1. Skeletal dysplasias resulting from
mutations in cartilage structural proteins
Qualitative (i.e., dominant-negative or antimorphic
mutations) defects in cartilage structural proteins result in a
diverse range of both recessive and dominant genetic skeletal
dysplasias (Table I). These
structural proteins include cartilage collagens (types II, IX and
XI), proteoglycans (aggrecan) and glycoproteins [cartilage
oligomeric matrix protein (COMP) and matrilin-3 (MATN3)] and many
of the mutations are predicted to affect the folding and/or
function of these important molecules. This review specifically
concentrates on those diseases resulting from mutations in the
non-collagenous glycoproteins, COMP and matrilin-3, which together
cause a continuum of phenotypes that are amongst the most common
autosomal dominant skeletal dysplasias.
 | Table IHuman genetic skeletal diseases
result from qualitative (anti-morphic) defects in cartilage
structural proteins. |
Table I
Human genetic skeletal diseases
result from qualitative (anti-morphic) defects in cartilage
structural proteins.
| Gene | Protein | Disease(s) | Genetic loci | Domain | Refs. |
|---|
| COMP | COMP |
Pseudoachondroplasia (AD)
Multiple epiphyseal dysplasia (AD) | PSACH
EDM1 | T3-7
repeats
C-terminal domain | (4,45) |
| MATN3 | Matrilin-3 | Multiple epiphyseal
dysplasia (AD) | EDM5 | vWFA | (18) |
COL9A1
COL9A2
COL9A3 | Type IX
collagen | Multiple epiphyseal
dysplasia (AD) | EDM6
EDM2
EDM3 | COL3 domain | (46–48) |
| COL2A1 | Type II
collagen | Diverse range of AD
and AR phenotypes collectively known as type II
collagenopathies | Various | Triple helical
region and C-propeptide | (49) |
COL11A1
COL11A2 | Type XI
collagen | Diverse range of AD
and AR phenotypes collectively known as type XI
collagenopathies | Various | | (50) |
| COL10A1 | Type X
collagen | Metaphyseal
chondrodysplasia, type Schmid (AD) | MCDS | Carboxyl-terminal
non-collagenous domain (NC1) | (51) |
| ACAN | Aggrecan |
Spondyloepimetaphyseal dyslasia
(AR)
Osteochondritis dissecans (AD)
Short stature, accelerated bonematuration (AD) | SEMD
OCD | G3 C-type lectin
domain | (52–54) |
The pseudoachondroplasia (PSACH) and multiple
epiphyseal dysplasia (MED) disease spectrum: the generation and
study of genetic mouse models. Originally believed to be
different but clinically related phenotypes, it was gene and
mutation identification in the 1990s that demonstrated that PSACH
and some forms of MED were allelic diseases resulting from
mutations in COMP (1).
However, MED is genetically heterogeneous and autosomal dominant
forms can also result from mutations in the genes encoding
matrilin-3 and type IX collagen (2) (Table
I).
Despite the first mutations being identified in
COMP, MATN3 and the type IX collagen genes in the six
years between 1995 and 2001 (Table
I), very little progress has been made in understanding disease
mechanisms despite numerous cell culture studies (reviewed in ref.
3). A lack of relevant
pathological tissues (such as cartilage growth plate), and the
obvious limitations of cell culture models to study long bone
growth, meant that only small incremental increases in knowledge
were possible.
Over the past eight years, however, a number of
different genetic approaches have been used to generate mouse
models of PSACH-MED that recapitulate the various phenotypes and
allow disease mechanisms to be studied in detail in vivo
(Table II). Moreover, the
generation of novel phenocopies to model specific disease
mechanisms have confirmed the importance of endoplasmic reticulum
(ER) stress and chondrocyte proliferation as key modulators of
growth plate dysplasia. Finally, new insight into related
musculoskeletal complications (such as myopathy and tendinopathy),
which may be of clinical utility, has also been gained through the
in-depth analysis of targeted mouse models of the PSACH-MED disease
spectrum.
| Table IIMouse models of the PSACH-MED disease
spectrum and novel phenocopies to model ER stress in the cartilage
growth plate. |
Table II
Mouse models of the PSACH-MED disease
spectrum and novel phenocopies to model ER stress in the cartilage
growth plate.
| Disease | Gene | Mutation | Approach taken | Promoter | Refs. |
|---|
| PSACH | COMP | D469del | Transgenic (rat
COMP cDNA) | Col2a1 | (20) |
| PSACH | COMP | D469del | Transgenic (human
COMP gene) | Native | (3) |
| PSACH | COMP | D469del | Transgenic (human
COMP cDNA) | Col2a1 | (3) |
| PSACH | COMP | D469del | Transgenic
inducible overexpression (human COMP cDNA) | Col2a1 and
tetracycline responsive element | (21,22,44) |
| PSACH | COMP | D469del | Knock-in | Native | (9,15,42) |
| PSACH-MED | COMP | T585M | Knock-in | Native | (7,9,38,41) |
| MED | MATN3 | V194D | Knock-in | Native | (9,19,23,42) |
|
Chondrodysplasia | TG | Rdw
(G2320R) | Transgenic
phenocopy | Col2a1 | (31) |
|
Chondrodysplasia | TG | Cog
(L2293P) | Transgenic
phenocopy | Col2a1 | (55) |
2. Knock-in mouse models provide new insight
into disease mechanisms
The application of homologous recombination and
Cre-lox technology has allowed the generation of a series of
mouse lines that genetically modelled phenotypes within the
PSACH-MED disease spectrum, thereby allowing key pathological
findings to be described and disease mechanisms to be studied in
detail (Tables II and III).
| Table IIIKey pathological and quantitative
measures of disease severity in various knock-in mouse models of
PSACH-MED and novel ER stress phenocopies. |
Table III
Key pathological and quantitative
measures of disease severity in various knock-in mouse models of
PSACH-MED and novel ER stress phenocopies.
| Model | Age at onset | Final reduction in
bone length
| Cell proliferation
| Apoptosis
| Protein
retention | ER stress | Stress pathway |
|---|
| 3 weeks | 6 weeks | 9 weeks | 3 weeks | 3 weeks |
|---|
|
CompD469del | ~6 weeks | No differences | ↓ 6% | ↓ 5% | ↓ 17% | ↑ 90-fold
(Pz)
↑ 5-fold (Hz) | Yes | Yes | Novel
(APR/EOR) |
|
CompT585M | ~9 weeks | No differences | No differences | ↓ 5% | ↓ 24% | ↑ 3-fold
(Rz)
↑ 12-fold (Pz)
↑ 3-fold (Hz) | Slight | Mild | Mild UPR |
|
Matn3V194D | ~2 weeks | ↓ 12% | ↓ 13% | ↓ 12% | ↑ 16% | Spatially
dysregulated | Yes | Yes | UPR |
|
Col2-TgRdw | At birth | ↓ 8% | ↓ 5% | ↓ 5% | ↑ 21% | No differences | Yes | Yes | UPR |
|
Col2-Tgcog | ~3 weeks | ↓ 7% | ↓ 4% | ↓ 4% | ↑ 12% | No differences | Yes | Yes | Novel |
In the first instance, an allelic series of PSACH
models were generated representing the two major classes of
COMP mutations: the type III repeat (T3) region and the
carboxyl-terminal domain (CTD) (2,4).
Type III repeat region mutations account for approximately 85% of
all COMP-related PSACH-MED, whereas CTD mutations represent 15% of
cases (2). The mutations that
were selected to represent these two different classes were the
common in-frame deletion of an aspartic acid residue from the
C-type motif of repeat T37 (p.Asp469del), which accounts
for 30% of all PSACH, and the recurrent p.Thr585Met missense
mutation in the CTD (2,4). The p.Thr585Met mutation has been
reported in four different studies and results in a phenotypic
spectrum ranging from MED to mild PSACH (4). Previous studies using cell models
consistently demonstrated the retention of mutant Asp469del COMP in
the ER (3). By contrast,
p.Thr585Met, along with several other CTD mutations, was
efficiently secreted into the culture media of various cell models
(5,6). These early cell model studies have
suggested different disease mechanisms between these two classes of
mutations that justified validation and in-depth analysis using
relevant mouse models.
The p.Thr585Met (CompT585M)
model of mild PSACH/severe MED
Individuals with p.Thr585Met [and several other CTD
mutations (4)] often present with
a mild form of PSACH and this was reflected in the
CompT585M mouse model which showed only a 5%
reduction in bone length at 9 weeks of age (7). This is consistent with previous
family studies, which demonstrated that individuals with the
Thr585Met mutation may have average stature, despite having
significant epiphyseal and metaphyseal changes in the large joints
(8).
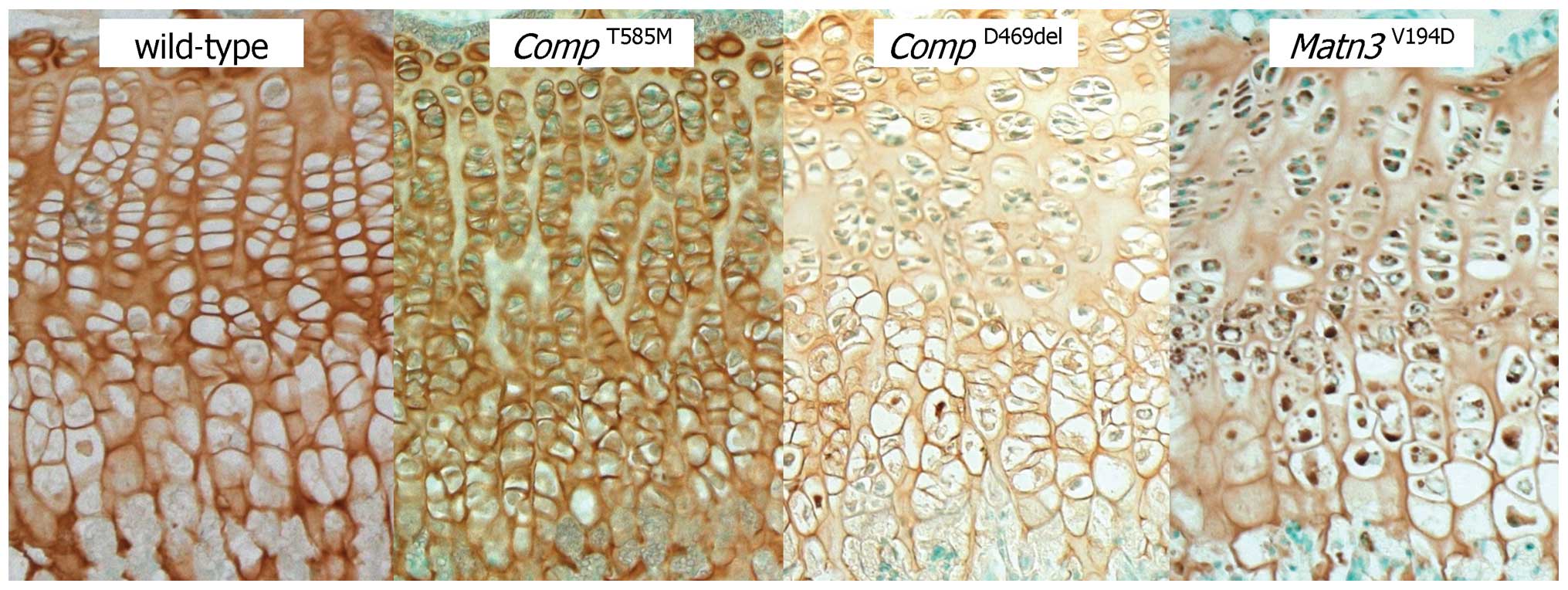
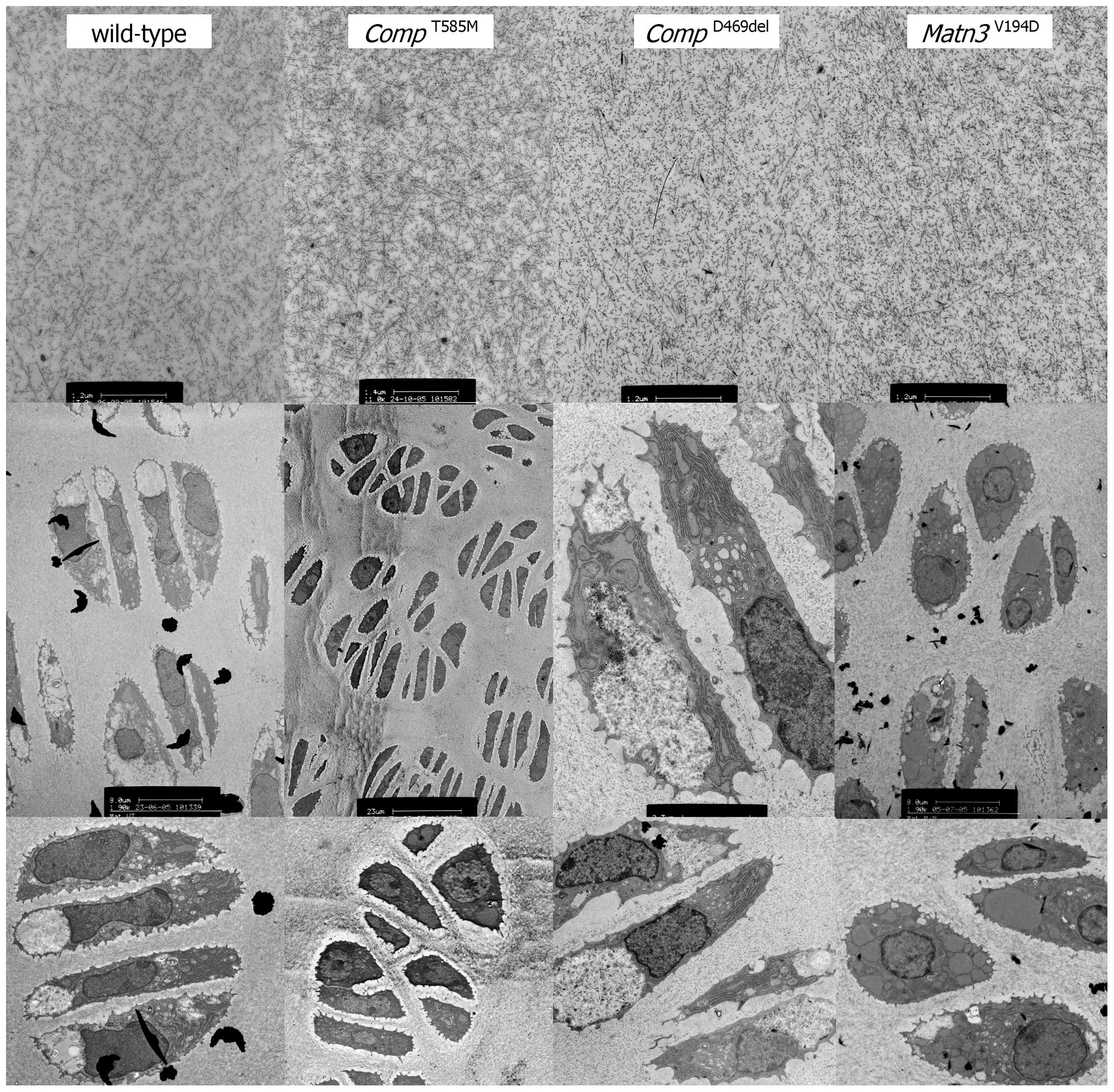
Immunohistochemistry and electron microscopy
confirmed a lack of mutant protein retention in the ER (Figs. 1 and 2), which is consistent with previous
cell culture studies (5,6). However, there were severe
disruptions to the morphology of the growth plate and to the size,
shape and arrangement of individual chondrocytes and chondrons
(7). The composition of the
growth plate extracellular matrix (ECM) was also disrupted with
changes to the localisation of several ECM proteins and the
under-sulfation of proteoglycans (7). Proteomic interrogation confirmed
changes to the extractability of several ECM proteins, including
collagens [α1(XIV), α1(IX), α1(XI), α1(XII) and α6(VI)], laminin β2
and fibronectin (9). Finally,
there was a significant reduction in chondrocyte proliferation that
was accompanied by increased and spatially dysregulated apoptosis
(7) (Table III).
The p.Asp469del (CompD469del)
model of typical severe PSACH
p.Asp469del is the most common COMP mutation
and invariably results in typically severe PSACH (4,5).
Numerous cell culture studies have been undertaken on this
archetypal mutation, which have consistently shown that p.Asp469del
results in the retention of mutant COMP protein within the rough ER
(rER) of cells, along with matrilin-3 and collagen type IX
(10,11). Furthermore, cartilage samples from
PSACH patients appear to have a disorganised ECM and there is an
increase in cell death in vivo (12) that can be recapitulated in
vitro by several different cell culture models (13,14).
CompD469del mice were normal at
birth, but grew slower than their wild-type littermates and
developed a progressive short-limb dwarfism, which was
characterised by a 5–6% reduction in final tibia and femur lengths.
Moreover, the mutant mice developed a hip dysplasia that was
characterised by an increase in the angle of deflection from the
vertical of the tuberosity of the ischium (15). The reductions in the final bone
lengths of CompD469del mice were less than would
be expected for a phenotypically exact model of typical severe
PSACH and may be best explained by the mixed genetic background of
the mutant mouse strain.
Notwithstanding the relatively mild reduction in
final bone lengths of CompD469del mice, there was
significant growth plate pathology that was illustrative of the
hallmarks of PSACH (15)
(Table III). In particular,
chondrocyte columns were reduced in number and poorly organised in
the growth plates of CompD469del mice, whilst
mutant COMP was retained within the ER of chondrocytes at all
stages of differentiation (15)
(Fig. 1). The composition and
appearance of the ECM was disrupted, with changes to the
localisation of several ECM proteins, including the co-retention of
matrilin-3 and type IX collagen (15). Proteomic interrogation confirmed
changes to the extractability of several ECM proteins, including
throm-bospondin-3 and -4, several collagens [α1(XIV), α1(IX) and
α1(VI)], tenascin-C and -X and epiphycan (9). Most noticeably, chondrocyte
proliferation was significantly reduced and apoptosis was greatly
increased in the proliferative zone and spatially dysregulated
throughout the entire growth plate (15) (Table III).
The p.Val194Asp (Matn3V194D)
model of moderate MED
Missense mutations and small in-frame deletions in
the von Willebrand Factor A (vWFA) domain of matrilin-3 cause
moderate to severe forms of MED (2). The various missense mutations are
distributed between the β-sheet (~85%) and α-helical (~15%) regions
of the vWFA domain. Mutations in the β-sheet regions primarily
disrupt the folding of the vWFA domain, resulting in the retention
of mutant protein in the ER of cells (16). By contrast, some α-helical
mutations allow the correct folding and secretion of mutant
matrilin-3 (17); however, there
are exceptions to these generalisations. The MATN3 mutation
(p.Val.194Asp) selected for mouse model generation was the first
EDM5 mutation to be identified and is an archetypal example
(18).
Matn3V194D mice were normal at
birth but developed short-limbed dwarfism that was characterised by
a final reduction in tibia length of 12–13% (19). There was a significant retention
of mutant matrilin-3 in the growth plate chondrocytes, which were
abnormal in shape and resulted in poorly organised chondrons and
chondrocyte columns (Figs. 1 and
2). Chondrocyte proliferation was
significantly reduced and whilst there was no overall increase in
apoptosis it was noticeably dysregulated with increased cell death
occurring throughout the entire hypertrophic zone (19) (Table III). Changes to the appearance
of the cartilage ECM may be partly attributable to differences in
the extractability of various collagens [α1(XIV) and α3(VI)],
osteomodulin, biglycan, tenascin-C and epiphycan (9).
3. Transgenic approaches for generating
mouse models
Several transgenic approaches have also been used to
generate mouse models of the common PSACH mutation, which do not
rely on the targeted integration of a single copy of the mutant
allele (Table II).
Standard transgenic approaches
In the first example, a transgenic mouse model was
generated by Schmitz et al, in which rat Asp469del COMP cDNA
expression was achieved through the Col2a1 promoter
(20). Mutant COMP was
over-expressed by ~40% and transgenic mutant male mice were up to
8% shorter than transgenic wild-type mice at 6 months of age. In an
attempt to exacerbate this relatively mild phenotype, the
transgenic mutant mice were crossed onto a Comp-null line to
ensure that only rat COMP Asp469del was expressed by chondrocytes.
This cross resulted in a male-specific increase in disease severity
that was characterised by a further decrease in body and femur
lengths. Chondrocytes within the proliferative zone showed the
retention of the transgenic mutant COMP, the levels of which were
further increased on the Comp-null background (20). Moreover, there were abnormal
changes to the morphology of the cartilage ECM, including the loss
of proteoglycans and changes to the localisation of other ECM
molecules, such as type VI collagen, aggrecan and matrilin-1.
Finally, increased apoptosis was observed in combination with
distinct areas of hypocellularity and these key pathological
features are now recognised as hallmark descriptors of the PSACH
growth plate in vivo (20).
Subsequently, Posey et al hypothesised that
the overexpression of Asp469del COMP would best recapitulate the
PSACH disease phenotype in a mouse model and therefore used the
inducible overexpression of mutant Comp that was driven from
the Col2a1 promoter (21).
One reason for taking this approach was as a consequence of limited
phenotypic and pathological effects observed in earlier PSACH
transgenic mice models (3), which
highlights the unpredictability of standard transgenic approaches
for developing mouse models of human genetic skeletal diseases
(GSDs) (Table II).
In the first model, a BAC clone containing human
COMP and its native promoter was used to generate two
founders (1 male and 1 female) of which only the female
demonstrated a clinical phenotype and both founders died
prematurely (3). In a second
approach, Asp469del COMP expression from human cDNA was driven by a
mouse Col2a1 promoter and enhancer. Genomic analysis
demonstrated that 12–20 copies of the transgene had integrated into
mouse chromosome 10. Tibia measurements confirmed a 6% reduction in
final length and there was some disorganization of the growth
plate, with fewer chondrocytes organised into columns of chondrons;
however, in contrast to the study by Schmitz et al (20), no intracellular retention of
mutant Asp469del COMP was observed in growth plate chondrocytes
(3).
Bigenic inducible overexpression of
mutant Comp Asp469del
The most recent transgenic approach involved the
generation of bigenic mice in which the expression of Comp
Asp469del was controlled by a tetracycline responsive element
promoter, which was itself under the control of the tetracycline
trans-activator coding sequence with a Col2a1 promoter to
ensure cartilage-specific expression (21). Whilst Col2a1-driven
overexpression of Comp Asp469del was assumed to be robust,
the levels of mutant Comp mRNA were only determined at E15
and shown to be 4- to 6-fold higher than endogenous Comp
mRNA (21). Unfortunately, the
relative levels of Comp Asp469del mRNA and protein were not
determined at later time-points, thus limiting the interpretation
of phenotypic and pathological readouts, particularly when these
analyses were performed postnatally. Nevertheless, key pathological
features such as mutant COMP retention, growth plate dysplasia and
marginally increased chondrocyte apoptosis were identified in this
mouse model (21). Subsequent
developmental analysis of this mouse model (22) confirmed growth plate dysplasia by
3 weeks of age that was characterised by disorganization, reduced
thickness and distinct areas of hypocellularity. Mutant Asp469del
COMP retention was shown to be progressive until 2 weeks of age.
Analysis of chondrocyte death by terminal
deoxynucleotidyltransferase-mediated dUTP nick-end labelling
(TUNEL) confirmed that abnormally increased apoptosis was occurring
in the hypertrophic zone by 2 weeks of age, and increased further
with age (~3–4-fold); however, the precise localisation of
increased apoptosis within the different growth plate zones was not
determined. Long bone growth was significantly reduced by 12% in
the tibia; however, skull and snout lengths were also shown to be
smaller than wild-type controls, indicating that intramembraneous
ossification was also disrupted by the transgenic overexpression of
mutant COMP, which is not consistent with other mouse models or
individuals with PSACH. Moreover, the pathophysiological relevance
of Comp Asp469del overexpression is not known and whilst
strong phenotypic effects were observed, the genetic pathways that
are induced by this transgenic approach may not necessarily be
representative of the human disease.
4. Activation of canonical and/or novel ER
stress pathways is genotype-specific
The expression of CompD469del and
Matn3V194D results in ER retention of the
relevant mutant proteins and the co-retention of interacting
partners that ultimately results in ER stress (15,19); however, the downstream stress
pathways that are activated are genotype-specific.
For example, the expression of
Matn3V194D induces the activation of the
canonical unfolded protein response (UPR), which is characterised
by the upregulation of binding immunoglobulin protein (BiP) (a
sentinel marker of UPR) and a broad range of chaperones, foldases
and protein disulphide isomerases (23). Moreover, the downstream splicing
of X-box binding protein 1 (Xbp1) in Matn3V194D
chondrocytes (and also in cell models) is symbolic of classical
UPR; however, this prolonged ER stress does not result in a
C/EBP-homologous protein (CHOP)-mediated increase in apoptosis
(23) (and our unpublished
observations).
In contrast to mutant matrilin-3, the ER retention
of CompD469del does not induce a canonical UPR,
but a novel form of ER stress that is characterised by changes in
the expression of groups of genes implicated in oxidative stress,
cell cycle regulation and apoptosis (15). Similar UPR-independent pathways,
such as the ER overload response (EOR), appear to be associated
with the aggregation of mutant proteins in the ER that eventually
induce toxic gain-of-function (24). This has been noted with
serpinopathies in which the aggregation of insoluble misfolded
α1-antitrypsin triggers an alternative ER stress response that is
independent of the UPR and involves the activation of nuclear
factor-κB (NF-κB) signalling (25). Moreover, it has been proposed that
an aggregating protein response (APR) is activated by glycine
substitutions in the triple helical regions of type I collagen that
also do not induce canonical UPR (26,27).
Our own studies have identified differences in the
molecular organization of the mutant COMP and matrilin-3 proteins
that are retained in the ER of mouse chondrocytes and cell culture
models (9,28). Mutant matrilin-3 forms non-native
disulphide bonded aggregates, due to a delay in the folding of the
single vWFA domain, which may render it inaccessible to
degradation. By contrast, mutant D469del COMP does not appear to
aggregate and is retained in the ER in its apparent native state of
either tetramers or pentamers (9). This difference in the molecular
organization of mutant protein aggregates may play a role in
determining which ER stress pathways are activated.
These unexpected findings demonstrate that ER
stress, as a result of mutant protein misfolding, and the genetic
pathways that are activated in an attempt to restore ER
homeostasis, are likely to be more diverse in mouse models and
human genetic diseases than in the more commonly studied
chemically-induced ER stress experimental systems (29). However, despite the downstream
activation of several potential pathways, such as UPR, APR and/or
EOR, a common consequence appears to be the reduction in
chondrocyte proliferation (Table
III).
5. Common genetic pathways identified in the
different mouse models of Asp469del COMP pseudoachondroplasia
Despite differences in the relative severity of
growth plate dysplasia, chondrocyte cell death and long bone growth
between the different Comp Asp469del mouse models (Table II), transcriptomic profiling has
nevertheless implicated several genetic pathways in common
(15,22).
The microarray analysis of mRNAs isolated from
chondrocytes of the CompD469del knock-in mouse
model showed a complex disease profile with expression changes in
groups of genes implicated in oxidative stress, cell cycle
regulation and apoptosis, which is consistent with the chondrocyte
and growth plate pathology (15).
Moreover, this study demonstrated that reduced cell proliferation
and increased dysregulated chondrocyte apoptosis was associated
with the downregulation of peroxiredoxin 2, which is a gene
important for protecting cells against oxidative damage and also in
regulating apoptosis (15).
Similarly, transcriptomic analysis of mRNAs isolated from the
chondrocytes of the bigenic transgenic mouse identified cell cycle
regulation, inflammation, oxidative stress and DNA damage as the
major classes of genes implicated in disease pathogenesis (22).
6. Missense mutations in matrilin-3 and COMP
cause changes in the extractability of other cartilage proteins and
influence ECM organization
Analysis of the cartilage proteome of
Matn3V194D, CompD469del and
CompT585M mice has identified differences in the
extractability of numerous structural ECM components relative to
the controls (9). The extraction
of type IX collagen [a fibril-associated collagen with interrupted
triple helices (FACIT)] was increased in
CompD469del and CompT585M mice,
which may alter the integrity of the II/IX/XI heterofibrillar
collagen network. Other changes in the extractability of FACIT
collagens were observed in Matn3V194D (i.e., the
decreased extraction of collagen types XII and XIV),
CompD469del (decrease in types IX and XIV) and
CompT585M (decrease in types IX and XII, but
increase in type XIV) cartilage, which further implied disruption
to ECM organization as a consequence of Comp and
Matn3 mutations that correlated with the observations of
altered ECM morphology detected by electron microscopy (7,15,19) (Fig.
2).
In addition to these commonalities between mutant
mouse models, discrete patterns of ECM protein extraction were also
observed (9). Most notably, the
extraction of tenascin X was greatly reduced from
CompD469del cartilage; however, the functional
significance of this, and also the increases in tenascin C
extractability observed in CompD469del and
Matn3V194D mice, remains to be determined.
7. Novel phenocopies of chondrocyte-specific
ER stress provide information regarding intracellular disease
mechanisms and the influence of chondrocyte proliferation
Defining the relative contributions of intracellular
disease mechanisms (ER stress) and extracellular defects (ECM
disruption) to the initiation and progression of growth plate
dysplasia is experimentally challenging. Therefore, to directly
test the role of ER stress in growth plate dysplasia and reduced
long bone growth, independent of disruptions to the cartilage ECM,
a novel transgenic approach was taken. This approach was previously
established and validated as an innovative tool to dissect disease
mechanisms in metaphyseal chondrodysplasia type Schmid, which can
be caused by missense mutations in the gene encoding type X
collagen (30).
To delineate the relative influence of ER stress to
the development of chondrodysplasia, the expression of a G2320R
mutant form of thyroglobulin (Rdw) was targeted primarily to
resting and proliferating chondrocytes using the Col2a1
promoter (31). The expression
and retention of this mutant exogenous protein in the rER of growth
plate chondrocytes resulted in chronic cell stress and reduced bone
growth, but without inducing any disruptions to the architecture
and overall organization of the ECM. More significantly, however,
decreased bone growth appeared to be the direct result of reduced
chondrocyte proliferation in the growth plates in transgenic
mice.
8. Reduced chondrocyte proliferation is a
shared cellular response to all forms of ER stress and defines a
common denominator of PSACH-MED pathology
The pivotal role of ER stress in the initiation of
growth plate dysplasia and abnormal bone growth was revealed
through targeted knock-in mouse models and novel transgenic
phenocopies (Table II). Whilst
it has been previously suggested that increased and dysregulated
chondrocyte apoptosis is the major cause of reduced bone growth in
PSACH (22), our recent analysis
of the TgRdw transgenic phenocopy has
demonstrated that reduced chondrocyte proliferation alone is
sufficient to cause a significant reduction in bone length in the
absence of increased apoptosis or extensive disruption to cartilage
ECM composition and assembly (31). We therefore hypothesise that
modulating chondrocyte proliferation is likely to exert the
greatest influence on long bone growth in PSACH-MED and this may
represent an attractive target for therapy. Indeed, dysregulated
cyclin D1 signaling during the G1 phase of the cell-cycle causes
reduced chondrocyte proliferation in a mouse model of diastrophic
dysplasia due to a p.A386V substitution in the eighth transmembrane
domain of solute carrier family 26 member 2 (Slc26a2)
(32). Moreover, recent mouse
model studies have demonstrated that long bone length can be
restored in achondroplasia by targeting the mitogen-activated
protein kinase (MAPK) pathway, which is activated by the tyrosine
kinase receptor FGFR3 mutations in achondroplasia, and
causes a disruption to normal chondrocyte differentiation (33). Finally, transgenic expression of
mutant forms of type II collagen causes ER-stress induced
chondrodysplasia (34,35), which can also defined by reduced
chondrocyte proliferation and may be associated with disrupted cell
polarity and the abnormal organization and morphology of the
primary cilia (35).
9. Genetic background influences phenotypic
severity in the CompT585M mouse model of
PSACH-MED by impacting on chondrocyte proliferation and
apoptosis
Several clinical genetic studies have demonstrated
that there is considerable intra- and interfamilial variability in
disease presentation in both PSACH and MED, which strongly points
to the existence of genetic modifiers of phenotypic severity. This
is best highlighted by several MATN3 mutations, such as
Arg121Trp, which are associated with marked interfamilial
variability in the radiographic phenotype of patients (36). Patients carrying the T585M COMP
mutation also show variability in final adult height (8).
In-bred mouse strains are a useful tool which can be
used to study complex disease traits when the influence of a
genetic modifier is suspected (37). Interestingly, crossing the
CompT585M knock-in mice (originally on a mixed
genetic background) onto a C57BL6/J inbred background has been
shown to increase the severity of the phenotype by further
disrupting chondrocyte proliferation, apoptosis and matrix
deposition (38). Moreover,
mutant COMP was retained inside the rER of chondrocytes, which was
a feature not observed in the original mouse model (7). Dixon and Dixon (37) demonstrated a similar phenotypic
variability in their model of craniofacial abnormalities in
Treacher Collins Syndrome that is genetic background-dependent.
This feature renders inbred mouse strains a valuable genetic model
for quantitative trait loci (QTL) studies and the analysis of
genetic modifiers co-segregating with various disease traits.
10. Insight into additional musculoskeletal
complications of PSACH-MED revealed for the first time through the
in-depth analysis of mouse models
Several skeletal dysplasias, including PSACH and
MED, often result from mutations in the extracellular proteins that
are synthesised by cells of the mesenchymal lineage (39). It is therefore not surprising that
patients may present with additional related musculoskeletal
complications (40). Mild
PSACH/MED patients are sometimes diagnosed with mild myopathy or a
‘neuromuscular problem of unknown etiology’ prior to the
confirmation of a chondrodysplasia. Indeed, we have previously
reported several patients who initially presented with excessive
fatigue during walking; difficulty rising from a squatting position
and mildly elevated serum creatine kinase levels (39). These individuals were initially
referred to neurological clinics for assessment, prior to the
diagnosis of a skeletal dysplasia following radiographic
evaluation. The analysis of muscle biopsies often proved
inconclusive (40). An in-depth
analysis of the CompT585M model of mild PSACH/MED
(39,41) revealed that the mild myopathy
observed in patients, and recapitulated in this mouse model, was
actually the result of an underlying tendinopathy that was
manifesting primarily at the myotendinous and perimyseal junctions;
this finding further correlated with joint laxity, which is another
prominent clinical feature of PSACH and MED patients. Subsequent
phenotypic analysis of the other two PSACH/MED mouse models
(D469del COMP and V194D matrilin-3) revealed that both the myopathy
and underlying tendinopathy were associated with mutant COMP, and
not mutant matrilin-3, indicating that the neuromuscular
complications in PSACH and MED are not just a side effect of the
short limbed dwarfism alone (42). This in-depth phenotypic analysis
of relevant genetic models of the PSACH-MED disease spectrum has
yielded important information related to the specific site of the
musculoskeletal complications and their underlying cause; both of
which will be value aids for the diagnosis and management of these
conditions.
11. Strengths and weaknesses of different
genetic approaches to model skeletal diseases
The various transgenic approaches described in this
review have both strengths and weaknesses as with all model
systems. Homologous recombination allows the correct spatial and
temporal expression of the mutant allele at levels comparable to
the endogenous allele; however, mice are often required to be
homozygous for the mutant allele to achieve a recognisable and
quantifiable phenotype. This is in contrast to individuals with
autosomal dominant PSACH and MED who only need to be heterozygous
for the mutation. Posey et al (43) proposed this as weakness of the
homologous recombination approach and suggested that inducible
transgenic overexpression is required to produce a phenotype in
mice that are heterozygous for the mutant allele; however, the
random integration of multiple copies of the mutant transgene into
the mouse genome is also not representative of the human condition.
Furthermore, the inducible expression of mutant COMP also has its
limitations since it is impossible to regulate the amount of dox
ingested by individual transgenic mice and determining the levels
of mutant COMP expression in the cartilage of each mouse is not
feasible. Nevertheless, despite the differences of the two
transgenic approaches, both mutant mouse lines demonstrated
hallmark features of PSACH, which included mutant COMP retention,
increased and dysregulated apoptosis with corresponding areas of
hypocellularity (15,22,44). Moreover, cell proliferation was
markedly reduced in both mouse models, which has recently been
identified as a quintessential feature of growth plate dysplasia
and reduced bone growth (11).
The use of endogenous Matn3 and Comp
promoters by the homologus recombination approach (7,15,19) has also allowed the in-depth
analysis of a range of other musculoskeletal tissues that is not
possible with Col2a1-driven transgenic overexpression. These
mouse phenotyping studies have confirmed the importance of
tendinopathy and mild myopathy to the overall pathophysiology of
PSACH and MED that will ultimately provide greater understanding of
the clinical presentation of these GSDs.
The development and deep phenotyping of ‘ER-stress
phenocopies’ (30,31) is a novel genotype-independent
approach for determining the role of ER stress in GSDs. The use of
the Col2a1 promoter generated a model that exhibited ER
stress in a broad range of cartilaginous tissues including the
growth plate and articular cartilages. The resulting phenotype,
whilst not phenocopying any specific disease, provided a
‘generalised chondrodysplasia’ in which to identify fundamental
disease mechanisms. In contrast, the use of the Col10a1
promoter, which specifically targeted expression to hypertrophic
chondrocytes, accurately phenocopied metaphyseal chondrodysplasias
type Schmid. In conclusion, this genetic approach demonstrated that
ER stress can be accurately modelled in either a single cell type
(or differentiation state) or in an entire tissue and will provide
a powerful technique for determining the role of ER stress in a
broad group of inherited connective tissues diseases.
In conclusion, these different genetic mouse models
(transgenic overexpression expression, knock-in by homologous
recombination and novel phenocopies) of the PSACH-MED phenotypic
spectrum have recapitulated key aspects of disease pathology and
identified new fundamental mechanisms that have both temporal and
spatially context within the cartilage growth plate. The
acquisition of this new knowledge would not have been possible
through the use of cell culture models, which do not recapitulate
the dynamic properties of the cartilage growth plate.
Acknowledgments
Some of the work presented in this review was funded
by the Wellcome Trust (no. 084353/Z/07/Z) and the European
Community’s Seventh Framework Programme under grant agreement no.
602300 (SYBIL).
Abbreviations:
|
COMP
|
cartilage oligomeric matrix
protein
|
|
GSD
|
genetic skeletal disease
|
|
PSACH
|
pseudoachondroplasia
|
|
MED
|
multiple epiphyseal dysplasia
|
|
ER
|
endoplasmic reticulum
|
|
UPR
|
unfolded protein response
|
|
EOR
|
ER overload response
|
|
APR
|
aggregated protein response
|
|
ECM
|
extracellular matrix
|
|
CTD
|
carboxyl-terminal domain
|
|
vWFA
|
von Willebrand Factor A
|
|
Xbp1
|
X-box binding protein 1
|
|
BiP
|
binding immunoglobulin protein
|
|
CHOP
|
C/EBP-homologous protein
|
|
FACIT
|
fibril-associated collagens with
interrupted triple helices
|
References
|
1
|
Briggs MD, Hoffman SM, King LM, Olsen AS,
Mohrenweiser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines
ES, et al: Pseudoachondroplasia and multiple epiphyseal dysplasia
due to mutations in the cartilage oligomeric matrix protein gene.
Nat Genet. 10:330–336. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jackson GC, Mittaz-Crettol L, Taylor JA,
Mortier GR, Spranger J, Zabel B, Le Merrer M, Cormier-Daire V, Hall
CM, Offiah A, et al: Pseudoachondroplasia and multiple epiphyseal
dysplasia: A 7-year comprehensive analysis of the known disease
genes identify novel and recurrent mutations and provides an
accurate assessment of their relative contribution. Hum Mutat.
33:144–157. 2012. View Article : Google Scholar :
|
|
3
|
Posey KL, Yang Y, Veerisetty AC, Sharan SK
and Hecht JT: Model systems for studying skeletal dysplasias caused
by TSP-5/COMP mutations. Cell Mol Life Sci. 65:687–699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Briggs MD, Brock J, Ramsden SC and Bell
PA: Genotype to phenotype correlations in cartilage oligomeric
matrix protein associated chondrodysplasias. Eur J Hum Genet.
22:1278–1282. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Spitznagel L, Nitsche DP, Paulsson M,
Maurer P and Zaucke F: Characterization of a
pseudoachondroplasia-associated mutation (His587–>Arg) in the
C-terminal, collagen-binding domain of cartilage oligomeric matrix
protein (COMP). Biochem J. 377:479–487. 2004. View Article : Google Scholar
|
|
6
|
Schmitz M, Becker A, Schmitz A, Weirich C,
Paulsson M, Zaucke F and Dinser R: Disruption of extracellular
matrix structure may cause pseudoachondroplasia phenotypes in the
absence of impaired cartilage oligomeric matrix protein secretion.
J Biol Chem. 281:32587–32595. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Piróg-Garcia KA, Meadows RS, Knowles L,
Heinegård D, Thornton DJ, Kadler KE, Boot-Handford RP and Briggs
MD: Reduced cell proliferation and increased apoptosis are
significant pathological mechanisms in a murine model of mild
pseudoachondroplasia resulting from a mutation in the C-terminal
domain of COMP. Hum Mol Genet. 16:2072–2088. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Briggs MD, Mortier GR, Cole WG, King LM,
Golik SS, Bonaventure J, Nuytinck L, De Paepe A, Leroy JG,
Biesecker L, et al: Diverse mutations in the gene for cartilage
oligomeric matrix protein in the pseudoachondroplasia-multiple
epiphyseal dysplasia disease spectrum. Am J Hum Genet. 62:311–319.
1998. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bell PA, Wagener R, Zaucke F, Koch M,
Selley J, Warwood S, Knight D, Boot-Handford RP, Thornton DJ and
Briggs MD: Analysis of the cartilage proteome from three different
mouse models of genetic skeletal diseases reveals common and
discrete disease signatures. Biol Open. 2:802–811. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hecht JT, Hayes E, Snuggs M, Decker G,
Montufar-Solis D, Doege K, Mwalle F, Poole R, Stevens J and Duke
PJ: Calreticulin, PDI, Grp94 and BiP chaperone proteins are
associated with retained COMP in pseudoachondroplasia chondrocytes.
Matrix Biol. 20:251–262. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hecht JT, Hayes E, Haynes R and Cole WG:
COMP mutations, chondrocyte function and cartilage matrix. Matrix
Biol. 23:525–533. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hecht JT, Makitie O, Hayes E, Haynes R,
Susic M, Montufar-Solis D, Duke PJ and Cole WG: Chondrocyte cell
death and intracellular distribution of COMP and type IX collagen
in the pseudoachondroplasia growth plate. J Orthop Res. 22:759–767.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duke J, Montufar-Solis D, Underwood S,
Lalani Z and Hecht JT: Apoptosis staining in cultured
pseudoachondroplasia chondrocytes. Apoptosis. 8:191–197. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hashimoto Y, Tomiyama T, Yamano Y and Mori
H: Mutation (D472Y) in the type 3 repeat domain of cartilage
oligomeric matrix protein affects its early vesicle trafficking in
endoplasmic reticulum and induces apoptosis. Am J Pathol.
163:101–110. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Suleman F, Gualeni B, Gregson HJ, Leighton
MP, Piróg KA, Edwards S, Holden P, Boot-Handford RP and Briggs MD:
A novel form of chondrocyte stress is triggered by a COMP mutation
causing pseudoachondroplasia. Hum Mutat. 33:218–231. 2012.
View Article : Google Scholar :
|
|
16
|
Cotterill SL, Jackson GC, Leighton MP,
Wagener R, Mäkitie O, Cole WG and Briggs MD: Multiple epiphyseal
dysplasia mutations in MATN3 cause misfolding of the A-domain and
prevent secretion of mutant matrilin-3. Hum Mutat. 26:557–565.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fresquet M, Jowitt TA, Ylöstalo J, Coffey
P, Meadows RS, Ala-Kokko L, Thornton DJ and Briggs MD: Structural
and functional characterization of recombinant matrilin-3 A-domain
and implications for human genetic bone diseases. J Biol Chem.
282:34634–34643. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chapman KL, Mortier GR, Chapman K,
Loughlin J, Grant ME and Briggs MD: Mutations in the region
encoding the von Willebrand factor A domain of matrilin-3 are
associated with multiple epiphyseal dysplasia. Nat Genet.
28:393–396. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leighton MP, Nundlall S, Starborg T,
Meadows RS, Suleman F, Knowles L, Wagener R, Thornton DJ, Kadler
KE, Boot-Handford RP, et al: Decreased chondrocyte proliferation
and dysregulated apoptosis in the cartilage growth plate are key
features of a murine model of epiphyseal dysplasia caused by a
matn3 mutation. Hum Mol Genet. 16:1728–1741. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schmitz M, Niehoff A, Miosge N, Smyth N,
Paulsson M and Zaucke F: Transgenic mice expressing D469Delta
mutated cartilage oligomeric matrix protein (COMP) show growth
plate abnormalities and sternal malformations. Matrix Biol.
27:67–85. 2008. View Article : Google Scholar
|
|
21
|
Posey KL, Veerisetty AC, Liu P, Wang HR,
Poindexter BJ, Bick R, Alcorn JL and Hecht JT: An inducible
cartilage oligomeric matrix protein mouse model recapitulates human
pseudoachondroplasia phenotype. Am J Pathol. 175:1555–1563. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Posey KL, Coustry F, Veerisetty AC, Liu P,
Alcorn JL and Hecht JT: Chop (Ddit3) is essential for D469del-COMP
retention and cell death in chondrocytes in an inducible transgenic
mouse model of pseudoachondroplasia. Am J Pathol. 180:727–737.
2012. View Article : Google Scholar :
|
|
23
|
Nundlall S, Rajpar MH, Bell PA, Clowes C,
Zeeff LA, Gardner B, Thornton DJ, Boot-Handford RP and Briggs MD:
An unfolded protein response is the initial cellular response to
the expression of mutant matrilin-3 in a mouse model of multiple
epiphyseal dysplasia. Cell Stress Chaperones. 15:835–849. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pahl HL and Baeuerle PA: The ER-overload
response: Activation of NF-kappa B. Trends Biochem Sci. 22:63–67.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ekeowa UI, Freeke J, Miranda E, Gooptu B,
Bush MF, Pérez J, Teckman J, Robinson CV and Lomas DA: Defining the
mechanism of polymerization in the serpinopathies. Proc Natl Acad
Sci USA. 107:17146–17151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chessler SD and Byers PH: BiP binds type I
procollagen pro alpha chains with mutations in the
carboxyl-terminal propeptide synthesized by cells from patients
with osteogenesis imperfecta. J Biol Chem. 268:18226–18233.
1993.PubMed/NCBI
|
|
27
|
Ishida Y, Yamamoto A, Kitamura A, Lamandé
SR, Yoshimori T, Bateman JF, Kubota H and Nagata K: Autophagic
elimination of misfolded procollagen aggregates in the endoplasmic
reticulum as a means of cell protection. Mol Biol Cell.
20:2744–2754. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hartley CL, Edwards S, Mullan L, Bell PA,
Fresquet M, Boot-Handford RP and Briggs MD: Armet/Manf and Creld2
are components of a specialized ER stress response provoked by
inappropriate formation of disulphide bonds: Implications for
genetic skeletal diseases. Hum Mol Genet. 22:5262–5275. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kaufman RJ: Orchestrating the unfolded
protein response in health and disease. J Clin Invest.
110:1389–1398. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rajpar MH, McDermott B, Kung L, Eardley R,
Knowles L, Heeran M, Thornton DJ, Wilson R, Bateman JF, Poulsom R,
et al: Targeted induction of endoplasmic reticulum stress induces
cartilage pathology. PLoS Genet. 5:e10006912009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gualeni B, Rajpar MH, Kellogg A, Bell PA,
Arvan P, Boot-Handford RP and Briggs MD: A novel transgenic mouse
model of growth plate dysplasia reveals that decreased chondrocyte
proliferation due to chronic ER stress is a key factor in reduced
bone growth. Dis Model Mech. 6:1414–1425. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
De Leonardis F, Monti L, Gualeni B, Tenni
R, Forlino A and Rossi A: Altered signaling in the G1 phase
deregulates chondrocyte growth in a mouse model with proteoglycan
undersulfation. J Cell Biochem. 115:1779–1786. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yamashita A, Morioka M, Kishi H, Kimura T,
Yahara Y, Okada M, Fujita K, Sawai H, Ikegawa S and Tsumaki N:
Statin treatment rescues FGFR3 skeletal dysplasia phenotypes.
Nature. 513:507–511. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liang G, Lian C and Huang D, Gao W, Liang
A, Peng Y, Ye W, Wu Z, Su P and Huang D: Endoplasmic reticulum
stress-unfolding protein response-apoptosis cascade causes
chondrodysplasia in a col2a1 p.Gly1170Ser mutated mouse model. PLoS
One. 9:e868942014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Arita M, Fertala J, Hou C, Steplewski A
and Fertala A: Mechanisms of aberrant organization of growth plates
in conditional transgenic mouse model of spondyloepiphyseal
dysplasia associated with the R992C substitution in collagen II. Am
J Pathol. 185:214–229. 2015. View Article : Google Scholar
|
|
36
|
Jackson GC, Barker FS, Jakkula E,
Czarny-Ratajczak M, Mäkitie O, Cole WG, Wright MJ, Smithson SF,
Suri M, Rogala P, et al: Missense mutations in the beta strands of
the single A-domain of matrilin-3 result in multiple epiphyseal
dysplasia. J Med Genet. 41:52–59. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dixon J and Dixon MJ: Genetic background
has a major effect on the penetrance and severity of craniofacial
defects in mice heterozygous for the gene encoding the nucleolar
protein Treacle. Dev Dyn. 229:907–914. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Piróg KA, Irman A, Young S, Halai P, Bell
PA, Boot-Handford RP and Briggs MD: Abnormal chondrocyte apoptosis
in the cartilage growth plate is influenced by genetic background
and deletion of CHOP in a targeted mouse model of
pseudoachondroplasia. PLoS One. 9:e851452014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Piróg KA and Briggs MD: Skeletal
dysplasias associated with mild myopathy-a clinical and molecular
review. J Biomed Biotechnol. 2010:6864572010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jackson GC, Marcus-Soekarman D,
Stolte-Dijkstra I, Verrips A, Taylor JA and Briggs MD: Type IX
collagen gene mutations can result in multiple epiphyseal dysplasia
that is associated with osteochondritis dissecans and a mild
myopathy. Am J Med Genet A. 152A:863–869. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Piróg KA, Jaka O, Katakura Y, Meadows RS,
Kadler KE, Boot-Handford RP and Briggs MD: A mouse model offers
novel insights into the myopathy and tendinopathy often associated
with pseudoachondroplasia and multiple epiphyseal dysplasia. Hum
Mol Genet. 19:52–64. 2010. View Article : Google Scholar
|
|
42
|
Piróg KA, Katakura Y, Mironov A and Briggs
MD: Mild myopathy is associated with COMP but not MATN3 mutations
in mouse models of genetic skeletal diseases. PLoS One.
8:e824122013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Posey KL, Alcorn JL and Hecht JT:
Pseudoachondroplasia/COMP - translating from the bench to the
bedside. Matrix Biol. 37:167–173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Posey KL, Coustry F, Veerisetty AC, Liu P,
Alcorn JL and Hecht JT: Chondrocyte-specific pathology during
skeletal growth and therapeutics in a murine model of
pseudoachondroplasia. J Bone Miner Res. 29:1258–1268. 2014.
View Article : Google Scholar :
|
|
45
|
Hecht JT, Nelson LD, Crowder E, Wang Y,
Elder FF, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere
M, et al: Mutations in exon 17B of cartilage oligomeric matrix
protein (COMP) cause pseudoachondroplasia. Nat Genet. 10:325–329.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Paassilta P, Lohiniva J, Annunen S,
Bonaventure J, Le Merrer M, Pai L and Ala-Kokko L: COL9A3: A third
locus for multiple epiphyseal dysplasia. Am J Hum Genet.
64:1036–1044. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
47
|
Muragaki Y, Mariman EC, van Beersum SE,
Perälä M, van Mourik JB, Warman ML, Olsen BR and Hamel BC: A
mutation in the gene encoding the alpha 2 chain of the
fibril-associated collagen IX, COL9A2, causes multiple epiphyseal
dysplasia (EDM2). Nat Genet. 12:103–105. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Czarny-Ratajczak M, Lohiniva J, Rogala P,
Kozlowski K, Perälä M, Carter L, Spector TD, Kolodziej L, Seppänen
U, Glazar R, et al: A mutation in COL9A1 causes multiple epiphyseal
dysplasia: Further evidence for locus heterogeneity. Am J Hum
Genet. 69:969–980. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
49
|
Terhal PA, Nievelstein RJ, Verver EJ,
Topsakal V, van Dommelen P, Hoornaert K, Le Merrer M, Zankl A,
Simon ME, Smithson SF, et al: A study of the clinical and
radiological features in a cohort of 93 patients with a COL2A1
mutation causing spondyloepiphyseal dysplasia congenita or a
related phenotype. Am J Med Genet A. 167:461–475. 2015. View Article : Google Scholar
|
|
50
|
Majava M, Hoornaert KP, Bartholdi D, Bouma
MC, Bouman K, Carrera M, Devriendt K, Hurst J, Kitsos G, Niedrist
D, et al: A report on 10 new patients with heterozygous mutations
in the COL11A1 gene and a review of genotype-phenotype correlations
in type XI collagenopathies. Am J Med Genet A. 143A:258–264. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ho MS, Tsang KY, Lo RL, Susic M, Mäkitie
O, Chan TW, Ng VC, Sillence DO, Boot-Handford RP, Gibson G, et al:
COL10A1 nonsense and frame-shift mutations have a gain-of-function
effect on the growth plate in human and mouse metaphyseal
chondrodysplasia type Schmid. Hum Mol Genet. 16:1201–1215. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tompson SW, Merriman B, Funari VA,
Fresquet M, Lachman RS, Rimoin DL, Nelson SF, Briggs MD, Cohn DH
and Krakow D: A recessive skeletal dysplasia, SEMD aggrecan type,
results from a missense mutation affecting the C-type lectin domain
of aggrecan. Am J Hum Genet. 84:72–79. 2009. View Article : Google Scholar :
|
|
53
|
Stattin EL, Wiklund F, Lindblom K,
Onnerfjord P, Jonsson BA, Tegner Y, Sasaki T, Struglics A,
Lohmander S, Dahl N, et al: A missense mutation in the aggrecan
C-type lectin domain disrupts extracellular matrix interactions and
causes dominant familial osteochondritis dissecans. Am J Hum Genet.
86:126–137. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nilsson O, Guo MH, Dunbar N, Popovic J,
Flynn D, Jacobsen C, Lui JC, Hirschhorn JN, Baron J and Dauber A:
Short stature, accelerated bone maturation, and early growth
cessation due to heterozygous aggrecan mutations. J Clin Endocrinol
Metab. 99:E1510–E1518. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kung LH, Rajpar MH, Preziosi R, Briggs MD
and Boot-Handford RP: Increased classical endoplasmic reticulum
stress is sufficient to reduce chondrocyte proliferation rate in
the growth plate and decrease bone growth. PLoS One.
10:e01170162015. View Article : Google Scholar : PubMed/NCBI
|