Introduction
Cerebral edema is a leading cause of mortality after
an ischemic stroke. The disruption of the blood-brain barrier (BBB)
caused by cerebral ischemia can initiate the development of
cerebral edema and the progression of brain injuries. The
disruption of BBB integrity is one of the most crucial initiating
factors in cerebral ischemia/reperfusion (I/R) injury (1). Water content progressively increases
within 2 days of an occlusion and then gradually decreases for up
to 14 days following an ischemic stroke (2). The vascular leakage associated with
an ischemic insult may lead to a narrowing of the vascular lumen
and to an increase in the viscosity of the blood, which results in
the progression of brain edema and in the impairment of
microvascular perfusion, which in turn contribute to the subsequent
neuronal injury (3). It has been
known for several years that vascular endothelial growth factor
(VEGF) is a secreted mitogen that is associated with angiogenesis
and is also a potent vascular permeability factor (4,5).
In rats, transient and permanent middle cerebral artery (MCA)
occlusions initiate the expression of VEGF in the ischemic brain
(6–8). Astrocyte-derived VEGFA has been
shown to be involved in BBB leakage (9). The early post-ischemia
administration of recombinant human VEGF (rhVEGF) to ischemic rats
has been shown to significantly increase BBB leakage, hemorrhagic
transformation and the size of ischemic lesions (10). In the ischemic brain, the
inhibition of VEGF during the acute stage of a stroke may reduce
the permeability of the BBB and the risk of hemorrhagic
transformation following focal cerebral ischemia.
Src family kinases serve as intermediate steps in
several cell signaling pathways and propagate signals directed
toward the cytoskeleton, membrane and nuclear targets. Following a
cerebral ischemic injury, continuous cytokine and growth factor
stimulation can overactivate Src (11,12). Additionally, a previous study
demonstrated that a Src inhibitor was effective when injected as
late as 6 h after an ischemic injury (13). In that study, the inhibition of
Src did not affect the resting cerebral blood flow or the perfusion
of the ischemic hemisphere after the stroke (13). Accumulating evidence suggests that
activated Src kinase regulates VEGFA production through the signal
transducer and activator of transcription 3 (STAT3) pathway.
Constitutively activated STAT3 has been reported to upregulate
VEGFA expression and thereby induce tumor angiogenesis (14); phosphorylated Src thus aggravates
BBB leakage following ischemia through the upregulation of VEGFA
expression.
Claudin-5 in endothelial cells is a major structural
component of the BBB formed by cells within the neurovascular unit
(NVU). Claudin-5 may play a key role in the formation of the
paracellular pores or channels that mediate the selective ion
permeability of the BBB (15). It
has previously been reported that dynamic changes occur at the
level of tight junction protein expression in the BBB in response
to traumatic brain injury. VEGFA specifically downregulates
claudin-5 protein and mRNA expression in cultures of brain
microvascular endothelial cells (16). In the mouse cerebral cortex, the
microinjection of VEGFA has been shown to disrupt claudin-5 and
induce the loss of barrier function. The authors of that study
suggested that the downregulation of claudin-5 by VEGFA is the
mechanism for the BBB breakdown (16). PP2 is a Src protein tyrosine
kinase (PTK) inhibitor that decreases the infarct volume and
improves neurological scores following MCA occlusion (26). These data suggest that the
VEGFA-mediated decrease in claudin-5 may be involved in the
disruption of the BBB following ischemia. The protective effects of
PP2 during ischemic injury are thus due to the increase in
claudin-5 expression and the subsequent protection of the BBB.
However, the mechanisms through which PP2 reduces
infarct size and preserves neural function remain unclear. In the
present study, we evaluated the protective effects of PP2 on the
BBB during an ischemic insult. We aimed to perform an extensive
analysis of the changes occurring in the protein levels of VEGFA,
phosphorylated (p)-Src and claudin-5 and in the mRNA levels of
claudin-5 and to evaluate the effects of PP2 on claudin-5 protein
expression and the leakage of fibrinogen through the BBB following
I/R injury in rats.
Materials and methods
Reagents and antibodies
The Src PTK inhibitor,
4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
(PP2), was obtained from Merck Calbiochem Co. (Darmstadt, Germany).
The primary antibodies used were rabbit anti-Src and anti-p-Src
(pY418) (Epitomics, an Abcam Co. Burlingame, CA, USA), mouse
anti-VEGF (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
claudin-5 monoclonal antibody (CLDN5; Invitrogen, Life
Technologies, Carlsbad, CA, USA), rabbit anti-glial fibrillary
acidic protein (GFAP; Boster, Wuhan, China) and rabbit polyclonal
antibody to β-actin (Beijing Biotech Co., Ltd.). The following
secondary antibodies were used: Alexa Fluor 594- conjugated
AffiniPure goat anti-rabbit IgG, Alexa Fluor 488- conjugated
AffiniPure goat anti-mouse IgG [Beijing Zhongshan Golden Bridge
Biotechnology Co. Ltd. (ZsBio), Beijing, China], goat anti-rabbit
or goat anti-mouse secondary antibody (Histostain-Plus kits;
ZsBio). A QuantiFast SYBR-Green PCR kit (Qiagen, Hilden, Germany)
was used for the quantitative (real-time) polymerase chain reaction
(PCR) assays.
Animals
All the experimental procedures were approved by the
Administrative Panel on Laboratory Animal Care of Chongqing Medical
University (no. SCXK2007-000341). A total of 155 Sprague-Dawley
rats (weighing 250–280 g) were used in this study. The rats were
randomly divided into the following 4 groups: the sham-operated
group (Sham), the I/R group, the vehicle (DMSO)-treated group (V)
and the PP2-treated group (PP2). Transient middle cerebral artery
occlusion (MCAO) was induced via intraluminal nylon filament
intrusion as previously described by Longa et al (17). Briefly, the rats were anesthetized
with an intraperitoneal injection of 3.5% chloral hydrate (350
mg/kg). A midline incision was made in the neck, and the right
external carotid artery (ECA) was sequentially exposed and
dissected. The distal portion of the ECA was ligated with sutures,
and the branches between the ECA and ICA were also cauterized.
After an incision was made in the ECA, a monofilament nylon suture
was inserted from the ECA into the right internal carotid artery to
occlude the origin of the right MCA. The sham-operated rats
underwent identical surgeries with the exception that the suture
was not inserted. The rectal temperature was maintained at
37.0±0.5°C with a heating pad and a heating lamp. Laser-Doppler
flowmetry (Perimed, Stockholm, Sweden) was used to confirm the
induction of ischemia and reperfusion in the rats. The Src family
tyrosine kinase inhibitor, PP2, was dissolved in saline containing
1% dimethyl sulfoxide (DMSO). The PP2-treated rats were
administered PP2 (1.0 mg/kg) (18), and the vehicle-treated rats were
administered the same volume of the vehicle (DMSO) in the
peritoneal space after 30 min of MCAO. After 120 min of occlusion,
the suture was removed to allow reperfusion, the ECA was ligated
and the wound was sutured.
Neurological evaluation
The neurological function of each animal was
assessed using a set of modified neurological severity scores
(mNSSs) at 1, 3, and 7 days post-reperfusion. The mNSS is a
composite measurement of motor, sensory, reflex and balance
statuses (19). The neurological
deficit was graded on a scale of 0 (normal) to 18 (maximal
deficit). One point was awarded for the inability to perform the
test or for the lack of a tested reflex. Therefore, higher scores
indicated a more severe injury.
Quantitative reverse transcription PCR
(RT-qPCR)
The peri-infarct tissues that were supplied by the
MCA were excised from the brain tissue on ice, snap-frozen in
liquid nitrogen and stored at −80°C. Total RNA was isolated using
TRIzol reagent (Takara, Dalian, China) according to the
instructions of the manufacturer. With a PrimeScript RT Reagent kit
(Takara), 1 μg of RNA was reverse transcribed, and genomic DNA was
eliminated by the addition of DNase. The primers for the PCR assays
were supplied by Sangon Biotech (Shanghai, China) and were as
follows: claudin-5, 5′-GGCGATTACGACAAGAAGAACT-3′ (sense) and
5′-CCCGAACCCAACCTAACTT-3′ (antisense); β-actin,
5′-CCCATCTATGAGGGTTACGC-3′ (sense) and 5′-TTTAATGTCACGCACGATTTC-3′
(antisense). RNA was quantified using the QuantiFast SYBR-Green PCR
kit (Qiagen). The PCR assays were performed in an Eppendorf
Mastercycler PCR system according to the following protocol: 5 min
hold at 95°C, followed by 10 sec at 95°C and 30 sec at 60°C (40
cycles). The transcript levels were standardized with β-actin and
calculated using the ΔΔCT method.
Western blot analyses of proteins and
brain microvasculature fractionation
The ischemic brain tissue and time-matched tissue
samples from the sham-operated rats were dissected. The protein
samples were prepared using a commercially available kit
(Beyotime). The proteins were separated by 10% SDS-PAGE and
transferred onto polyvinylidene fluoride membranes. The membranes
were blocked with 5% BSA for 1 h and were then incubated overnight
at 4°C with primary antibodies. The membranes were washed prior to
incubation with the secondary antibodies for 75 min at 37°C. After
extensive washing, specific immunoreactivities were visualized via
a chemiluminescence ECL plus system and quantified by scanning
densitometry with a Bio-Image Analysis System (Bio-Rad, Hercules,
CA, USA). Isolation of the brain microvasculature fraction was
performed using a modified procedure that was similar to that of a
previous study (20). The brain
tissues from the ischemic hemispheres were homogenized in 5 ml
Dulbecco’s modified Eagle’s medium (DMEM) and centrifuged at 3,000
rpm for 5 min. The homogenates were resuspended and then treated
with 0.005% (wt/vol) dispase at 37°C for 2 h. The homogenates were
centrifuged at 3,000 rpm for 5 min, and the pellets were
re-suspended in a dextran solution (Mw 70,000; 10% wt/vol; Sigma)
and centrifuged at 3,000 rpm at 4°C for 10 min. The pellets were
then re-suspended in phosphate-buffered saline (PBS) and
centrifuged at 1,000 rpm for 5 min. The final pellet was
re-suspended in RIPA lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl,
1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 2 mM sodium
pyrophosphate, 25 mM β-glycerophosphate, 1 mM EDTA, 1 mM
Na3VO4, and 0.5 μg/ml leupeptin) and
centrifuged at 12,000 rpm for 20 min at 4°C; the supernatant was
removed for claudin-5 analysis.
Immunohistochemistry (IHC) and
immunofluorescence
At various time points following reperfusion, the
rats were deeply anesthetized with chloral hydrate and perfused
through the left ventricle with ice-cold PBS and 4%
paraformaldehyde. The brains were removed and post-fixed in 4%
paraformaldehyde. The brain tissues were embedded in
tissue-freezing medium and sectioned into 10 μm-thick slices. The
remaining brain tissues were paraffin-embedded and cut into 7 μm
coronal serial sections. For IHC staining, endogenous peroxidase
was blocked by incubating the slides with 3% hydrogen peroxide for
20 min. The sections were rinsed 3 times for 5 min each with PBS.
Non-specific binding sites were blocked by incubation with 10%
control goat serum for 30 min. The brain slices were incubated with
primary antibodies overnight at 4°C. The slides were rinsed 3 times
for 5 min each with PBS and then incubated with secondary
antibodies at room temperature for 30 min. The sections were rinsed
3 times for 5 min each with PBS and then incubated with an
avidin-HRP complex solution for 30 min. The staining was visualized
by developing the sections in a solution containing 5%
3,3′-diaminobenzidine (DAB) and 3% hydrogen peroxide in PBS. The
sections were counterstained, dehydrated and mounted with
coverslips. Sections that were stained without primary antibodies
were used as negative controls. Five non-overlapping images in the
peri-infarct area were acquired and analyzed using Image Pro Plus
6.0 software to measure the optical density of the positively
stained areas.
Double immunofluorescence labeling was performed for
claudin-5/fibrinogen and claudin-5/GFAP. The brain cryosections (10
μm) were pre-incubated in cold acetone and then rinsed in PBS 3
times for 5 min each. Following heat-induced antigen retrieval in a
microwave in a citrate buffer (pH 6.0), the sections were blocked
with 10% goat serum and were then incubated with a combination of
mouse anti-claudin-5 antibodies and rat anti-GFAP antibodies or
rabbit anti-fibrinogen antibodies and mouse anti-claudin-5
antibodies at 4°C. The sections were then rinsed and incubated with
a mixture of Alexa Fluor 594 goat anti-rabbit IgG and Alexa Fluor
488 goat anti-mouse IgG for 2 h at room temperature. Images were
acquired using a confocal microscope (Leica TCS Sp2; Leica
Microsystems, Wetzlar, Germany). Lesions that were dual-labeled for
claudin-5 and fibrinogen or GFAP were used to quantify the number
of vessels in the entire lesion area that exhibited BBB breakdown.
The data are presented either as percentages of the area of
double-positive claudin-5 and fibrinogen staining compared to the
total area of claudin-5 positivity or as percentages of cells that
were double-positive for claudin-5 and GFAP compared to the total
number of GFAP-positive cells.
Statistical analyses
The values are presented as the means ± standard
deviation (SD), and statistical analyses were performed using a
one-tailed unpaired t-test for 2 groups or an ANOVA for comparisons
of multiple groups. Statistical analyses were performed on the
normalized real-time PCR and western blot analysis data to
determine significant differences. An ANOVA and Tukey’s post-hoc
test were conducted to determine the differences in the
neurological scores. A value of p<0.05 was considered to
indicate a statistically significant difference.
Results
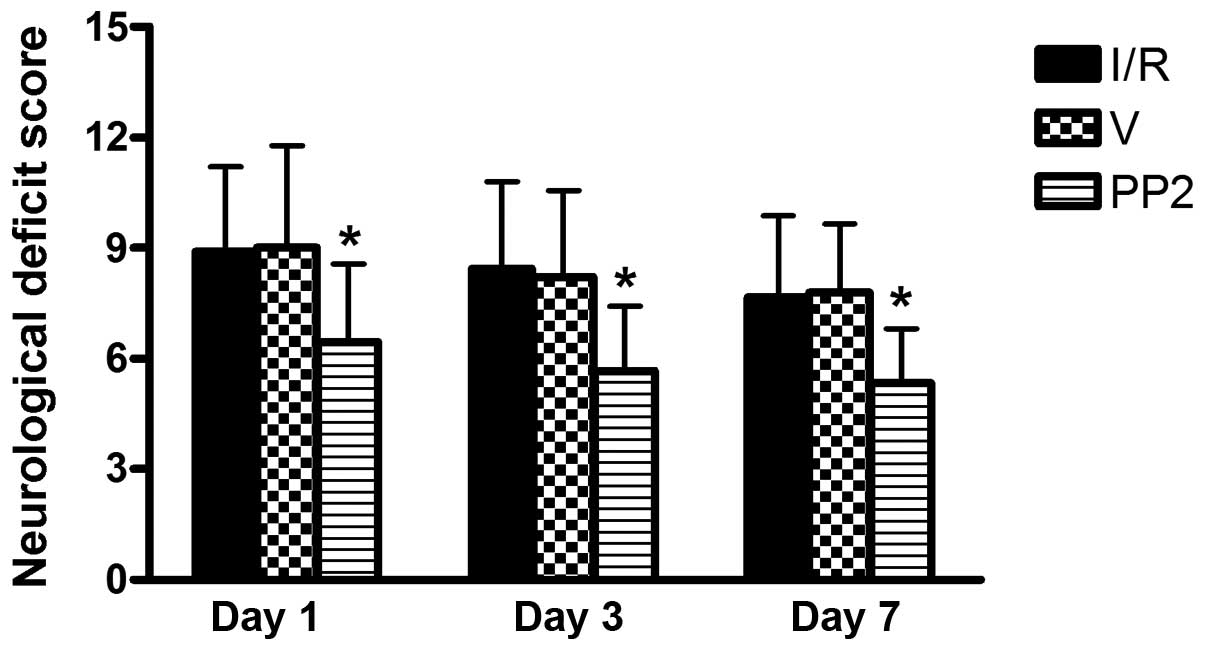
Improvement of the neural deficit scale
scores in the PP2-treated group
To assess the immediate impact of PP2 treatment on
functional recovery, we evaluated the average mNSSs for the Sham,
I/R, V and PP2 groups at 1, 3 and 7 days following reperfusion. In
the Sham group, no neurological deficit was found. No significant
differences were observed between the I/R group and the V group at
any of the examined time points. At 1 day post-reperfusion, the
scores in the I/R, V and PP2 groups indicated moderate injuries;
however, the PP2 group had a significantly lower mean score
(6.4±2.01) than did the I/R and V groups (8.9±2.3 and 9.0±2.17,
respectively, p<0.05). On days 3 and 7, the neurological scores
of the PP2 group remained significantly lower than those of the I/R
and V groups (Fig. 1).
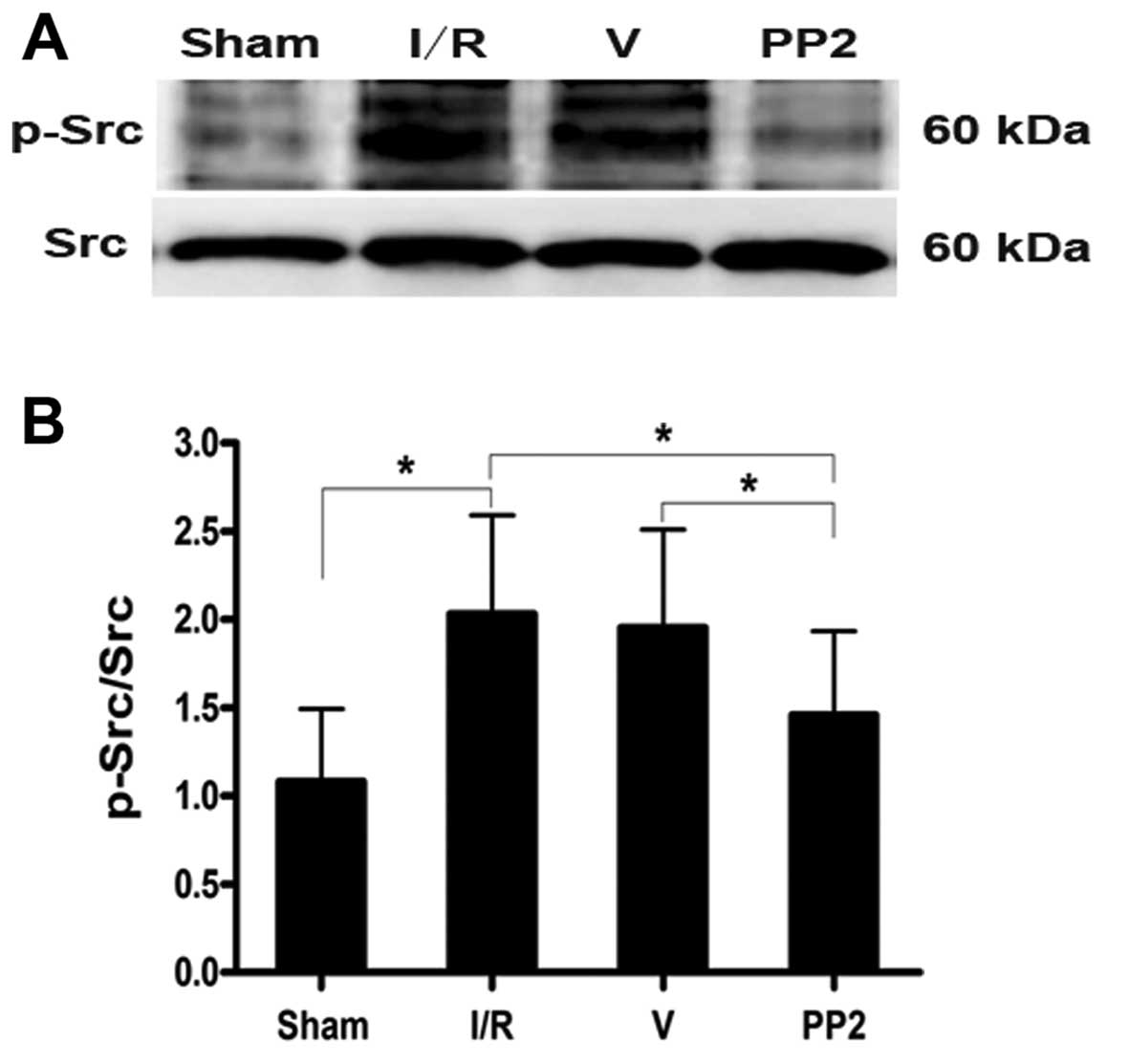
Expression of p-Src protein and the
effects of PP2 on p-Src expression
Abundant Src immunoreactivity was detected (Fig. 2A) in the neurons of both the
cortices and hippocampi of the animals in the Sham, I/R, V and PP2
groups (Fig. 2A-a–d,
respectively) and can be seen as the yellowish-brown color in the
micrographs. We also observed Src immunoreactivity in the
astrocytes and cerebral perivascular tissue. The distribution of
Src immunoreactivity was not altered following cerebral I/R injury
(Fig. 2A-b–d). The
Src-immunostained cells did not accumulate in the peri-infarct
areas (Fig. 2A-b–d), and the
Src-immunostained cells of the animals in the PP2 group did not
differ from those of the animals in the Sham or the other groups
(Fig. 2A-d). Western blot
analyses were performed to further quantify the alterations in
p-Src that were induced by I/R insults. p-Src was present at very
low levels under normal conditions, and p-Src protein levels began
to increase at 6 h, reached their peak value at 24 h, and remained
at a high levels until 48 h after the ischemic insult. The tyrosine
p-Src level in the rats in the I/R group was twice that of the rats
in the Sham group at 24 h after the ischemic insults (Fig. 2B and C). Furthermore, the marked
increase in p-Src levels that was induced by ischemia was
significantly inhibited by PP2 at 24 h after the ischemic stroke.
The PP2 group had a markedly lower level of the tyrosine p-Src than
did the I/R and V groups (Fig. 3;
p<0.05).
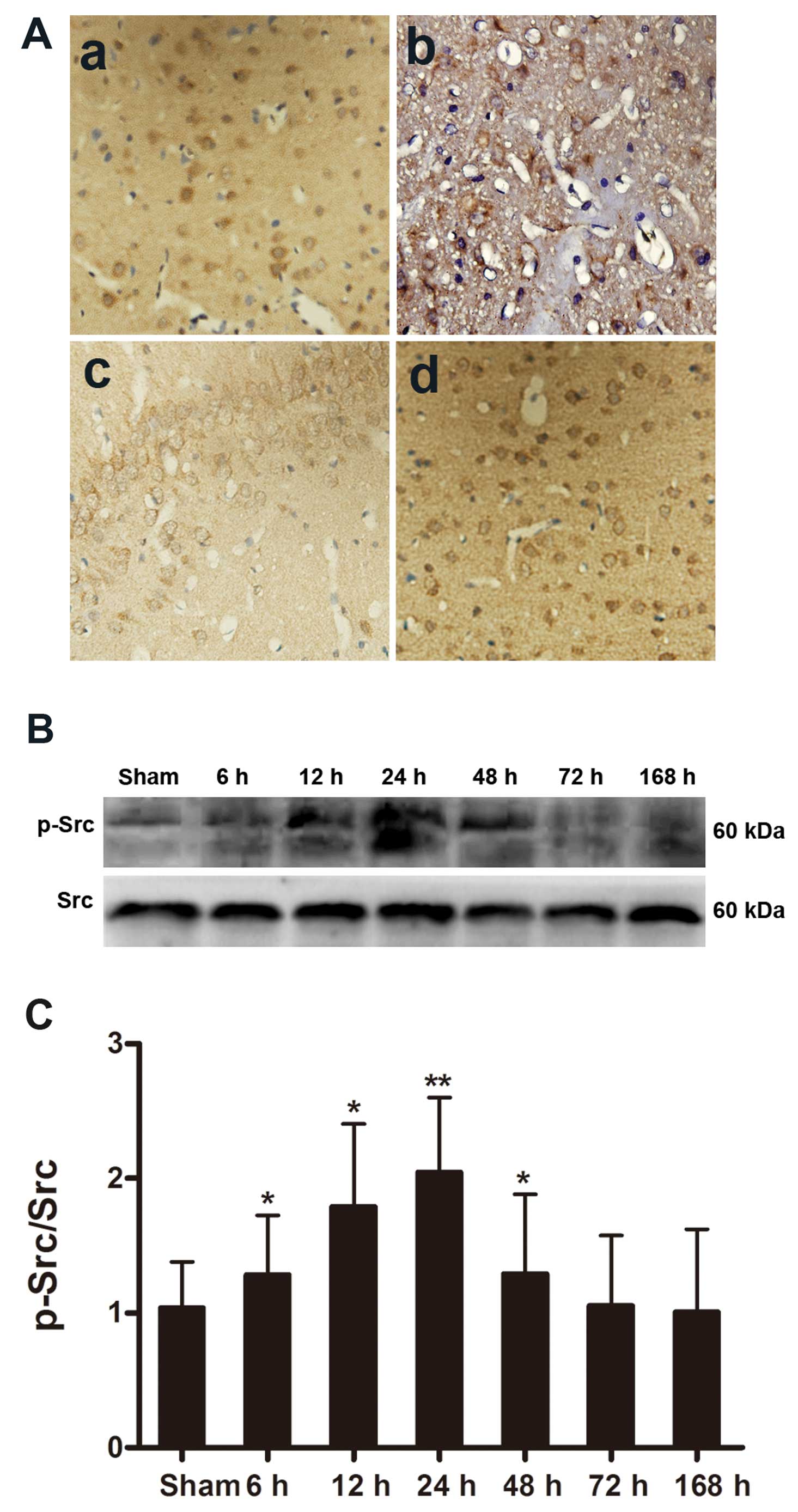 | Figure 2Distribution and expression of the
Src protein analyzed by immunohistochemistry and analysis of the
expression of p-Src protein by western blot analysis following
ischemia. (A) (a-d) Src immunoreactivity was detected in the
cortical neurons, astrocytes and perivascular tissues of rats in
the sham-operated (Sham), ischemia/reperfusion (I/R), vehicle
(DMSO)-treated (V) and PP2-treated (PP2) groups following
reperfusion. (d) The Src-immunopositive cells in the PP2 group did
not differ from those of the Sham or other groups. (a) Sham group;
(b) I/R group; (c) V group; (d) PP2 group. Original magnification
of the images, ×400. (B) Ipsilateral hemisphere tissue at various
time points (Sham, 6, 12, 24, 48, 72 and 168 h) following ischemia
and reperfusion. p-Src production was determined by western blot
analysis. (C) The data are displayed as the values corresponding to
the p-Src bands normalized to the total Src in the same blots.
p-Src protein levels began to increase at 6 h, peaked at 24 h, and
remained elevated until 48 h (*p<0.05 and
**p<0.01, significant differences compared to the
Sham group, n=6). |
Expression of VEGFA protein and the
suppressive effects of PP2 on the expression of VEGFA after I/R
injury
VEGFA protein expression was measured by western
blot analysis and immunohistochemical staining as shown in Fig. 4A and E. We found that the VEGFA
expression level was very low under normal conditions and that
VEGFA immunoreactivity was observed in the astrocytes and
peri-vascular tissue of the rats in the Sham group. VEGFA
expression began to increase at 6 h, peaked at 24 h, and remained
at a high level until 1 week following ischemic stimulation
(Fig. 4B). The VEGFA protein
level increased to 2-fold that of the Sham group at 12 and 24 h
post-reperfusion, and the level of VEGFA at 24 h was slightly
higher than that at 12 h (Fig. 4A and
E). Furthermore, this increase in VEGFA protein expression was
significantly inhibited by PP2 at 24 h after ischemia (Fig. 4C–F). The VEGFA expression level in
the PP2 group was significantly lower than that in the I/R and V
groups, and the number of VEGFA-positive cells in the PP2 group was
much lower than that in the I/R and V groups. The intensities of
VEGFA immunostaining did not differ between the I/R group, the V
group and the PP2 group 7 days after ischemia. Increases in the
VEGFA immunoreactivity in hypertrophic astrocytes were observed in
the penumbra area at 24 h after ischemia, and these increases
persisted for at least 1 week (Fig.
4E–H).
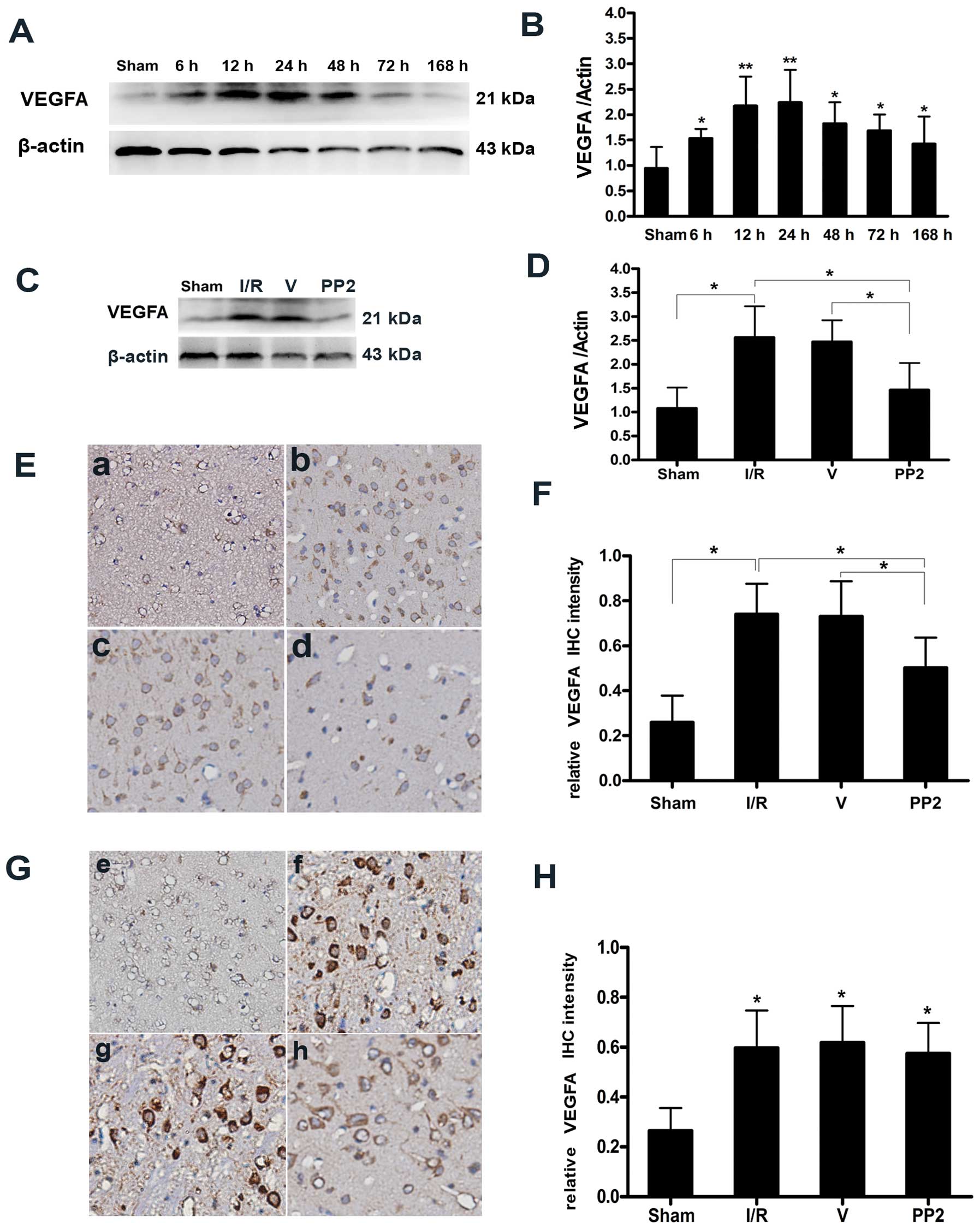 | Figure 4Expression of vascular endothelial
growth factor A (VEGFA) protein and the suppressive effects of PP2
on the expression of VEGFA. (A) Representative western blot of
VEGFA indicating that VEGFA expression was increased in the
ipsilateral hemispheres at various time points (6, 12, 24, 48, 72
and 168 h) after ischemia/reperfusion (I/R) injury. (B) Values
corresponding to the VEGFA bands were normalized to the
sham-operated (Sham) group. VEGFA protein levels began to increase
at 6 h, peaked at 24 h, and remained elevated until 7 days
(*p<0.05 and **p<0.01, significant
differences compared with the Sham group, n=6). (C) Representative
immunoblots of VEGFA from the Sham group, the I/R group, the
vehicle (DMSO)-treated (V) group, and the PP2 group at 24 h
following ischemic stroke indicating that the expression of VEGFA
was strongly downregulated by PP2. (D) Quantitative determinations
of the immunoblots revealed that VEGFA expression was markedly
decreased by PP2 (*p<0.05, n=6). (E)
Immunohistochemical staining for VEGFA in the penumbral areas at 24
h post-reperfusion. (F) Histogram of the optical intensities of the
VEGFA-positive staining in the peri-infarct regions of the PP2
group were markedly decreased at 24 h (*p<0.05, n=6).
(G) Immunohistochemical staining for VEGFA in the penumbral areas
at 7 days post-reperfusion. (H) Histogram indicating that there
were no differences in the optical intensities for VEGFA-positive
staining in the peri-infarct regions of the ischemic hemispheres 7
days post-reperfusion across the 3 groups subjected to I/R
(*p<0.05, n=6). (E and G) (a and e) Sham group; (b
and f) I/R group; (c and g) V group; (d and h) PP2 group. The
images shown in (b-d) were acquired 24 h after ischemia, and those
in (f-h) were acquired 168 h after ischemia. The original
magnification of the iamnges was ×400. |
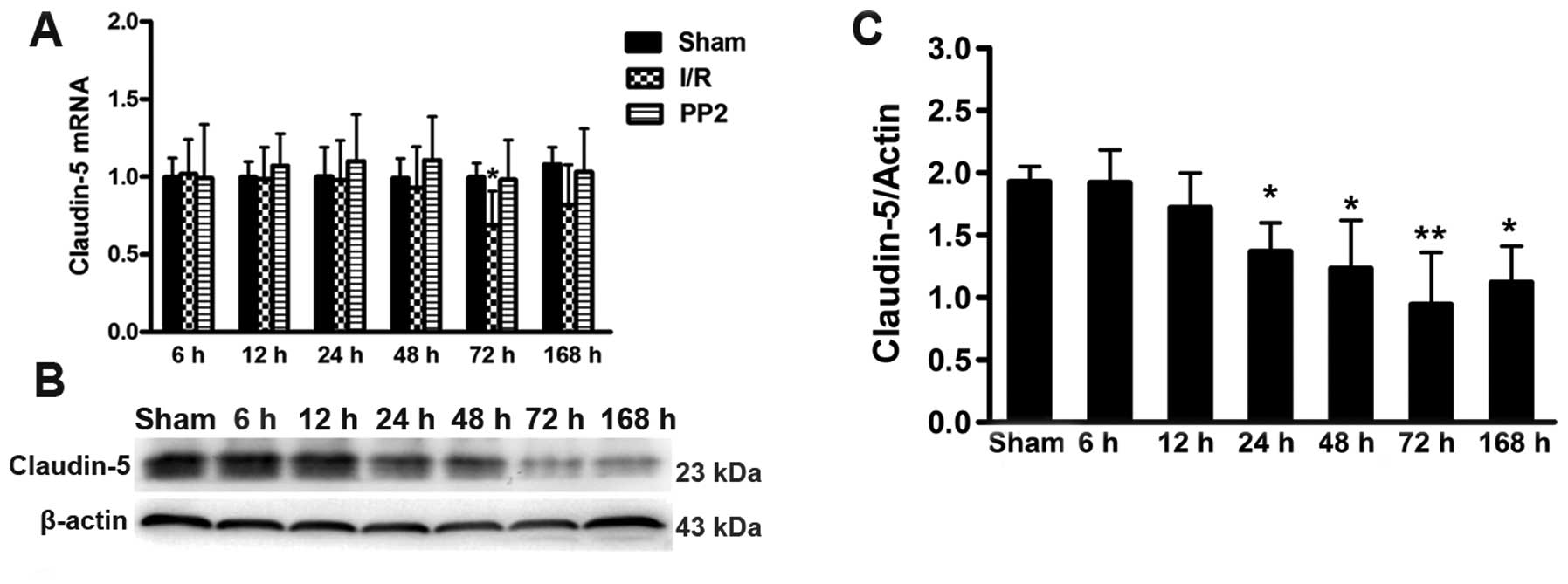
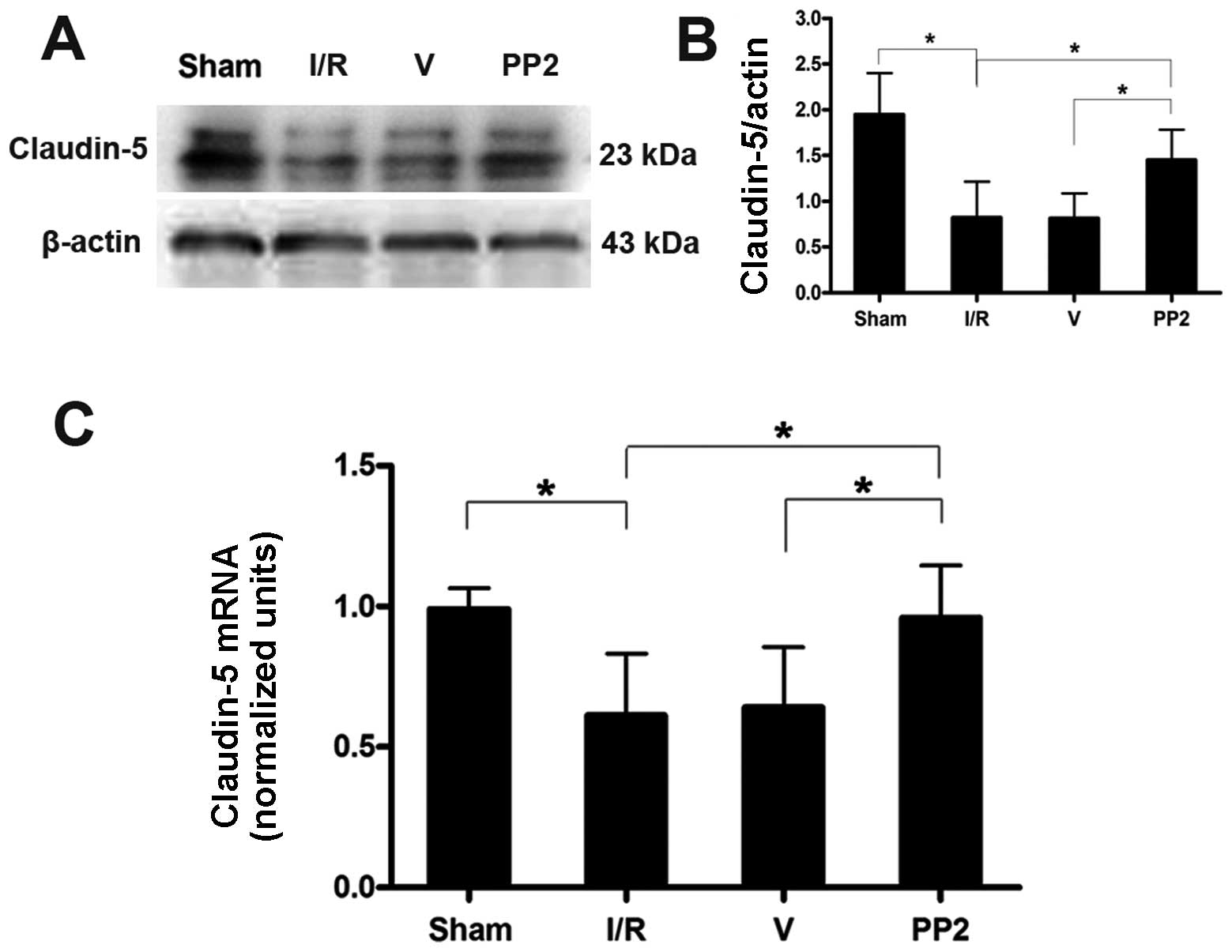
Expression of claudin-5 and the effects
of PP2 on the expression of claudin-5
Transcriptional analyses of the tight junction
protein, claudin-5, were performed. An ischemic insult induced the
inhibition of mRNA synthesis 72 h after the I/R injury. As
expected, the Src inhibitor, PP2, blocked the decrease in the
claudin-5 transcript level (Fig.
5C). Further experiments revealed that claudin-5 protein
expression was consistent with the changes in the nucleic acid
transcripts; however, the decrease in claudin-5 expression
continued for 1 week following reperfusion (Fig. 5A and B). The results from RT-qPCR
and western blot analysis were compared amongst all the groups at
72 h after the ischemic injuries. These comparisons demonstrated
that PP2 prevented the decrease in claudin-5 protein and mRNA
expression and protected the BBB (Fig. 6).
Protective effects of PP2 on the BBB
following ischemic insult
As the disruption of the BBB that was induced by
ischemia increased, claudin-5 expression decreased. Confocal
imaging of the brain tissue from the rats in the Sham group
revealed that the astrocytic endfeet around the blood vessels were
completely separate from claudin-5, and no extravasation of
fibrinogen was evident. The ischemic injuries induced the loss of
claudin-5 immunoreactivity and increased the immunostaining for
fibrinogen. When these changes were quantified, the analyses
revealed a significant correlation between the disruption of the
BBB in the rats following ischemic insult and the decrease in
claudin-5 immunoreactivity; i.e., we found that the severity of the
BBB disruption was proportional to the decrease in claudin-5
immunoreactivity, as well as to the increase in the extravasation
of fibrinogen. Fibrinogen was primarily exuded at 72 h; PP2
protected the BBB from this disruption and significantly blocked
the leakage of fibrinogen. The increased disruption of the BBB was
accompanied by the co-localization of claudin-5 and GFAP in the
ischemic hemisphere at 72 h. Quantitative analysis revealed a
strong association between claudin-5 immunoreactivity and GFAP
staining, and the more intense co-localization of claudin-5 and
GFAP immunoreactivity correlated with more severe BBB disruptions.
The increase in the co-localization of claudin-5 and GFAP was
prevented by PP2; consequently, the integrity of the BBB was
protected (Fig. 7).
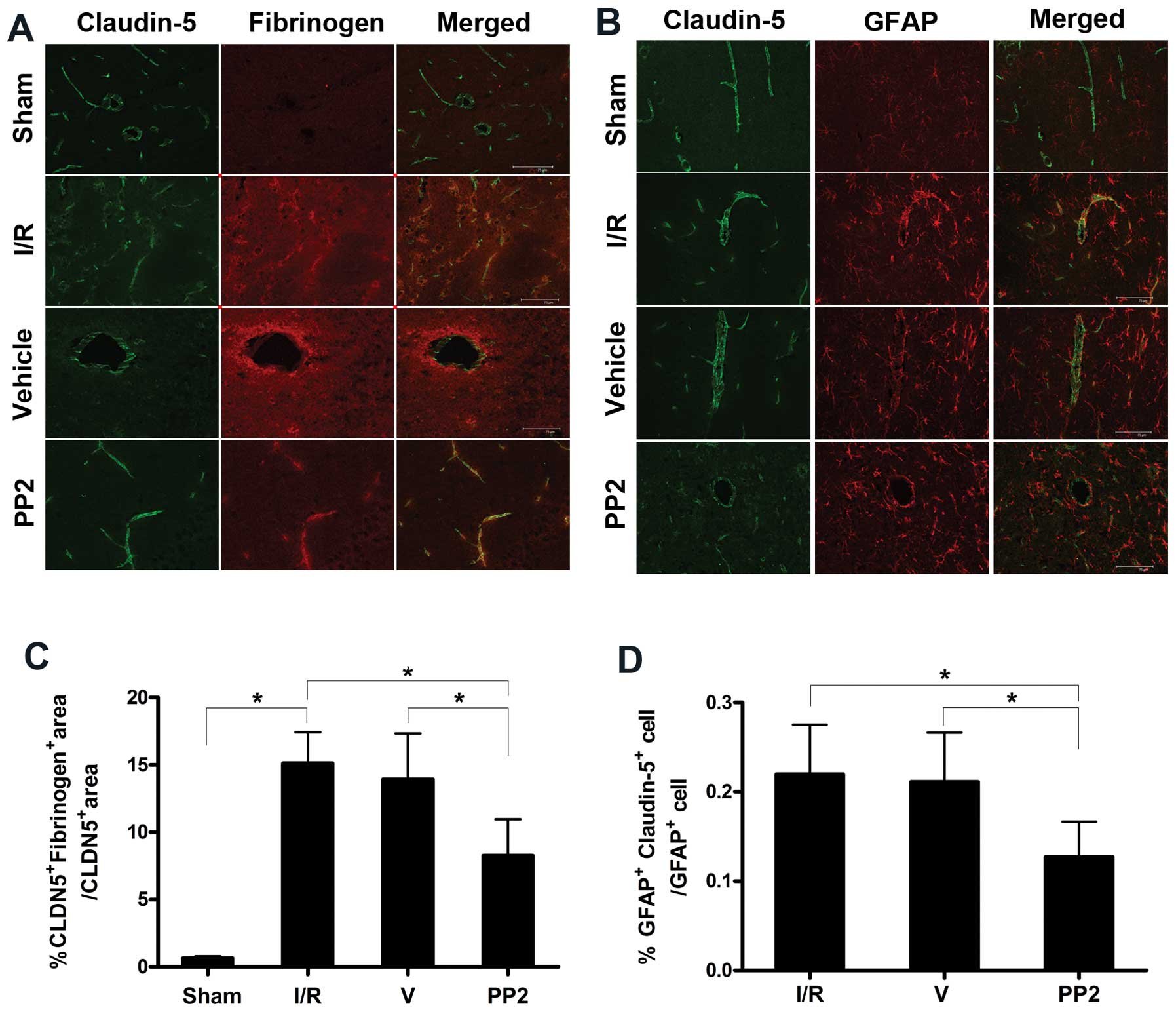 | Figure 7Protective effects of PP2 on
endothelial tight-junctions. (A) Confocal micrographs revealing
claudin-5 and fibrinogen immunoreactivity in the sham-operated
(Sham), ischemia/reperfusion (I/R), vehicle (DMSO)-treated (V) and
PP2 groups. The greater the fibrinogen extravasation in the I/R and
V groups, the lower the claudin-5 levels, resulting in simultaneous
severe blood-brain barrier (BBB) disruptions. The decrease in
claudin-5 was blocked, and the extravasation of fibrinogen was
markedly reduced, as observed in the confocal micrographs taken of
the penumbral areas in the PP2 group 72 h after reperfusion. (B)
Confocal micrographs from the Sham group indicating that the
claudin-5 in the blood vessels was separated from the astrocytes;
the confocal images from the penumbral areas of the rats in the I/R
group reveal fragmentation and degeneration of the claudin-5
immunoreactivity. The merged immunostaining for claudin-5 and GFAP
illustrates the close proximity of the astrocytic end-feet to the
vessels in the I/R group. The proximity of the astrocytic endfeet
to the vessels in the PP2 group was obviously decreased, and the
integrity of the endothelial tight-junctions was significantly
preserved. (C) The significant protective effect of PP2 on the BBB
was confirmed via quantitative analysis (*p<0.05,
ANOVA; n=6) (D) Quantitative analyses revealed that the
double-staining for GFAP and claudin-5 in cells was markedly
reduced by PP2 (*p<0.05, ANOVA; n=6) (scale bars, 75
μm). |
Discussion
In vivo, the BBB, with its neuronal and
non-neuronal components, represents the NVU (21). The NVU is characterized by a
functional interaction between the brain endothelial cells,
astrocytes, pericytes, microglia and neurons. Ischemic injuries
induce disruptions of the BBB that may cause numerous severe
clinical complications, such as the formation of brain edema and
neuroinflammation (22,23). Accumulating evidence indicates
that tight junction proteins may play a pivotal role in the
maintenance of the physical barrier function of the BBB (15,35). The Src family kinases mediate
signaling activity in response to a variety of growth factors,
including VEGF (24,25). Available evidence suggests that
the Src kinase regulates VEGF-mediated vascular permeability in the
brain following stroke and that the suppression of Src activity
decreases vascular permeability and thereby minimizes brain injury.
To assess the extent of the injuries in this study, we evaluated
the average NSSs of the rats in the Sham, I/R, V and PP2 groups at
1, 3 and 7 days post-reperfusion; the Src inhibitor, PP2, markedly
improved the neurological function of the rats in the PP2 group. No
significant differences were observed between the I/R and V groups
at the time points examined. The results of the present study are
consistent with those of a previous study that found that Src
inhibitors have neuroprotective effects (13). The protective effects of PP2
against ischemic injury may be mediated through its ability to
protect the BBB and reduce the infarct area.
In the NVU, p-Src not only regulates cells, but also
influences the surrounding environment. The results of our study
revealed that Src was widely expressed in neurons, brain
endothelial cells and astrocytes. p-Src is the activated form of
Src and plays an important role in transcellular transduction. We
observed that large quantities of Src were phosphorylated following
ischemia, consistent with earlier observations. It has been
reported that PP2 significantly reduces the infarct size compared
with the controls following transient focal brain ischemia
(26); however, the mechanisms
underlying this effect have remained elusive. Takenaga et al
(27) reported that the levels of
p-Src reached their peak 6 h after ischemic injury; our study
revealed that the time at which the levels of p-Src protein peaked
was 24 h after ischemia. The ischemic penumbra is constantly
changing and expanding from the central infarct area to the
surrounding area. This discrepancy may be the result of differences
in the sampling of the tissues examined. In our study, we examined
samples of peri-infarct brain tissue that were distinct from the
brain capillary fragments. Korematsu et al (28) reported that the occlusion of the
rat MCA for 1 h led to a significant increase in protein tyrosine
phosphorylation in the microglia in the insulted cerebral cortex 3
h post-reperfusion. Increased protein tyrosine phosphorylation has
also been found in the rat retina following I/R injury, and the
VEGF levels in the damaged tissue are also increased. Following an
ischemic insult, astrocytes become hypertrophied and proliferative
(29). It has previously been
demonstrated that cellular (c)-Src is activated by the
hypoxia-induced upregulation of VEGFA expression and that
constitutive viral (v)-Src increases the VEGF mRNA levels (30). VEGFA was produced in response to
an ischemic injury after p-Src was upregulated due to the hypoxemic
stimulation (31). p-Src has been
implicated in the production of astrocyte-derived VEGFA following
ischemia (26,32). In our study, VEGFA was elevated at
6 h after reperfusion and peaked at 24 h, which was compatible with
the changes in p-Src levels following the ischemic injury. Notably,
recent studies have indicated that Src-induced VEGF upregulation
requires the persistent activation of STAT3. For example, STAT3 has
been shown to be necessary for VEGF upregulation through v-Src as
the blockade of STAT3 signaling inhibits VEGF expression that is
induced by Src tyrosine kinase activity (14). In the present study, the Src
inhibitor, PP2, markedly reduced the production of p-Src and
produced a similar inhibition in the increase in the VEGFA protein
level. Quantitative analyses revealed that PP2 markedly decreased
VEGFA expression, which verified that PP2 not only inhibited p-Src
production, but also inhibited the expression of VEGFA.
Immunohistochemical staining of VEGFA in the penumbral areas at 24
h post-reperfusion proved that VEGFA expression was markedly
reduced; however, there were no differences in the optical
intensities of the VEGFA-positive immunohistochemical stains in the
peri-infarct regions of the ischemic hemispheres 1 week
post-reperfusion among the 3 ischemic groups. Furthermore, our
findings support the notion that PP2 inhibits the expression of
VEGFA following ischemia. Our results revealed that the neurons and
astrocytes had much stronger effects on the disruption of the BBB
than did the endothelial cells, and these findings represent
another discrepancy between this and previously reported data
(27).
It is well established that claudin-5 is a pivotal
component of the tight junctions between brain endothelial cells
that effectively separate the brain from the cerebral blood flow.
Claudin-5 was selected for our study due to previous observations
that claudin-5 is present in large amounts in brain endothelial
cells and is an endothelial cell-specific component of tight
junction strands (33).
Astrocyte-derived VEGFA has been implicated in the breakdown of the
BBB during inflammation of the central nervous system. The
VEGFA-induced downregulation of claudin-5 is a significant
determinant of the increased paracellular permeability of brain
microvascular endothelial cells (16). In the present study, we found that
the claudin-5 mRNA levels in the rats in the I/R group were
significantly lower than those of the rats in the Sham group at 72
h after ischemia; moreover, claudin-5 protein expression was
downregulated from 48 h after the ischemic insult, and the decrease
in claudin-5 protein expression was most evident at 72 h. The
inhibitor PP2 blocked the downregulation of claudin-5 transcription
at 72 h and similarly blocked the decrease in claudin-5 protein
levels 72 h after ischemia. In transient ischemic experiments,
Huang et al (34) observed
that the opening of the BBB appears in the first half-hour of
reperfusion; this is followed by a partial closing and then a
delayed opening that occurs between 22 and 46 h post-reperfusion.
In our study, a decrease in claudin-5 protein expression was
detected at 48 h that was compatible with the findings of these
studies. Although this bimodal increase in BBB permeability is
widely believed to occur in the I/R model, the time points for the
peak BBB permeability have not been completely defined. Jiao et
al (35) found that 2 peaks
in BBB permeability appeared after reperfusion, at 3 and 72 h
post-reperfusion, following 2 h of focal ischemia. This result is
consistent with our finding of a second BBB permeability peak at 72
h. However, Jiao et al (35) found a decrease in claudin-5
protein beginning at 6 h after reperfusion, which is different from
our results. This difference may be attributable to the different
tissue samples examined and the diverse trial methods employed. Liu
et al (36) found that
ischemia triggers 2 rapid and parallel processes that cause the
early disruption of the BBB: matrix metalloproteinase-2
(MMP-2)-mediated occludin degradation and caveolin-1
(Cav-1)-mediated redistribution of claudin-5 from the cytoskeleton
to the cytosol in the ischemic cerebral microvessels. During the
early phase of BBB breakdown, Nag et al (37) observed a significant increase in
caveolin-1 expression at the lesion site following injury at 12 h
and on day 2, while claudin-5 expression was decreased only on day
2.
Fibrinogen leakage is a well-documented and
validated marker of a breakdown in the BBB (38,39). In our study, confocal imaging of
the peri-infarct regions of the ipsilateral hemispheres indicated
that ischemia caused a loss of the claudin-5 immunoreactivity in
the blood vessels and disrupted the appearance of the astrocytic
foot processes around the endothelial cells. Following ischemia,
claudin-5 immunostaining decreased, the BBB disruption increased
and the extravasation of fibrinogen became more intensive and
extensive; at 72 h, the extravasation of fibrinogen was massive.
PP2 protected the BBB from disruption and significantly reduced the
leakage of fibrinogen. Claudin-5 appeared in linear rows, which
have previously been referred to as having a ‘zip-locked’
appearance (40). The astrocytic
endfeet around the blood vessels were completely separated from the
claudin-5 in the images from the Sham group. In the present study,
we observed GFAP-positive immunoreactivity that co-localized with
diffuse cytoplasmic claudin-5 immunostaining of the ischemic
penumbra. The BBB was extensively disrupted, and the
co-localization of claudin-5 and GFAP was obviously present in the
ischemic hemisphere at 72 h. The observed fibrinogen leakage at 72
h supports the theory that the water content progressively
increases within 2 days and that the second peak of opening the BBB
appears within 3 days. PP2 protected the BBB from the disruption
caused by VEGFA and decreased the co-localization of GFAP and
claudin-5. PP2 markedly reduced the area of co-localization and
significantly protected the BBB against ischemic insult.
The main causes of the delayed opening that occurred
at 72 h were the VEGFA-mediated decrease in claudin-5 at the
transcription and protein levels; however, other factors cannot be
excluded. As previously demonstrated, since the inflammatory
response that occurs secondarily to ischemia and hypoxia following
reperfusion had fully developed, many additional factors were
involved, including cytokines and infiltrating leukocytes (41). Therefore, the possibility that PP2
blocked MMP or eNO expression and activation cannot be completely
excluded (29,42). Moreover, future studies are
planned to elucidate the mechanisms of the effects of VEGFA on the
expression and transcription of claudin-5. The BBB opening that
occurs in claudin-5-deficient mice is selective for small molecules
(<800 D) (43), whereas the
BBB disruption that follows ischemia is more severe and less
size-selective. The loss of claudin-5 is important for the
initiation of barrier disruption in vivo, whereas the loss
of occludin (OCLN) that has been reported (37,44) may exacerbate the phenotype. Our
results suggest that the VEGFA-induced downregulation of claudin-5
constitutes an important mechanism of BBB disruption following an
ischemic insult and that the inhibitor of Src exerts a protective
effect against I/R injury. These findings may indicate a potential
target for therapeutic intervention.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 81271307) and by the
National Key Clinical Specialties Construction Program of
China.
References
|
1
|
Ayata C and Ropper AH: Ischaemic brain
oedema. J Clin Neurosci. 9:113–124. 2002. View Article : Google Scholar
|
|
2
|
Hatashita S and Hoff JT: Role of
blood-brain barrier permeability in focal ischemic brain edema. Adv
Neurol. 52:327–333. 1990.PubMed/NCBI
|
|
3
|
Ames A III, Wright RL, Kowada M, Thurston
JM and Majno G: Cerebral ischemia. II. The no-reflow phenomenon. Am
J Pathol. 52:437–453. 1968.PubMed/NCBI
|
|
4
|
Dvorak HF, Brown LF, Detmar M and Dvorak
AM: Vascular permeability factor/vascular endothelial growth
factor, microvascular hyperpermeability, and angiogenesis. Am J
Pathol. 146:1029–1039. 1995.PubMed/NCBI
|
|
5
|
Dobrogowska DH, Lossinsky AS, Tarnawski M
and Vorbrodt AW: Increased blood-brain barrier permeability and
endothelial abnormalities induced by vascular endothelial growth
factor. J Neurocytol. 27:163–173. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kovács Z, Ikezaki K, Samoto K, Inamura T
and Fukui M: VEGF and flt. Expression time kinetics in rat brain
infarct. Stroke. 27:1865–1873. 1996.PubMed/NCBI
|
|
7
|
Hayashi T, Abe K, Suzuki H and Itoyama Y:
Rapid induction of vascular endothelial growth factor gene
expression after transient middle cerebral artery occlusion in
rats. Stroke. 28:2039–2044. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lennmyr F, Ata KA, Funa K, Olsson Y and
Terént A: Expression of vascular endothelial growth factor (VEGF)
and its receptors (Flt-1 and Flk-1) following permanent and
transient occlusion of the middle cerebral artery in the rat. J
Neuropathol Exp Neurol. 57:874–882. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang ZG, Zhang L, Tsang W, et al:
Correlation of VEGF and angiopoietin expression with disruption of
blood-brain barrier and angiogenesis after focal cerebral ischemia.
J Cereb Blood Flow Metab. 22:379–392. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang ZG, Zhang L, Jiang Q, et al: VEGF
enhances angiogenesis and promotes blood-brain barrier leakage in
the ischemic brain. J Clin Invest. 106:829–838. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levy DE and Darnell J: Stats:
transcriptional control and biological impact. Nat Rev Mol Cell
Biol. 3:829–838. 2002.
|
|
12
|
Turkson J: STAT proteins as novel targets
for cancer drug discovery. Exp Opin Ther Targets. 8:409–422. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Paul R, Zhang ZG, Eliceiri BP, et al: Src
deficiency or blockade of Src activity in mice provides cerebral
protection following stroke. Nat Med. 7:222–227. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Niu G, Wright KL, Huang M, et al:
Constitutive Stat3 activity up-regulates VEGF expression and tumor
angiogenesis. Oncogene. 21:2000–2008. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Anderson JM: Molecular structure of tight
junctions and their role in epithelial transport. News Physiol Sci.
16:126–130. 2001.PubMed/NCBI
|
|
16
|
Argaw AT, Gurfein BT, Zhang Y, Zameer A
and John GR: VEGF-mediated disruption of endothelial CLN-5 promotes
blood-brain barrier breakdown. Proc Natl Acad Sci USA.
106:1977–1982. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu DZ, Ander BP, Xu H, et al: Blood-brain
barrier breakdown and repair by Src after thrombin-induced injury.
Ann Neurol. 67:526–533. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen J, Li Y, Wang L, et al: Therapeutic
benefit of intravenous administration of bone marrow stromal cells
(MSCs) after cerebral ischemia in rats. Stroke. 32:1005–1011. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Campbell M, Hanrahan F, Gobbo OL, et al:
Targeted suppression of claudin-5 decreases cerebral oedema and
improves cognitive outcome following traumatic brain injury. Nat
Commun. 3:8492012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zlokovic BV: The blood-brain barrier in
health and chronic neurodegenerative disorders. Neuron. 57:178–201.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rosenberg GA and Yang Y: Vasogenic edema
due to tight junction disruption by matrix metalloproteinases in
cerebral ischemia. Neurosurg Focus. 22:E42007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shichita T, Sugiyama Y, Ooboshi H, et al:
Pivotal role of cerebral interleukin-17-producing gammadeltaT cells
in the delayed phase of ischemic brain injury. Nat Med. 15:946–950.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Johnson FM, Saigal B, Talpaz M and Donato
NJ: Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses
invasion and induces cell cycle arrest and apoptosis of head and
neck squamous cell carcinoma and non-small cell lung cancer cells.
Clin Cancer Res. 11:6924–6932. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Desai CJ, Sun Q and Zinn K: Tyrosine
phosphorylation and axon guidance: of mice and flies. Curr Opin
Neurobiol. 7:70–74. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lennmyr F, Ericsson A, Gerwins P, Akterin
S, Ahlström H and Terént A: Src family kinase-inhibitor PP2 reduces
focal ischemic brain injury. Acta Neurol Scand. 110:175–179. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takenaga Y, Takagi N, Murotomi K, Tanonaka
K and Takeo S: Inhibition of Src activity decreases tyrosine
phosphorylation of occludin in brain capillaries and attenuates
increase in permeability of the blood-brain barrier after transient
focal cerebral ischemia. J Cereb Blood Flow Metab. 29:1099–1108.
2009. View Article : Google Scholar
|
|
28
|
Korematsu K, Goto S, Nagahiro S and Ushio
Y: Microglial response to transient focal cerebral ischemia: an
immunocytochemical study on the rat cerebral cortex using
anti-phosphotyrosine antibody. J Cereb Blood Flow Metab.
14:825–830. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Okutani D, Lodyga M, Han B and Liu M: Src
protein tyrosine kinase family and acute inflammatory responses. Am
J Physiol Lung Cell Mol Physiol. 291:L129–L141. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mukhopadhyay D, Tsokias L, Zhou X, Foster
D, Brugge J and Sukhatme V: Hypoxic induction of human vascular
endothelial growth factor expression through c-Src activation.
Nature. 375:577–581. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bjorge JD, Jakymiw A and Fujita DJ:
Selected glimpses into the activation and function of Src kinase.
Oncogene. 19:5620–5635. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zan L, Zhang X, Xi Y, Wu H, Song Y, Teng
G, Li H, Qi J and Wang J: Src regulates angiogenic factors and
vascular permeability after focal cerebral ischemia-reperfusion.
Neuroscience. 262:118–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Morita K, Sasaki H, Furuse M and Tsukita
S: Endothelial claudin: claudin-5/TMVCF constitutes tight junction
strands in endothelial cells. Journal Cell Biol. 147:185–194. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang ZG, Xue D, Karbalai H, et al:
Biphasic opening of the blood-brain barrier following transient
focal ischemia: effects of hypothermia. Canad J Neurol Sci.
26:298–304. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiao H, Wang Z, Liu Y, Wang P and Xue Y:
Specific role of tight junction proteins claudin-5, occludin, and
ZO-1 of the blood-brain barrier in a focal cerebral ischemic
insult. J Mol Neurosci. 44:130–139. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu J, Jin X, Liu KJ and Liu W: Matrix
metalloproteinase-2-mediated occludin degradation and
caveolin-1-mediated claudin-5 redistribution contribute to
blood-brain barrier damage in early ischemic stroke stage. J
Neurosci. 32:3044–3057. 2012. View Article : Google Scholar
|
|
37
|
Nag S, Venugopalan R and Stewart DJ:
Increased caveolin-1 expression precedes decreased expression of
occludin and claudin-5 during blood-brain barrier breakdown. Acta
Neuropathol. 114:459–469. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brown H, Hien T, Day N, et al: Evidence of
blood-brain barrier dysfunction in human cerebral malaria.
Neuropathol Appl Neurobiol. 25:331–340. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kirk J, Plumb J, Mirakhur M and McQuaid S:
Tight junctional abnormality in multiple sclerosis white matter
affects all calibres of vessel and is associated with blood-brain
barrier leakage and active demyelination. J Pathol. 201:319–327.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tsukita S, Furuse M and Itoh M:
Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol.
2:285–293. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lo EH, Dalkara T and Moskowitz MA:
Mechanisms, challenges and opportunities in stroke. Nat Rev
Neurosci. 4:399–415. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang N, Ashrafpour H, Levine RH, Forrest
CR, Neligan PC, Lipa JE and Pang CY: Vasorelaxation effect and
mechanism of action of vascular endothelial growth factor-165 in
isolated perfused human skin flaps. J Surg Res. 172:177–186. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nitta T, Hata M, Gotoh S, et al:
Size-selective loosening of the blood-brain barrier in
claudin-5-deficient mice. J Cell Biol. 161:653–660. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Saitou M, Furuse M, Sasaki H, Schulzke JD,
Fromm M, Takano H, Noda T and Tsukita S: Complex phenotype of mice
lacking occludin, a component of tight junction strands. Mol Biol
Cell. 11:4131–4142. 2000. View Article : Google Scholar : PubMed/NCBI
|