Introduction
Diabetic nephropathy (DN) is the leading cause of
chronic kidney failure and end-stage renal disease worldwide, and
the prevalence has progressively increased in recent years
(1,2). DN is characterized by a decreased
glomerular filtration rate, proteinuria, mesangial expansion,
tubulointerstitial fibrosis and glomerulosclerosi (3). Hyperglycemia is the initiating
factor in the development and progression of diabetic renal injury
observed clinically in the DN (4). In certain patients,
tubulointerstitial fibrosis also appears early in diabetic kidney
injury, but it is more prominent in the later stages of the disease
and correlates closely with the decline in renal function (5,6).
Previous studies have suggested that hyperglycemia induced
epithelial-to-mesenchymal transition (EMT) of tubular cells, and is
an important mechanism of renal tubulointerstitial fibrosis in DN
(7–9). The specific therapeutic options to
inhibit the progression of chronic renal disease are not available
in the clinic. Modulation of EMT may offer a novel therapeutic
target to potentially inhibit renal fibrogenesis in the diabetic
kidney.
EMT is a highly regulated process that may require
the participation of growth factors or cytokines and integration of
multiple signal pathways, involving loss of epithelial cell
adhesion, de novo α-smooth muscle actin (α-SMA) expression
and actin reorganization, disruption of tubular basement membrane
and enhanced cell migration and invasion into the interstitium
(8,9). Previous studies have indicated that
high glucose (HG) levels induce a complex mixture of
proinflammatory and profibrotic stimuli during renal tubular
epithelial cells EMT in vivo (7), and HG can upregulate the expression
of transforming growth factor-β1 (TGF-β1), a strong inducer of the
EMT in the renal tubular epithelial cells (9,10).
In addition, HG-induced damage in DN is primarily from
mitochondrial superoxide overproduction, whose damage to proteins
is one of the major pathogenic mechanisms in numerous chronic
diseases including diabetes (11–13). HG, advanced glycation end
products, angiotensin II and TGF-β1 all increase intracellular
reactive oxygen species (ROS) and contribute to the development and
progression of diabetic renal injury (7,14).
ROS is associated with MAPK-mediated Smad activation during
HG-induced EMT in proximal tubular epithelial cells and
antioxidants effectively reversed HG-induced EMT in the renal
tubular epithelial cells and DN (6,9,15).
Zinc (Zn) is an essential element that mediates a
wide variety of physiological processes, including the enzymes
involved in cellular signaling pathways and transcription factors
(16,17). Evidence indicates that a low Zn
concentration has important implications for patients with DN
(10,11). The mechanisms of the protective
functions or function of Zn in the pathogenesis of DN, including
EMT in proximal tubular epithelial cells, vascular cell injury or
dysfunction, are not clear. Numerous studies have indicated that Zn
supplementation inhibits fibrosis, such as in myocardial, liver,
perivascular and cystic fibrosis (18–22). However, it is not known whether Zn
is involved in HG-induced EMT of the normal rat tubular epithelial
cell line NRK-52E. For this purpose, the effect of Zn was measured
on HG-induced EMT, cellular TGF-β1 and ROS production, as well as
PI-3K and MAPK activation in NRK-52E cells.
Materials and methods
Cell culture
NRK-52E cells were obtained from the American Type
Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s
modified Eagle’s medium (low glucose) (HyClone, Logan, UT, USA),
supplemented with 10% fetal calf serum (HyClone), glutamine (2 mM),
100 U/ml penicillin and 100 μg/ml streptomycin. Cells were
cultured at 37°C in a humidified atmosphere of 5% CO2 in
air and passaged twice a week. Cells were cultured at a density of
5×103 cells/well in 6-well culture plates. Near
confluent NRK-52E cells were subsequently transferred to serum-free
DMEM medium for overnight starvation prior to each experiment. In
the control groups, the NRK-52E cells were treated with serum-free
DMEM medium only. In certain other groups, the cells were
pretreated with 10 μM ZnSO4 for 24 h followed by
incubation of 30 mM HG for an addition 48 h. To deplete the
intracellular Zn stores, the Zn chelator N,N,N′,N′-tetrakis
(2-pyridylmethyl) ethylenediamine (TPEN) (1 μM) was added 12
h before the end of the 48-h incubation period with/without HG.
Assessment of cell viability
Cell viability was measured by the quantitative
colorimetric assay with
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay as described by Mossman (23) at 105 cells/ml in
96-well plates. Briefly, at the indicated time after treatment, 10
μl MTT (final concentration, 500 μg/ml) was added to
the medium and incubated at 37°C for 3 h. The MTT solution was
removed and 100 μl dimethyl sulfoxide (DMSO) was added to
dissolve the colored formazan crystals for 15 min. The absorbance
at 570 nm of each aliquot was measured using a Sunrise RC
microplate reader (Tecan Schweiz AG, Männedorf, Switzerland). Cell
viability was expressed as the ratio of the signal obtained from
treated cultures and control cultures.
Enzyme-linked immunosorbent assay
(ELISA)
The protein level of TGF-β1 was measured by a TGF-β1
ELISA kit (R&D Systems, Minneapolis, MN, USA). Briefly, the
NRK-52E cells were seeded at a density of 3×105
cells/well in a 12-well plate and cultured for 24 h. The cells were
subsequently treated as previously described. The supernatants were
collected from cultures of NRK-52E cells for ELISA testing.
Secreted TGF-β1 protein concentration per 105 cells was
measured and calculated from the standard curve by an ELISA kit.
Briefly, 100 μl samples were added and incubated for 1 h
with a plate shaker following washing with the washing buffer. An
enzyme-conjugated secondary antibody was added to the wells and was
incubated for 2 h before the substrate was added, and the reading
was assessed with an absorbance ELISA reader at the 450 nm
wavelength. All the procedures were performed at room
temperature.
Detection of intracellular ROS level
The ROS assay experiments were performed using the
reactive oxygen species assay kit (Beyotime Institute of
Biotechnology, Haimen, China) according to the manufacturer’s
instructions. Briefly, NRK-52E cells were treated as previously
described in the section of cells culture. Subsequently, cells
(5×106) were incubated with 10 μmol/l
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) probes at 37°C
for 30 min and washed with phosphate-buffered saline (PBS) 3 times
in order to remove the residual probes. DCFH-DA was deacetylated
intracellularly by non-specific esterase, which was further
oxidized by ROS to the fluorescent compound 2,7-dichlorofluorescein
(DCF). DCF fluorescence was detected by flow cytometer
(Becton-Dickinson, San Jose, CA, USA). The results were analyzed by
the CellQuest software (Becton-Dickinson).
Western blot analysis
Cells were pelleted by centrifugation at 125 g at
4°C for 10 min and subsequently washed with ice-cold PBS. Cells
were lysed using the radioimmunoprecipitation assay buffer and
phenylmethanesulfonylfluoride mixture (1:100), on ice for 30 min
with occasional vortexing. Lysed cells were sonicated and
centrifuged at 8,000 x g at 4°C for 5 min. The total protein
concentration measurement was performed with the Bradford method
(12). Protein samples were
boiled for 5 min and 50 μg of total protein was loaded on
the appropriate SDS-PAGE gel. The proteins on the gel were
subsequently transferred to a polyvinylidene fluoride membrane
using a Bio-Rad apparatus (Bio-Rad Laboratories, Hercules, CA, USA)
for 2 h at 4°C using 100 V. The protein-bound membrane was blocked
in 5% milk in tris-buffered saline (TBS) (containing 0.5% Tween-20)
at room temperature for 1 h and subsequently incubated with primary
antibodies. The primary antibodies used included rabbit polyclonal
anti-vimentin (1:400, sc-5565; Santa Cruz Biotechnology, Dallas,
TX, USA), mouse monoclonal anti-α-SMA (1:1,000, sc-324317; Sigma,
St. Louis, MO, USA), mouse monoclonal anti-β-actin (1:4,000,
sc-8432; Sigma), rabbit monoclonal anti-E-cadherin (1:1,000,
sc-7870; BD Biosciences, San Jose, CA, USA), rabbit monoclonal
anti-Akt (1:400, SAB4500797; Sigma), anti-phospho-Akt (1:400,
SAB4503853; Sigma), rabbit polyclonal anti-c-Jun N-terminal kinase
(JNK) (1:1,000, SAB4502398; Sigma), rabbit polyclonal anti-phospho
JNK (1:1,000, SAB4504449; Sigma) rabbit polyclonal anti-p38
(1:1,000, M0800; Zymed Laboratories, San Francisco, CA, USA),
rabbit polyclonal anti-phospho p38 (1:1,000, SAB4301534; Zymed
Laboratories), rabbit polyclonal
anti-extracellular-signal-regulated kinase (ERK)1/2 (1:800, M5670;
Sigma) and rabbit polyclonal anti-phospho ERK1/2 (1:800, E7028;
Sigma). Following completion of the primary antibody staining, the
membranes were washed several times with TBS/0.1% Tween-20, which
was followed by incubation with horseradish peroxidase-conjugated
secondary antibodies overnight at 4°C. The membrane was
subsequently developed with an enhanced chemiluminescence kit
(Walterson Biotechnology, Inc., Beijing, China) and the images were
captured with UVP (G:BOX EF, Chemi HR16; Syngene, Frederick, MD,
USA). The protein bands were quantified using the NIH ImageJ
version 1.44 densitometry software.
Statistical analyses
Data are expressed as the means ± standard error of
the mean. Variance was homogenous for use of standard analysis of
variance (ANOVA) methodology. Subsequent to establishing the
statistical significance by ANOVA, individual comparisons were
performed using the Tukey’s multiple comparison test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of Zn on the expression of
HG-induced EMT in NRK-52E cells
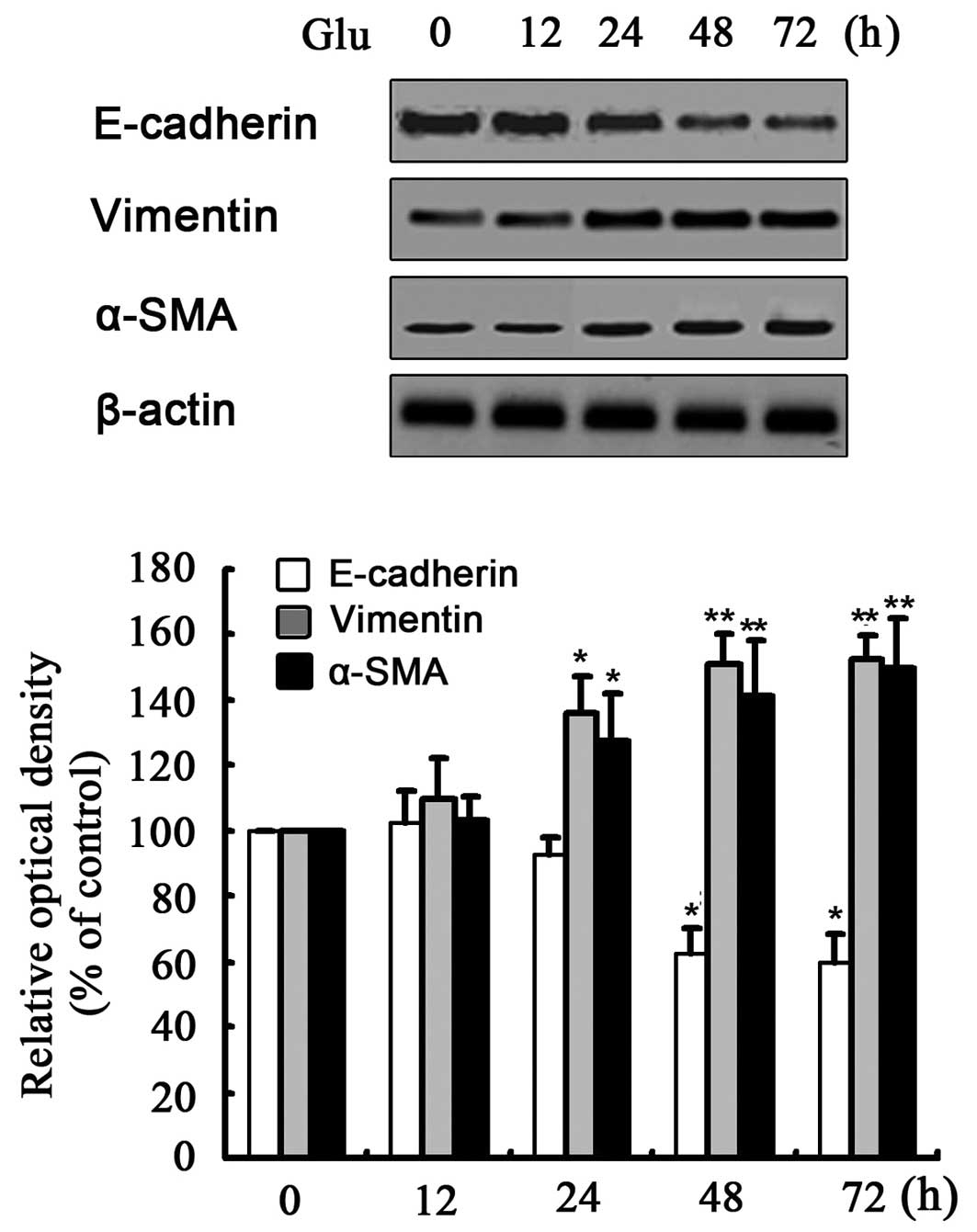
First, exposure of the NRK-52E cells to HG (30 mM
D-glucose) for 24–72 h decreased protein expression of E-cadherin
and increased the expression of α-SMA and vimentin (Fig. 1). Mannitol or L-glucose (30 mM)
did not change the expression of any of these markers, which
suggested that it was not the high osmolarity, but HG that induced
EMT in the NRK-52E cells (data not shown). Subsequently, the
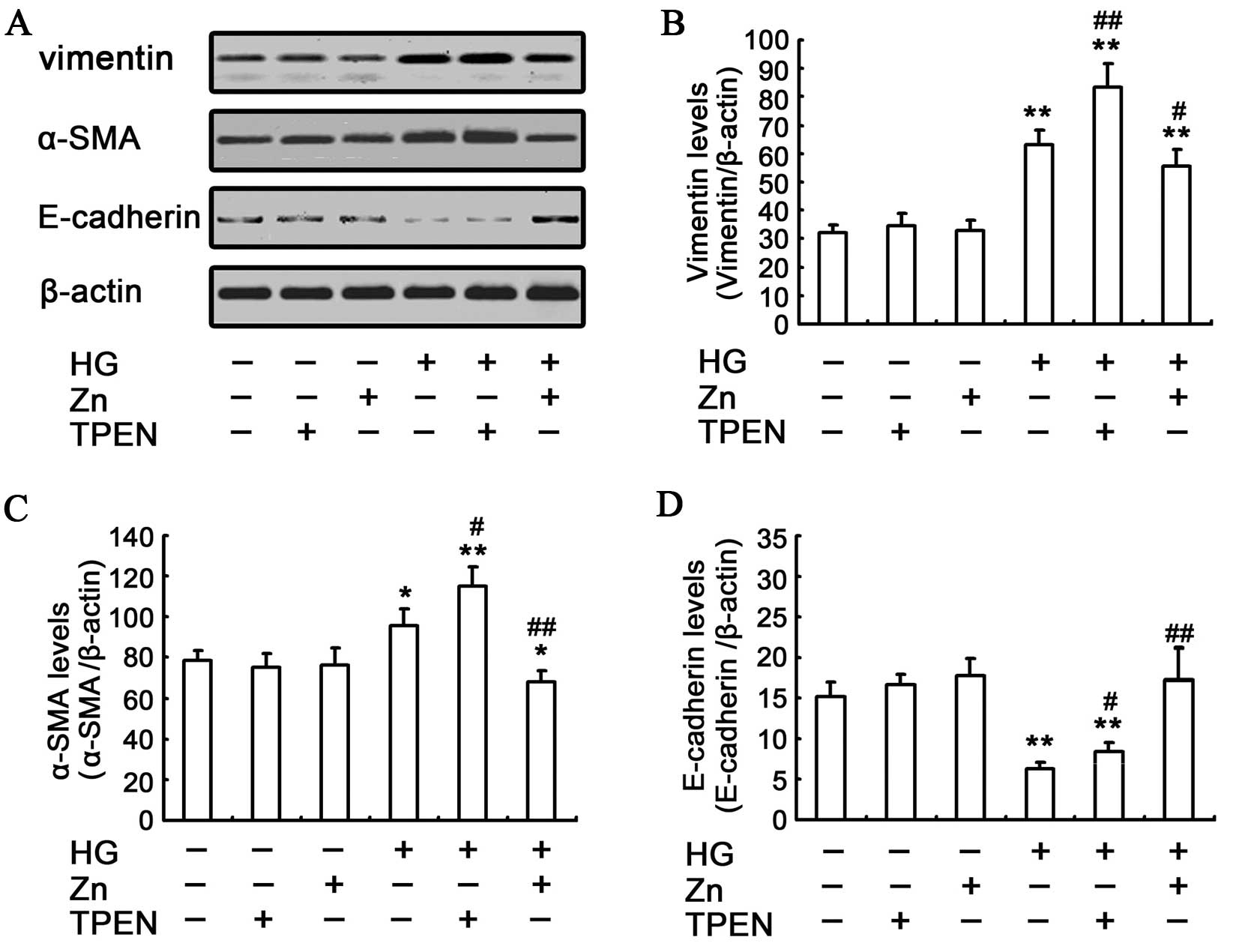
effects of Zn on HG-induced EMT of NRK-52E cells were assessed by
western blotting. The HG-induced EMT can be attenuated by
pre-treating the NRK-52E cells with 10 μM ZnSO4,
which was evidenced by the reduced upregulation of α-SMA and
vimentin, and the ameliorated expression of E-cadherin (Fig. 2). These results showed that the
physiologically optimal levels of Zn supplementation can reverse
HG-induced EMT in NRK-52E cells.
Effect of Zn on TGF-β1 expression in the
HG-treated NRK-52E cells
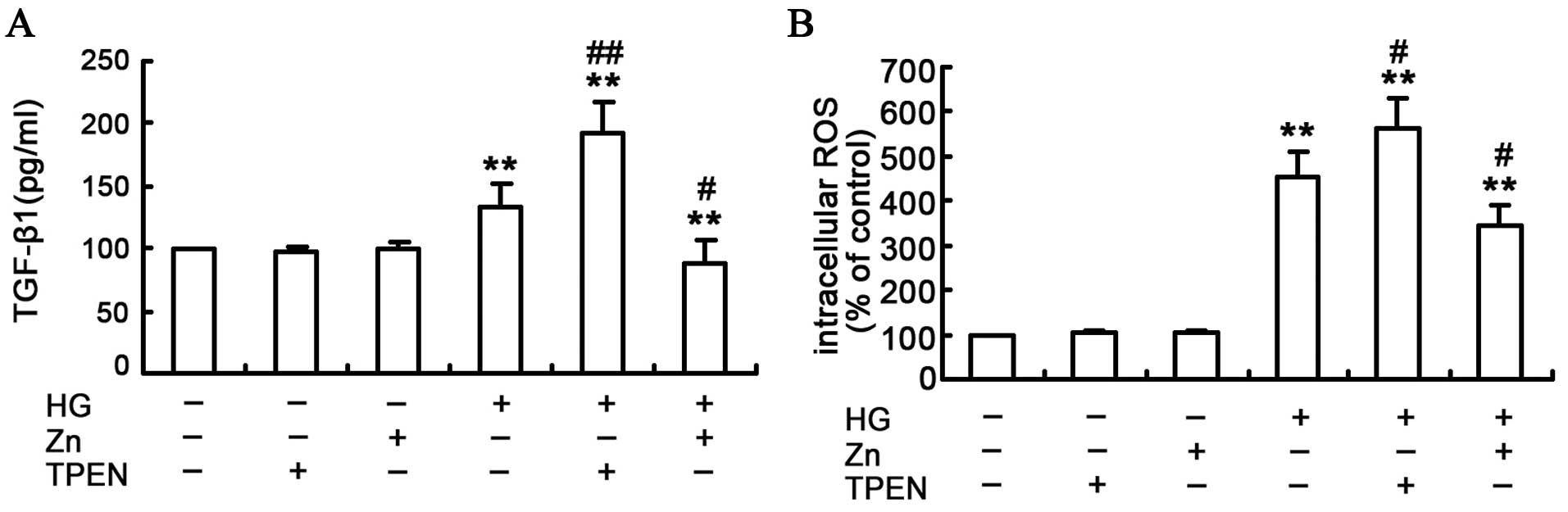
To assess the effect of Zn on the TGF-β1 expression
in the HG-treated NRK-52E cells, TGF-β1 protein was measured by the
ELISA assay (Fig. 3A). In
contrast to the control group, the TGF-β1 expression was
significantly higher in the HG-treated group, and the TGF-β1
expression was further enhanced in the TPEN/HG group. Conversely,
ZnSO4 treatment reduced the HG-induced TGF-β1 expression
in the NRK-52E cells. Furthermore, Zn or TPEN alone did not
significantly alter the TGF-β1 expression. Considering the above
findings, the present study indicates that Zn can attenuate
HG-induced TGF-β1 expression in the NRK-52E cells.
Effect of Zn on ROS production in the
HG-treated NRK-52E cells
ROS is the initial and primary event that
subsequently activates a number of other pathways implicated in the
development of EMT in renal tubular epithelial cells (24–26). Therefore, the effect of Zn was
examined on the HG-induced ROS induction in the NRK-52E cells by
measuring the intracellular ROS with DCF-DA staining. The result
indicated that depletion of Zn with TPEN, in conjunction with HG
treatment, resulted in a substantial increase of ROS production in
the NRK-52E cells (Fig. 3B). By
contrast, Zn pre-treatment significantly attenuated HG-induced ROS
production in the NRK-52E cells.
Effect of Zn supplementation on the
HG-induced PI3K/Akt signaling pathway
The PI3K signaling pathway is involved in EMT in the
NRK-52E cells (27,28). Having shown that Zn inhibited EMT,
whether Zn mediated its effects on EMT in the NRK-52E cells through
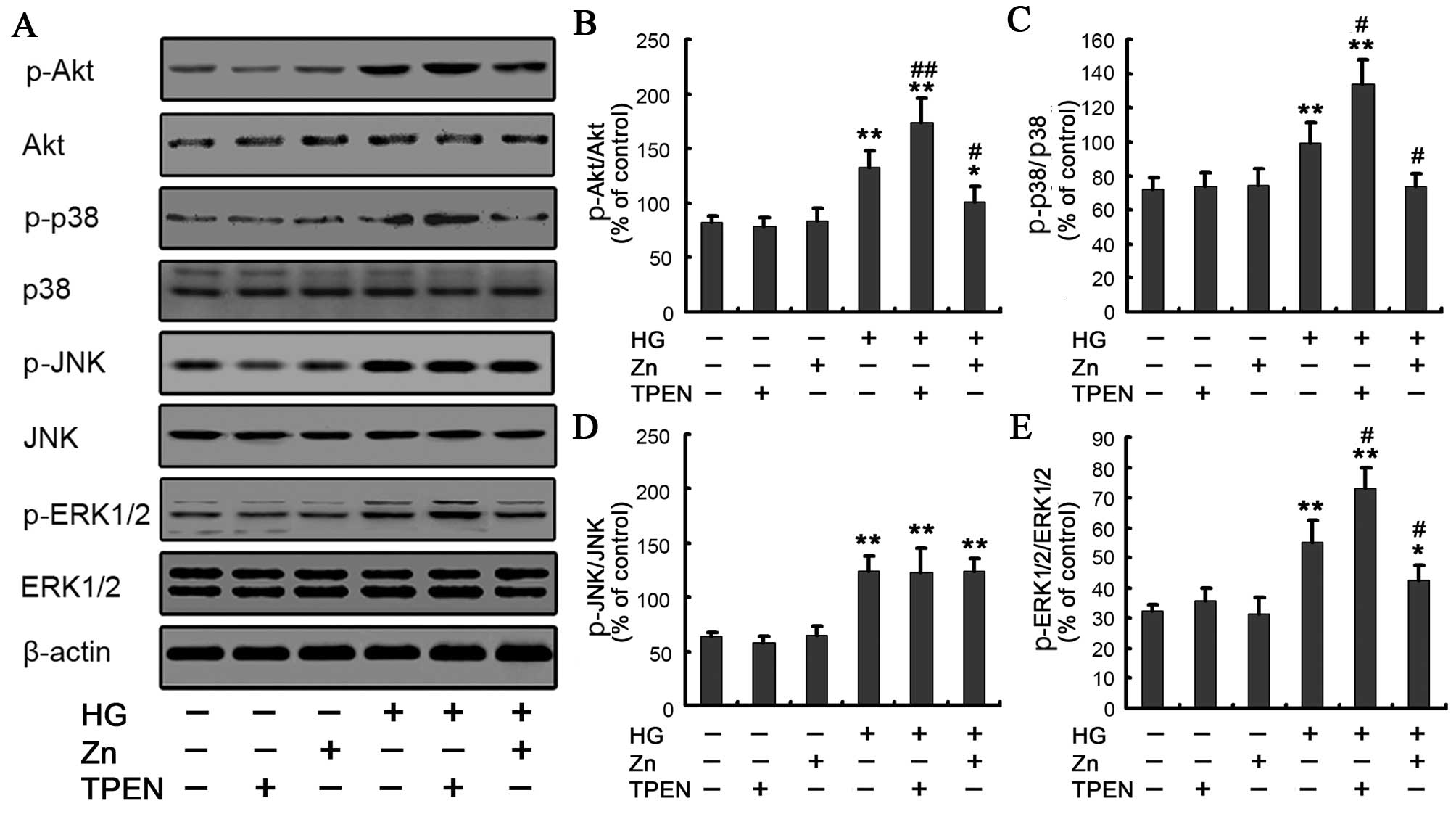
this pathway was determined under HG conditions by western
blotting. When the cells were exposed to HG for 48 h, Akt
phosphorylation increased compared to the control, whereas 10
μM ZnSO4 treatment significantly decreased the
expression of Akt phosphorylation (Fig. 4A and B). To further examine the
effect of Zn on HG-induced EMT, the NRK-52E cells were incubated
with or without 10 μM LY294002 [an inhibitor of upstream
enzyme PI3K, the concentration of LY294002 is from reference
(22)] for 1 h and were
subsequently exposed to 30 mM HG in the presence or absence of 10
μM ZnSO4 pretreatment for 24 h. The expected
results showed that the HG/Zn or HG/LY294002 group decreased the
expression of Akt phosphorylation and HG-induced EMT in the NRK-52E
cells (Fig. 5A). There was no
significant difference in the HG/Zn versus HG/LY294002 group. Taken
together, these results indicated that the regulation mechanism of
HG-induced EMT by Zn may be through abrogation of HG-induced
PI3K/Akt activation in the renal tubular epithelial cells.
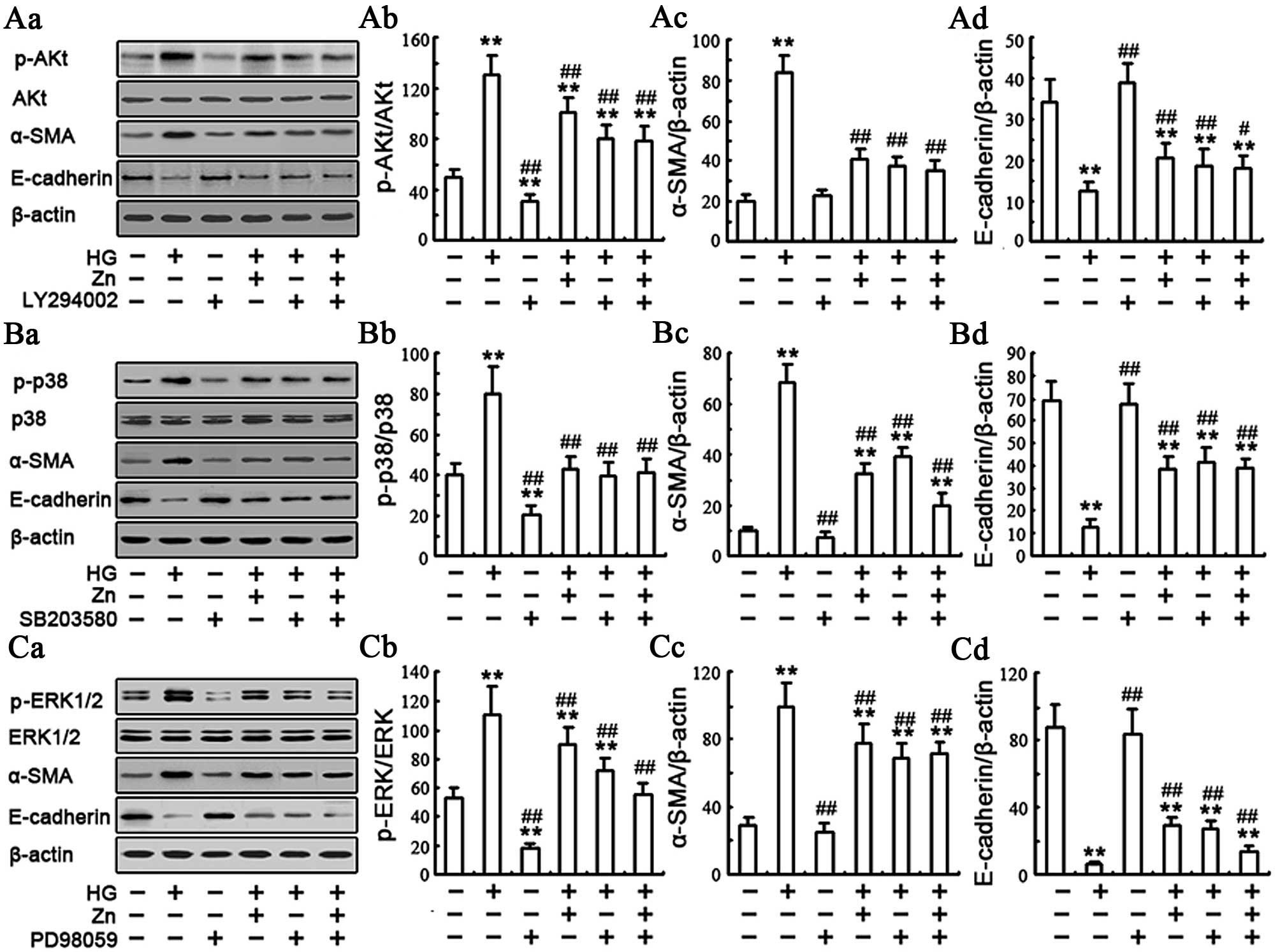 | Figure 5Zinc (Zn) inhibits the
phosphatidylinositol 3-kinase/Akt (PI3K/Akt) pathway and
mitogen-activated protein kinase (MAPK) protecting cells from
epithelial-to-mesenchymal transition (EMT). Cells were incubated
with or without 10 μM LY294002 for 1 h, 2 μM SB203580
for 1 h, 10 μM PD98059 for 1 h, respectively, and were
subsequently exposed to 30 mM high glucose (HG) for 48 h in the
presence or absence of 30 μM ZnSO4 pretreatment
for 24 h. (Aa–Ca) The expression of EMT proteins, total-Akt and
phospho (p)-Akt, total-p38 and p-p38, total-ERK and p-ERK were
assessed by western blot analysis. β-actin served as the loading
control. (Ab-d, Bb-d and Cb-d) Quantitative analysis was performed
by measuring the fluorescence intensity relative to the control.
Each value represents the mean ± standard error of the mean (n=10).
All the results were obtained from three independent experiments.
(**P<0.01, *P<0.05 vs. control;
##P<0.01, #P<0.05 vs. HG.) |
Effect of Zn on HG-induced MAPK signaling
pathway
The MAPK signaling pathway has been reported to be
involved in the EMT (24,29–31), but whether Zn executes its effect
on the EMT in the renal tubular epithelial cells through this
pathway remains unknown. Therefore, the effect of Zn
supplementation on the MAPK pathway, including JNK, p38 MAPK and
ERK, was examined in the NRK-52E cells. The activation of the JNK,
p38 MAPK and ERK pathways was analyzed by western blot analysis
with phospho-p38, phospho-JNK and phospho-ERK antibodies. Compared
to the control group, the phospho-p38, phospho-JNK and phospho-ERK
in the HG group increased to varying degrees (Fig. 4A and C–E). The data are consistent
with these earlier observations and provide a novel molecular
signaling mechanism in which the MAPK pathway mediates HG-induced
EMT in renal tubular epithelial cells (24). Of note, the TPEN/HG group, which
depleted Zn with TPEN in the HG-treated NRK-52E cells, showed a
robust increase of the phospho-p38, and phospho-ERK in comparison
with the HG group. Conversely, preincubation of the NRK-52E cells
with 10 μM ZnSO4 significantly inhibited
HG-induced expression of phospho-p38 MAPK and phospho-ERK. There
was no significant difference of the expression of phospho-JNK in
the HG/Zn versus HG group (Fig. 4A
and D). All these results suggested that Zn may be involved in
HG-induced EMT through regulation of the p38 MAPK and ERK pathways.
To further examine the involvement of the p38 MAPK and ERK pathways
in HG-induced EMT, the cells were incubated with or without 2
μM of p38 MAPK inhibitor SB203580 [concentration is from
(30)] or 10 μM of the ERK
inhibitor PD98059 for 1 h [concentration is from (22)], respectively, and subsequently
exposed to 30 mM of HG in the presence or absence of 10 μM
ZnSO4 pretreatment for 24 h. As shown in Fig. 5, HG evidently upregulated the
expression of α-SMA and downregulated the expression of E-cadherin.
As expected, when compared to the cells treated with HG alone,
co-treatment of 10 μM ZnSO4, SB203580 or PD98059
with HG significantly decreased the expression of α-SMA and
ameliorated the expression of epithelial protein E-cadherin to
varying degrees (Fig. 5B and C).
Furthermore, similar to SB203580 or PD98059, Zn treatment decreased
the HG-induced EMT and effectively inhibited p38 and ERK
phosphorylation (Fig. 5B and C).
Collectively, these results suggested that Zn protected the cells
from HG-induced EMT possibly through abrogation of HG-induced p38
MAPK and ERK activation.
Discussion
Several studies in animal models and few clinical
studies have demonstrated that Zn supplementation has a positive
effect of inhibiting fibrosis in chronic inflammatory diseases,
such as in liver, myocardial and cystic fibrosis (18,21,32). Conversely, previous studies have
indicated that Zn deficiency can accelerate the degradation of
E-cadherin and β-catenin proteins in lung and endothelial
epithelial cells and lead to damage of membrane barrier integrity
(33,34). The results in the present study
demonstrate that Zn pre-treatment provides effective protection
against HG-induced EMT in the renal tubular epithelial cells, as
evidenced by a decrease in upregulation of vimentin and α-SMA and
amelioration of E-cadherin associated with a transition in the
epithelial phenotype of the NRK-52E cells to a myofibroblastic
phenotype. The mechanism may be through abrogation of HG-induced
oxidative stress and PI3K/Akt, and MAPK (p38 MAPK and ERK)
activation in the NRK-52E cells. These results are the first to
demonstrate that the physiologically optimal levels of Zn inhibit
HG-induced EMT in the renal tubular epithelial cells.
TGF-β1, a strong profibrotic cytokine, as well as
the TGF-β/Smad pathway were extensively studied for the EMT in
previous years (14,15,35). TGF-β1 plays an important role in
changing the phenotype of renal epithelial cells, actions that
significantly contribute to the profibrotic actions of this growth
factor (35,36). In addition, there is sufficient
evidence that TGF-β1 signals through MAPKs and the activation of
p38 MAPK is required in TGF-β1-induced EMT in human proximal
tubular epithelial cells (7). A
previous study indicated that a Zn deficiency resulted in the
TGF-β1 induction in neurogenesis to regulate neuronal precursor
cell proliferation and survival by regulating the p53-dependent
molecular mechanism (37).
Another study demonstrated that TGF-β1 has stimulating and
inhibiting effects on osteoclast-like cell formation in mouse
marrow culture, and that Zn can inhibit the stimulatory effect of
TGF-β1 (38). Zn supplementation
decreases ethanol- and acetaldehyde-induced liver stellate cell
activation partly by inhibiting Smad signaling (39). In the present study, the
physiologically optimal levels of Zn supplementation were confirmed
to reduce the HG-induced TGF-β1 production from the NRK-52E
cells.
The role of Zn in modulating oxidative stress has
previously been recognized and Zn deficiency enhanced diabetic
renal damage, which is associated with oxidative stress (40). Previous evidence has demonstrated
that Zn deficiency can trigger oxidative stress and
oxidant-mediated damage to cell components, alterations of cell
functions and cell proliferation (41,42). HG, advanced glycation end
products, angiotensin II and TGF-β1 all increase ROS and contribute
to the development and progression of diabetic renal injury
(7,14). Numerous studies have confirmed
that EMT of tubular epithelial cells in DN patients is generally
regarded to be the result of hyperglycemia-induced oxidative
stress, as antioxidants effectively reverse the EMT in all tubular
epithelial cells (6,9,13,43). In the present study, Zn treatment
attenuated HG-induced ROS generation, whereas Zn depletion
increased HG-induced ROS generation, suggesting that
physiologically optimal levels of Zn inhibit the HG-induced EMT
possibly through abrogation of HG-induced oxidative stress in
NRK-52E cells.
The mechanisms by which HG and its metabolite
regulate E-cadherin, vimentin and α-SMA gene expression as
markers of EMT in the NRK-52E cells have not been completely
elucidated. Several studies have reported that the MAPK and PI-3K
pathways are involved in the pathology of various forms of kidney
injury, including renal fibrosis (8,24,27,28). Phosphorylation of Akt is
associated with a loss of cell-cell adhesion, a decrease in
cell-matrix adhesion, and induction of cell motility and other
characteristics of myofibroblasts (27,44). Furthermore, inhibition of PI3K/Akt
activity causes a decrease in GSK-3β phosphorylation attenuated
TGF-β1-mediated EMT in rat kidney epithelial cells (9,45).
A previous study demonstrated that cyclosporin A activated JNK
signaling in human renal epithelial cells and that JNK inhibition
reduced the cyclosporin A-induced E-cadherin downregulation, cell
migration and Snail-1 expression (46). The p38 MAPK activation is a key
modulator in the progression of renal diseases and is thought to
occur in HG-induced cell damage in renal tubular epithelial cells
(8). Elevated ERK activity can
enhance TGF-β1-mediated EMT in rat kidney epithelial cells, and ERK
inhibition reduces the induced EMT (24). In PI3K-inhibited NRK-52E cells,
the direct association between Akt and EMT was further confirmed.
Phosphorylation of Akt increased in HG-treated NRK-52E cells and Zn
supplementation decreased its level. The HG-mediated Akt
activation, the reduction in E-cadherin and the upregulation of
vimentin and α-SMA were reversed by a PI3K inhibitor, with no
significance between with the effect of Zn, which is consistent
with the results of the effect of Zn on HG-induced phosphorylation
of ERK and p38 MAPK in NRK-52E cells. The results provide a novel
molecular signaling mechanism in which Zn mediates HG-induced EMT
possibly through abrogation of HG-induced PI3K/Akt, ERK and p38
MAPK activation in the renal tubular epithelial cells.
In conclusion, the present study provides new
evidence regarding the association between Zn and EMT in NRK-52E
cells. The results reveal that the physiologically optimal levels
of Zn inhibit HG-induced EMT, most likely through inhibition of
ROS, TGF-β1 production, and PI3K/Akt, ERK and p38 MAPK signaling
pathways in NRK-52E cells. Given the important role that EMT plays
in the development and progression of interstitial fibrosis, the
identification of Zn as a key regulator of HG-induced EMT
represents an important finding. Further studies may confirm it as
a potentially important target for therapeutic intervention in an
attempt to limit EMT and with it the decline in renal function
observed in patients with DN.
Acknowledgments
The present study was supported by the National
Grand Fundamental Research 973 Program of China (grant no.
2012CB722405), the Natural Science Foundation of China (grant nos.
81170561, 81370517, 31171259 and 31271364), and the Shen Yang City
Science and Technology Program (grant no. F11-264-1-21).
Abbreviations:
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
HG
|
high glucose
|
|
MAPK
|
mitogen-activated protein kinase
|
|
JNK
|
jun N-terminal kinase
|
|
ERK
|
extracellular-signal-regulated
kinase
|
|
MTT
|
3-(4,5-dimethylthiazol-2-y)-2,5-diphenyltetrazolium bromide
|
|
DMEM
|
Dulbecco’s modified Eagle’s medium
|
|
DMSO
|
dimethyl sulfoxide
|
|
BSA
|
bovine serum albumin
|
|
SMA
|
smooth muscle cell actin
|
|
TPEN
|
N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine
|
|
FITC
|
fluorescein isothiocyanate
|
|
TGF
|
transforming growth factor
|
|
PBS
|
phosphate-buffered saline
|
|
DCF-DA
|
2,7-dichlorofluorescein diacetate
|
|
TBS
|
tris-buffered saline
|
References
|
1
|
Schena FP and Gesualdo L: Pathogenetic
mechanisms of diabetic nephropathy. J Am Soc Nephrol. 16(Suppl 1):
S30–S33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lapice E, Pinelli M, Riccardi G, et al:
Pro12Ala polymorphism in the PPARG gene contributes to the
development of diabetic nephropathy in Chinese type 2 diabetic
patients: comment on the study by Liu et al. Diabetes Care.
33:e1142010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ayodele OE, Alebiosu CO and Salako BL:
Diabetic nephropathy - a review of the natural history, burden,
risk factors and treatment. J Natl Med Assoc. 96:1445–1454.
2004.PubMed/NCBI
|
|
4
|
Yeh CH, Chang CK, Cheng KC, et al: Role of
bone morphogenetic proteins-7 (BMP-7) in the renal improvement
effect of DangGui (Angelica sinensis) in type-1 diabetic rats. Evid
Based Complement Alternat Med. 2011:7967232011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gilbert RE and Cooper ME: The
tubulointerstitium in progressive diabetic kidney disease: more
than an aftermath of glomerular injury? Kidney Int. 56:1627–1637.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Simonson MS: Phenotypic transitions and
fibrosis in diabetic nephropathy. Kidney Int. 71:846–854. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Burns WC, Twigg SM, Forbes JM, et al:
Connective tissue growth factor plays an important role in advanced
glycation end product-induced tubular epithelial-to-mesenchymal
transition: implications for diabetic renal disease. J Am Soc
Nephrol. 17:2484–2494. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lv ZM, Wang Q, Wan Q, et al: The role of
the p38 MAPK signaling pathway in high glucose-induced
epithelial-mesenchymal transition of cultured human renal tubular
epithelial cells. PLoS One. 6:e228062011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee YJ and Han HJ: Troglitazone
ameliorates high glucose-induced EMT and dysfunction of SGLTs
through PI3K/Akt, GSK-3beta, Snail1, and beta-catenin in renal
proximal tubule cells. Am J Physiol Renal Physiol. 298:F1263–F1275.
2009. View Article : Google Scholar
|
|
10
|
Karatug A, Kaptan E, Bolkent S, et al:
Alterations in kidney tissue following zinc supplementation to
STZ-induced diabetic rats. J Trace Elem Med Biol. 27:52–57. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dogukan A, Sahin N, Tuzcu M, et al: The
effects of chromium histidinate on mineral status of serum and
tissue in fat-fed and streptozotocin-treated type II diabetic rats.
Biol Trace Elem Res. 131:124–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Simonian MH and Smith JA:
Spectrophotometric and colori-metric determination of protein
concentration. Curr Protoc Mol Biol. Chapter 10: Unit 10.1A. 2006.
View Article : Google Scholar
|
|
13
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeisberg M and Kalluri R: The role of
epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med
(Berl). 82:175–181. 2004. View Article : Google Scholar
|
|
15
|
Sato M, Muragaki Y, Saika S, et al:
Targeted disruption of TGF-beta1/Smad3 signaling protects against
renal tubulointerstitial fibrosis induced by unilateral ureteral
obstruction. J Clin Invest. 112:1486–1494. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hills CE and Squires PE: TGF-beta1-induced
epithelial-to-mesenchymal transition and therapeutic intervention
in diabetic nephropathy. Am J Nephrol. 31:68–74. 2010. View Article : Google Scholar
|
|
17
|
Hills CE and Brunskill NJ: Intracellular
signalling by C-peptide. Exp Diabetes Res. 2008:6351582008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Takahashi M, Saito H, Higashimoto M, et
al: Possible inhibitory effect of oral zinc supplementation on
hepatic fibrosis through downregulation of TIMP-1: a pilot study.
Hepatol Res. 37:405–409. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang L, Zhou Z, Saari JT, et al:
Alcohol-induced myocardial fibrosis in metallothioneinnull mice:
prevention by zinc supplementation. Am J Pathol. 167:337–344. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gandhi MS, Deshmukh PA, Kamalov G, et al:
Causes and consequences of zinc dyshomeostasis in rats with chronic
aldosteronism. J Cardiovasc Pharmacol. 52:245–252. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Van Biervliet S, Vande Velde S, Van
Biervliet JP, et al: The effect of zinc supplements in cystic
fibrosis patients. Ann Nutr Metab. 52:152–156. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang X, Liang D, Guo B, et al: Zinc
inhibits high glucose-induced apoptosis in peritoneal mesothelial
cells. Biol Trace Elem Res. 150:424–432. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mossman BT: In vitro approaches for
determining mechanisms of toxicity and carcinogenicity by asbestos
in the gastrointestinal and respiratory tracts. Environ Health
Perspect. 53:155–161. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rhyu DY, Yang Y, Ha H, et al: Role of
reactive oxygen species in TGF-beta1-induced mitogen-activated
protein kinase activation and epithelial-mesenchymal transition in
renal tubular epithelial cells. J Am Soc Nephrol. 16:667–675. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang J and Liu Y: Dissection of key events
in tubular epithelial to myofibroblast transition and its
implications in renal interstitial fibrosis. Am J Pathol.
159:1465–1475. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ha H and Lee HB: Reactive oxygen species
and matrix remodeling in diabetic kidney. J Am Soc Nephrol.
14:S246–S249. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zeng R, Yao Y, Han M, et al: Biliverdin
reductase mediates hypoxia-induced EMT via PI3-kinase and Akt. J Am
Soc Nephrol. 19:380–387. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boca M, D’Amato L, Distefano G, et al:
Polycystin-1 induces cell migration by regulating
phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements
and GSK3beta-dependent cell cell mechanical adhesion. Mol Biol
Cell. 18:4050–4061. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu Q, Mao H, Nie J, et al: Transforming
growth factor {beta}1 induces epithelial-mesenchymal transition by
activating the JNK-Smad3 pathway in rat peritoneal mesothelial
cells. Perit Dial Int. 28(Suppl 3): S88–S95. 2008.PubMed/NCBI
|
|
30
|
Yang F, Chung AC, Huang XR, et al:
Angiotensin II induces connective tissue growth factor and collagen
I expression via transforming growth factor-beta-dependent and
-independent Smad pathways: the role of Smad3. Hypertension.
54:877–884. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yamashita M, Fatyol K, Jin C, et al: TRAF6
mediates Smad-independent activation of JNK and p38 by TGF-beta.
Mol Cell. 31:918–924. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
von Bulow V, Dubben S, Engelhardt G, et
al: Zinc-dependent suppression of TNF-alpha production is mediated
by protein kinase A-induced inhibition of Raf-1, I kappa B kinase
beta, and NF-kappa B. J Immunol. 179:4180–4186. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bao S and Knoell DL: Zinc modulates
cytokine-induced lung epithelial cell barrier permeability. Am J
Physiol Lung Cell Mol Physiol. 291:L1132–L1141. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mengheri E, Nobili F, Vignolini F, et al:
Bifidobacterium animalis protects intestine from damage induced by
zinc deficiency in rats. J Nutr. 129:2251–2257. 1999.PubMed/NCBI
|
|
35
|
Wang X, Pan X and Song J: AMP-activated
protein kinase is required for induction of apoptosis and
epithelial-to-mesenchymal transition. Cell Signal. 22:1790–1797.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lan HY: Tubular epithelial-myofibroblast
transdifferentiation mechanisms in proximal tubule cells. Curr Opin
Nephrol Hypertens. 12:25–29. 2003. View Article : Google Scholar
|
|
37
|
Corniola RS, Tassabehji NM, Hare J, et al:
Zinc deficiency impairs neuronal precursor cell proliferation and
induces apoptosis via p53-mediated mechanisms. Brain Res.
1237:52–61. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamaguchi M and Kishi S: Differential
effects of transforming growth factor-beta on osteoclast-like cell
formation in mouse marrow culture: relation to the effect of
zinc-chelating dipeptides. Peptides. 16:1483–1488. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Szuster-Ciesielska A, Plewka K, Daniluk J,
et al: Zinc supplementation attenuates ethanol- and
acetaldehyde-induced liver stellate cell activation by inhibiting
reactive oxygen species (ROS) production and by influencing
intracellular signaling. Biochem Pharmacol. 78:301–314. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Prasad AS: Clinical, immunological,
anti-inflammatory and antioxidant roles of zinc. Exp Gerontol.
43:370–377. 2008. View Article : Google Scholar
|
|
41
|
Ho E and Ames BN: Low intracellular zinc
induces oxidative DNA damage, disrupts p53, NFkappa B, and AP1 DNA
binding, and affects DNA repair in a rat glioma cell line. Proc
Natl Acad Sci USA. 99:16770–16775. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang X, Liang D, Guo B, et al: Zinc
transporter 5 and zinc transporter 7 induced by high glucose
protects peritoneal mesothelial cells from undergoing apoptosis.
Cell Signal. 25:999–1010. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kosugi T and Sato W: Midkine and the
kidney: health and diseases. Nephrol Dial Transplant. 27:16–21.
2012. View Article : Google Scholar
|
|
44
|
Agarwal E, Brattain MG and Chowdhury S:
Cell survival and metastasis regulation by Akt signaling in
colorectal cancer. Cell Signal. 25:1711–1719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kattla JJ, Carew RM, Heljic M, et al:
Protein kinase B/Akt activity is involved in renal TGF-beta1-driven
epithelial-mesenchymal transition in vitro and in vivo. Am J
Physiol Renal Physiol. 295:215–225. 2008. View Article : Google Scholar
|
|
46
|
Pallet N, Thervet E and Anglicheau D:
c-Jun-N-terminal kinase signaling is involved in
cyclosporine-induced epithelial phenotypic changes. J Transplant.
2012:3486042012.
|