4
|
Mink PJ, Kimmel CA and Li AA: Potential
effects of chlorpyrifos on fetal growth outcomes: implications for
risk assessment. J Toxicol Environ Health B Crit Rev. 15:281–316.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ridano ME, Racca AC, Flores-Martin J, et
al: Chlorpyrifos modifies the expression of genes involved in human
placental function. Reprod Toxicol. 33:331–338. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singh S, Kumar V, Thakur S, et al: DNA
damage and cholinesterase activity in occupational workers exposed
to pesticides. Environ Toxicol Pharmacol. 31:278–285. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sandahl JF, Baldwin DH, Jenkins JJ and
Scholz NL: Comparative thresholds for acetylcholinesterase
inhibition and behavioral impairment in coho salmon exposed to
chlorpyrifos. Environ Toxicol Chem. 24:136–145. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gupta SC, Mishra M, Sharma A, et al:
Chlorpyrifos induces apoptosis and DNA damage in Drosophila
through generation of reactive oxygen species. Ecotoxicol Environ
Saf. 73:1415–1423. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ojha A, Yaduvanshi SK, Pant SC, Lomash V
and Srivastava N: Evaluation of DNA damage and cytotoxicity induced
by three commonly used organophosphate pesticides individually and
in mixture, in rat tissues. Environ Toxicol. 28:543–552. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gralewicz S: Possible consequences of AChE
inhibition in organophosphate poisoning. A new approach to an old
problem. Med Pr. 48:469–472. 1997.(In Polish).
|
|
11
|
Fu Y, Li M, Liu C, et al: Effect of
atrazine and chlorpyrifos exposure on cytochrome P450 contents and
enzyme activities in common carp gills. Ecotoxicol Environ Saf.
94:28–36. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee WJ, Blair A, Hoppin JA, et al: Cancer
incidence among pesticide applicators exposed to chlorpyrifos in
the Agricultural Health Study. J Natl Cancer Inst. 96:1781–1789.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ventura C, Núñez M, Miret N, et al:
Differential mechanisms of action are involved in chlorpyrifos
effects in estrogen-dependent or -independent breast cancer cells
exposed to low or high concentrations of the pesticide. Toxicol
Lett. 213:184–193. 2012. View Article : Google Scholar
|
|
14
|
Ganz T: Hepcidin and iron regulation, 10
years later. Blood. 117:4425–4433. 2011.PubMed/NCBI
|
|
15
|
Jelić M1, Cvetković T, Djordjević V, et
al: Hepcidin and iron metabolism disorders in patients with chronic
kidney disease. Vojnosanit Pregl. 70:368–373. 2013.PubMed/NCBI
|
|
16
|
Pinnix ZK, Miller LD, Wang W, et al:
Ferroportin and iron regulation in breast cancer progression and
prognosis. Sci Transl Med. 2:43ra562010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sheftel A, Stehling O and Lill R:
Iron-sulfur proteins in health and disease. Trends Endocrinol
Metab. 21:302–314. 2010. View Article : Google Scholar
|
|
18
|
Frossard E, Bucher M, Machler F, Mozafar A
and Hurrell R: Potential for increasing the content and
bioavailability of Fe, Zn and Ca in plants for human nutrition. J
Sci Food Agric. 80:861–879. 2000. View Article : Google Scholar
|
|
19
|
Andrews NC: Forging a field: the golden
age of iron biology. Blood. 112:219–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hentze MW, Muckenthaler MU, Galy B and
Camaschella C: Two to tango: regulation of Mammalian iron
metabolism. Cell. 142:24–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Knutson MD: Iron-sensing proteins that
regulate hepcidin and enteric iron absorption. Annu Rev Nutr.
30:149–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ganz T and Nemeth E: Hepcidin and
disorders of iron metabolism. Annu Rev Med. 62:347–360. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Donovan A, Lima CA, Pinkus JL, et al: The
iron exporter ferroportin/Slc40a1 is essential for iron
homeostasis. Cell Metab. 1:191–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qu G, Zhang C, Yuan L, et al: Quantum dots
impair macrophagic morphology and the ability of phagocytosis by
inhibiting the Rho-associated kinase signaling. Nanoscale.
4:2239–2244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Prus E and Fibach E: Flow cytometry
measurement of the labile iron pool in human hematopoietic cells.
Cytometry A. 73:22–27. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hentze MW, Muckenthaler MU and Andrews NC:
Balancing acts: molecular control of mammalian iron metabolism.
Cell. 117:285–297. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nemeth E, Tuttle MS, Powelson J, et al:
Hepcidin regulates cellular iron efflux by binding to ferroportin
and inducing its internalization. Science. 306:2090–2093. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Singh B, Arora S, Agrawal P and Gupta SK:
Hepcidin: a novel peptide hormone regulating iron metabolism. Clin
Chim Acta. 412:823–830. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Doyard M, Fatih N, Monnier A, et al: Iron
excess limits HHIPL-2 gene expression and decreases osteoblastic
activity in human MG-63 cells. Osteoporos Int. 23:2435–2445. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Beutler E, Hoffbrand AV and Cook JD: Iron
deficiency and overload. Hematol Am Soc Hematol Educ Program.
2003:40–61. 2003. View Article : Google Scholar
|
|
31
|
Park CH, Valore EV, Waring AJ and Ganz T:
Hepcidin, a urinary antimicrobial peptide synthesized in the liver.
J Biol Chem. 276:7806–7810. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nicolas G, Viatte L, Bennoun M, Beaumont
C, Kahn A and Vaulont S: Hepcidin, a new iron regulatory peptide.
Blood Cells Mol Dis. 29:327–335. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gehrke SG, Kulaksiz H, Herrmann T, et al:
Expression of hepcidin in hereditary hemochromatosis: evidence for
a regulation in response to the serum transferrin saturation and to
non-transferrin-bound iron. Blood. 102:371–376. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kakhlon O and Cabantchik ZI: The labile
iron pool: characterization, measurement, and participation in
cellular processes(1). Free Radic Biol Med. 33:1037–1046.
2002.PubMed/NCBI
|
|
35
|
Breuer W, Shvartsman M and Cabantchik ZI:
Intracellular labile iron. Int J Biochem Cell Biol. 40:350–354.
2008. View Article : Google Scholar
|
|
36
|
Nishi K and Hundal SS: Chlorpyrifos
induced toxicity in reproductive organs of female Wistar rats. Food
Chem Toxicol. 62:732–738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xing H, Wu H, Sun G, Zhang Z, Xu S and Li
S: Alterations in activity and mRNA expression of
acetylcholinesterase in the liver, kidney and gill of common carp
exposed to atrazine and chlorpyrifos. Environ Toxicol Pharmacol.
35:47–54. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xing H, Li S and Wang X, Gao X, Xu S and
Wang X: Effects of atrazine and chlorpyrifos on the mRNA levels of
HSP70 and HSC70 in the liver, brain, kidney and gill of common carp
(Cyprinus carpio L.). Chemosphere. 90:910–916. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Acker CI, Souza AC, Dos Santos MP,
Mazzanti CM and Nogueira CW: Diphenyl diselenide attenuates hepatic
and hematologic toxicity induced by chlorpyrifos acute exposure in
rats. Environ Sci Pollut Res Int. 19:3481–3490. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tuzmen N, Candan N, Kaya E and Demiryas N:
Biochemical effects of chlorpyrifos and deltamethrin on altered
antioxidative defense mechanisms and lipid peroxidation in rat
liver. Cell Biochem Funct. 26:119–124. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Evstatiev R and Gasche C: Iron sensing and
signalling. Gut. 61:933–952. 2012. View Article : Google Scholar
|
|
42
|
Kong WN, Lei YH and Chang YZ: The
regulation of iron metabolism in the mononuclear phagocyte system.
Expert Rev Hematol. 6:411–418. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang Z, Zhang F, Guo X, An P, Tao Y and
Wang F: Ferroportin1 in hepatocytes and macrophages is required for
the efficient mobilization of body iron stores in mice. Hepatology.
56:961–971. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Galli A, Bergamaschi G, Recalde H, et al:
Ferroportin gene silencing induces iron retention and enhances
ferritin synthesis in human macrophages. Br J Haematol.
127:598–603. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang DL, Senecal T, Ghosh MC,
Ollivierre-Wilson H, Tu T and Rouault TA: Hepcidin regulates
ferroportin expression and intracellular iron homeostasis of
erythroblasts. Blood. 118:2868–2877. 2011. View Article : Google Scholar : PubMed/NCBI
|