Introduction
Epithelial-mesenchymal transition (EMT) is
characterized by the loss of epithelial morphology and the
acquisition of mesenchymal characteristics (1). EMT occurs during embryonic
development and is required for the formation of tissues and
organs. It also plays an important role in pathological processes,
such as organ fibrosis and cancer (2,3).
The activation of EMT in cancer cells may lead to tumor migration,
invasion and dissemination, which are cytological elements that
cause cancer metastasis (4).
Transforming growth factor-β1 (TGF-β1) is one of the
main EMT-inducing factors in both physiological and pathological
conditions (5), by activating
EMT-associated transcriptional regulators, such as Snail, Twist and
Zinc finger E-box binding homeobox 1 (ZEB1) (6). TGF-β1 exerts profound effects on the
growth and EMT-like responses of thyroid epithelial cells, oral
squamous cell carcinoma cells and pancreatic cancer cells (7–9).
The ability of TGF-β1 to induce EMT plays a critical role in the
acquisition of a migratory and invasive phenotype that correlates
with an enhanced metastatic potential in tumor cells (26). Epidermal growth factor (EGF) has
been shown to disrupt cell-cell junctions and induce EMT in a
number of cell lines (10–12).
EGF stimulates signaling pathways through the EGF receptor (EGFR),
which is associated with a more invasive behavior and a poor
prognosis (13,14). TGF-β1 and EGF have been implicated
in the process of EMT, and their corresponding intracellular
transduction pathways have been reported to form highly
interconnected networks; however, the mechanisms involved has not
been determined yet (15,16).
Hyaluronan (HA) is an ubiquitous component of the
pericellular matrix. Through its interaction with the cell surface
receptor, CD44, HA plays key regulatory roles in tissue homeostasis
and cancer progression (17).
Three isoforms of hyaluronan synthases (HAS) namely HAS1, HAS2 and
HAS3, have been identified thus far (18). CD44, an adhesion molecule that
binds to HA, has been implicated in cancer cell migration, invasion
and metastasis (19). CD44 is
composed of a common domain and a variable region of alternatively
spliced exons (20). The common
domain induces an extracellular region that interacts with its
extracellular matrix ligand, HA. CD44 is able to modulate
intracellular signaling through the formation of co-receptor
complexes with various receptor tyrosine kinases (21). Indeed, increasing evidence points
to a role for CD44 as a critical mediator of both growth factor-
and HA-induced invasive signaling in cancer cells (22,23).
In the present study, we aimed to investigate the
mechanisms through which TGF-β1 induces the EMT process by the
transactivation of EGF signaling. The HA-CD44-mediated TGF-β1 and
EGF signaling, and the co-localization of CD44/EGFR had an effect
on the activation of EGF signaling induced by TGF-β1 in lung and
breast cancer cells.
Materials and methods
Cell culture and reagents
The MCF-7 breast cancer cell line and the A549 lung
adenocarcinoma cell line were obtained from the Type Culture
Collection of the Chinese Academy of Sciences, Shanghai, China. The
cells were cultured in RPMI-1640 medium supplemented with 10% fetal
calf serum (HyClone, Logan, UT, USA) and 100 U/ml of penicillin and
100 mg/l of streptomycin at 37°C in a 5% CO2 humidified
atmosphere. Logarithmic phase cells were used in the experiments.
In some cases, 10% fetal calf serum was changed to serum-free
medium depending on the experiment. The cells were treated with EGF
at 10 ng/ml or TGF-β1 at 5 ng/ml for 24 h. EGF (AF-100-15) and
TGF-β1 (AF-100-18B) were purchased from PeproTech (Rocky Hill, NJ,
USA). 4-Methylumbelliferone (4-MU) was obtained from Sigma-Aldrich
(St. Louis, MO, USA).
Western blot analysis
For western blot analysis, the cells were harvested
in RIPA lysis buffer. Following incubation at 4°C for 30 min, the
lysate was centrifuged at 15,000×g for 10 min at 4°C. The protein
concentration was determined using a BCA assay (Pierce, Rorkford,
IL, USA). Samples were denatured by 5X sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer
for 5 min at 95°C and subjected to 10% SDS-PAGE. The separated
proteins were transferred onto PVDF membranes (Millipore, Bedford,
MA, USA) for 2 h at 4°C, blocked in 5% non-fat milk in
phosphate-buffered saline/Tween-20 and blotted with antibodies. The
following primary antibodies were used: E-cadherin (#3195s), ZEB-1
(#6935s), N-cadherin (#4061s), Snail (#5879s), vimentin (#7391s)
(all from Cell Signaling Technology, Beverly, MA, USA), Twist
(ab50581; Abcam, Cambridge, UK), EGFR (#4267s), phosphorylated
(p-)AKT (#4060s), p-extracellular signal-regulated kinase (ERK;
#4370s), p-Smad (#3101s), ERK (#4695), Smad (#5339s) (all from Cell
Signaling Technology), β-actin (Sigma-Aldrich), AKT (#2920s; Cell
Signaling Technology) and CD44 (sc-18849; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA).
Immunofluorescence staining
For immunofluorescence staining, the cultured cells
were fixed for 10 min in 4% paraformaldehyde, permeabilized with
0.1% Triton X-100 and incubated overnight at 4°C with rabbit
anti-human EGFR (#4267s; Cell Signaling Technology) and mouse
anti-human CD44 (sc-18849; Santa Cruz Biotechnology, Inc.).
Subsequently, the sections were rinsed and incubated with the
secondary antibody conjugated to the fluorescent dye Alexa
Fluor® 488 and Alexa Fluor® 594 (both from
Jackson ImmunoResearch, West Grove, PA, USA). The sections were
rinsed and incubated with 4′,6-diamidino-2-phenylindole
dihydrochloride (DAPI; Life Technologies, Grand Island, NY, USA). A
confocal laser scanning microscope (SP5; Leica, Wetzlar, Germany)
was used to visualize the immunofluoresence staining.
RNA isolation, cDNA synthesis and
RT-PCR
Total RNA was extracted from the cells using the
RNAiso Plus (Takara Bio, Shiga, Japan) according to the
manufacturer’s instructions. Reverse transcription was performed
with high capacity cDNA reverse transcripting kits (Takara Bio)
according to the manufacturer’s instructions. RT-PCR (98°C, 2 min;
36×98°C, 5 sec; 55°C, 30 sec) was carried out using 2X PCR Solution
Premix Taq (R004A; Takara Bio). The primer sequences were as
follows: TβR, 5′GGCCAAATATCCCAAACAGAT3′ and 5′AATCCAACTCCTTTGCCC3′;
HAS1, 5′-GACGTGCGGA TCCTTAACCC-3′ and 5′-CGTTGTACAGCCACTCACG
GAA-3′; HAS2, 5′-AAGGCCATTTTCAGAATCCAA-3′ and
5′-TGGCAGAATGAAAATAAACCCAT-3′; HAS3, 5′-CTTTCCTTCATCTCCCACGAAC-3′
and 5′-CAAGCCCT TAGCGAAGTCTG-3′.
shRNA transfection
shRNA targeting CD44 (shRNA-CD44) (GeneChem,
Shanghai, China) was transfected into the cells using Lipofectamine
LTX (Life Technologies, Carlsbad, CA, USA) according to the
manufacturer’s instructions. The shRNA sequence was
CCTCTGCAAGGCTTTCAAT, GCTCTGA GCATCGGATTTG and ATAGCACCTTGCCCACAAT.
There were 3 specific shRNAs, the inhibitory effect of which was
assessed by western blot analysis (data not shown). Briefly, the
most effective shRNA was incubated with PLUS reagent for 5 min,
following which, LTX reagent was added. A 30-min incubation at room
temperature ensued, and the complex was subsequently applied to the
cell culture medium. A negative control shRNA was purchased from
GeneChem, the sequence of which was TTCTCCGAACGTGTCACGT.
Cell migration and invasion assays
Cell migration and invasion assays were conducted
using 8 µm Transwell inserts (Corning Inc., Corning, NY,
USA) and 24-well BD BioCoat Matrigel Invasion Chambers (BD
Biosciences, San Jose, CA, USA) according to the manufacturer’s
recommendations. In brief, the cells were seeded into the upper
inserts (for migration assay, 5×104 cells/insert were
used, and for invasion assay, 1×105 cells/insert were
used) with RPMI-1640. The outer wells were filled with RPMI-1640
containing EGF and/or TGF-β1 as the chemoattractant. After 24 h of
incubation, the membranes with the migrated or invaded cells were
stained with hematoxylin, washed and then mounted on slides. The
entire membrane was counted under a light microscope (IX70;
Olympus, Tokyo, Japan). Data were expressed as the number of
invaded cells per well. Each assay was conducted in triplicate and
was repeated at least twice.
Co-immunoprecipitation (Co-IP) assay
The cells were lysed in ice-cold Co-IP lysis buffer
containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% NP-40, 0.25%
Na-deoxycholate, 1 mM EDTA, 50 mM NaF, 1 mM sodium orthovanadate,
0.1 mM PMSF and protease inhibitor cocktail, and were then
incubated on ice for 10 min. The insoluble material was pelleted at
13,000×g for 10 min at 4°C. The supernatant was pre-cleaned by
protein A/G PLUS-Agarose (Santa Cruz Biotechnology, Inc.) and the
aliquots were co-immunoprecipitated with either non-specific IgG or
specific antibody against CD44 in Co-IP lysis buffer at 4°C for 1
h, followed by incubation with protein A/G PLUS-Agarose beads for a
further 1 h at 4°C. The immunoprecipitated complexes were washed
with Co-IP washing buffer [200 mM Tris (pH 7.4), 150 mM NaCl, 0.5%
NP-40, and 1 mM EDTA] 5 times and once with Co-IP lysis buffer. The
precipitated proteins were then analyzed by western blot analysis.
The input was used as a positive control.
Statistical analysis
Data are presented as the means ± SD and were
analyzed by one-way analysis of variance (ANOVA). All the
experiments were independently carried out and repeated at least 3
times. A P-value <0.05 was considered to indicate a
statistically significant difference.
Results
TGF-β1 is an important inducer of EMT
compared with EGF
To determine the effects of TGF-β1 and EGF on
promoting EMT, we treated the A549 cells with TGF-β1 and EGF at
various concentrations (0, 5, 10 and 20 ng/ml) and found the
suitable concentrations required for each different cytokine.
Subsequently, the cells were exposed to TGF-β1 (5 ng/ml) and EGF
(10 ng/ml) for different periods of time (0, 24, 48 and 72 h). The
results revealed that TGF-β1 and EGF induced EMT in a dose- and
time-dependent manner (data not shown).
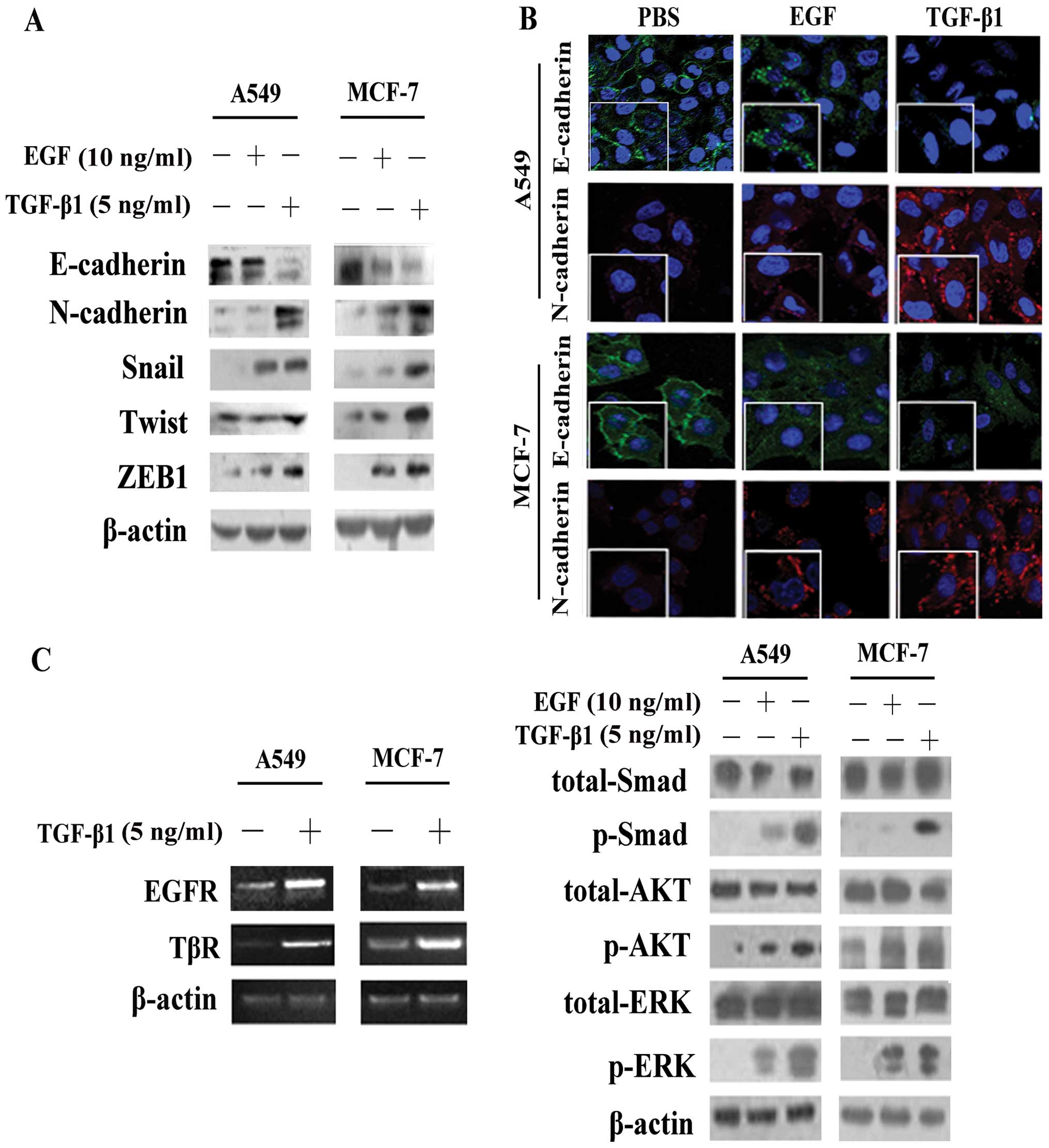
Significant alterations in the expression of
EMT-associated proteins (E-cadherin, N-cadherin, Snail, Twist and
ZEB1) were observed following stimulation of the cells with TGF-β1
and EGF, although it was clearly evident that TGF-β1 induced EMT to
a greater extent, as its effects on the epxression of these
proteins were more potent (Fig. 1A
and B).
The results of RT-PCR revealed that, in the A549 and
MCF-7 cells, the expression levels of both EGFR andTGF-β receptor
(TβR) were significantly increased following stimulation with
TGF-β1 compared with no stimulation (Fig. 1C). We then separately investigated
the activation of pathways following exposure to TGF-β1 or EGF for
4 h. Our data demonstrated that TGF-β1 not only induced the
activation of the Smad pathway, but also induced the
phosphorylation of AKT and ERK (Fig.
1C), which are commonly considered to be downstream molecules
of the EGF/EGFR pathway (13).
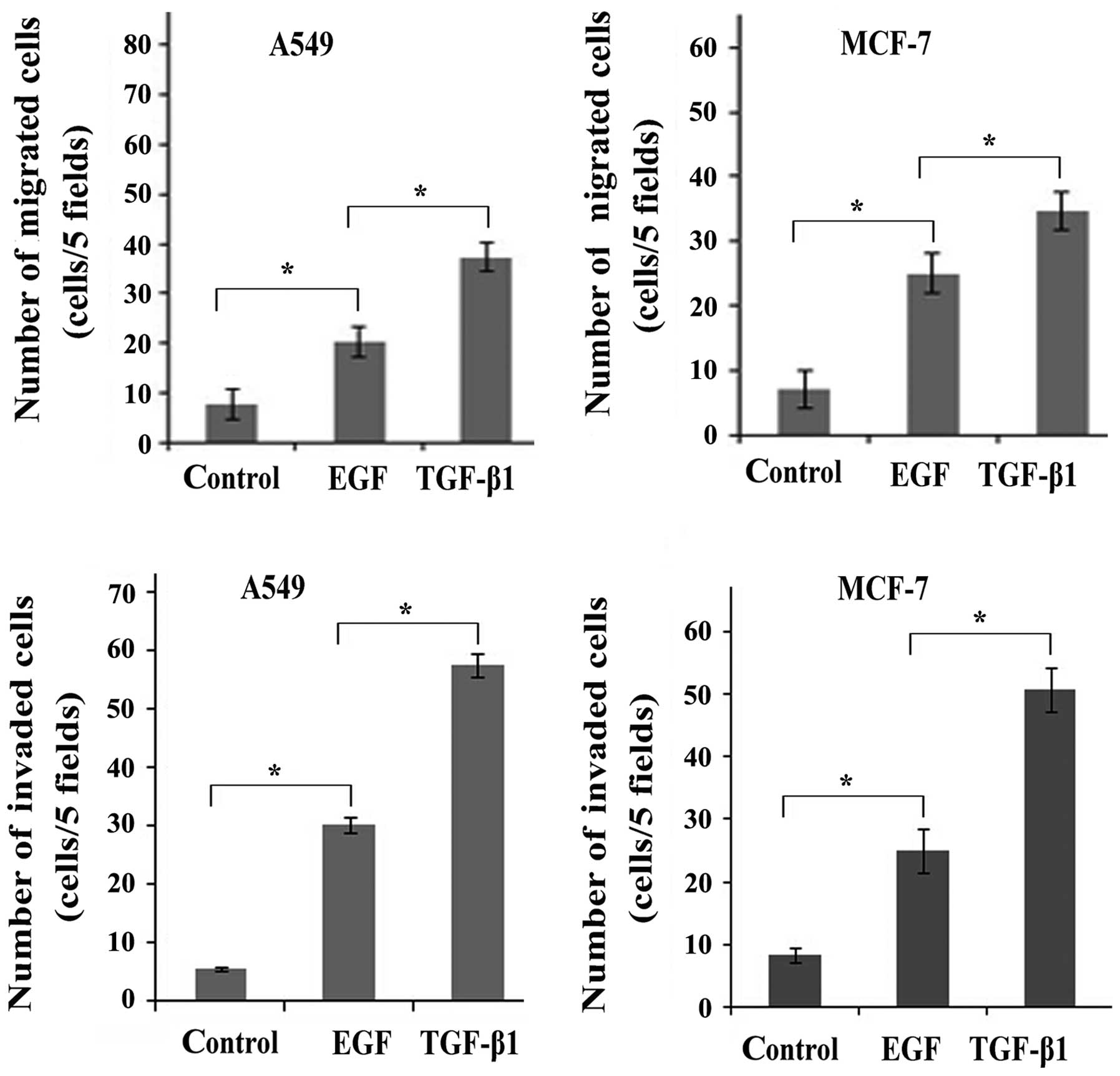
The transition between epithelial and mesenchymal
cell phenotypes is not only characterized by the expression of EMT
markers, but also the biological-functional and behavioral
phenotypes (15). As expected, a
larger number of both A549 and MCF-7 cells acquired migration and
invasion ability following stimulation with TGF-β1 or EGF than the
untreated control cells (Fig. 2),
suggesting that stimulation with TGF-β1 and EGF led to an enhanced
migratory and invasive potential of the cancer cells. In addition,
a larger number of migrated/invaded cells was observed following
stimulation with TGF-β1 than following stimulation with EGF. Taken
together, these findings demonstrated that TGF-β1 induced EMT to a
greater extent when compared with EGF. It was also suggested that
TGF-β1 transactivates EGF signaling by activating EGFR.
TGF-β1 induces EGFR and CD44 expression
and co-localization
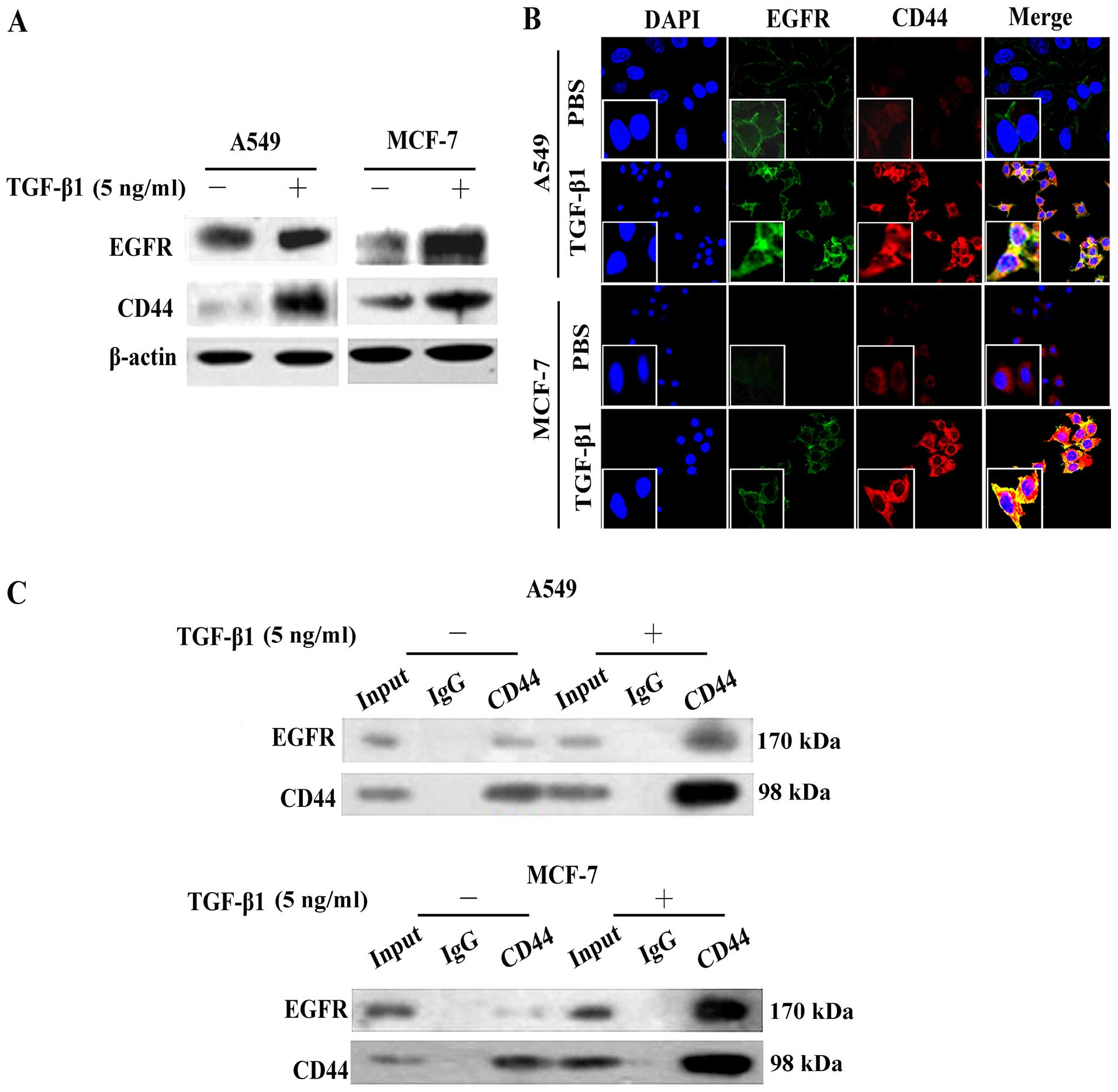
In accordance with the results of RT-PCR (Fig. 1C), western blot analysis also
demonstrated that EGFR cell surface expression levels increased
following stimulation of the A549 and MCF-7 cells with TGF-β1
(Fig. 3A). We found that the CD44
expression levels were also increased. Moreover, the expression
levels and cellular localization of EGFR and CD44 following
stimulation with TGF-β1 were assessed by confocal laser scanning
microscopy using the A549 and MCF-7 cells. As shown in Fig. 3B, without TGF-β1 stimulation, the
EGFR (green) and CD44 (red) proteins were weakly expressed around
the cytomembrane. However, following stimulation with TGF-β1, both
the EGFR and CD44 expression levels were upregulated and showed an
obvious co-localization, with visible yellow signals (merged image)
(Fig. 3B).
In order to explore the role of EGFR/CD44 in
TGF-β1-induced EMT and verify the interaction between EGFR and
CD44, we performed Co-IP assay. CD44 antibody was used to
co-immunoprecipitate EGFR protein from the A549 and MCF-7 cell
extracts. Compared with the negative band in the control groups
(Fig. 3C, IgG lane) and the weak
band in the normal extracts (Fig.
3C, input lane), EGFR protein was successfully
immunoprecipitated and was enriched by CD44 antibody (Fig. 3C; represented by a dark band, CD44
lane). These results suggest that TGF-β1 stimulates CD44 and
promotes CD44-EGFR complex formation.
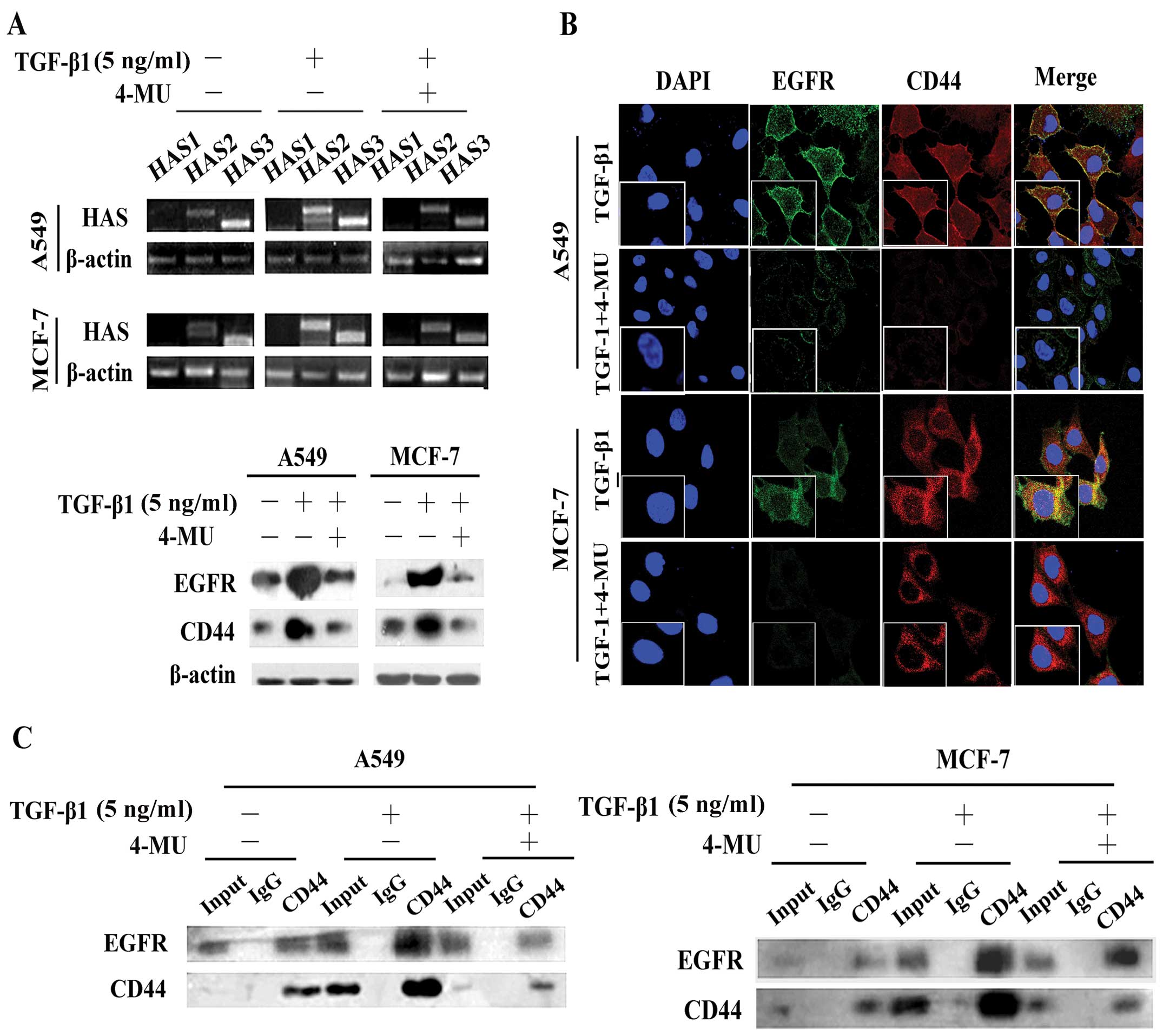
Inhibition of HAS by 4-MU abolishes
TGF-β1-induced CD44/EGFR expression and co-localization
Hyaluronan synthesis and degradation are regulated
by HAS (15). We found that
stimulation with TGF-β1 increased both HAS2 and HAS3 expression at
the mRNA level (Fig. 4A),
suggesting that HAS2 and HAS3 are involved in the TGF-β1-mediated
effects on HA production. Moreover, the upregulation of HAS2 was
more obvious than that of HAS3 in the A549 and MCF-7 cells, whereas
the mRNA expression levels of HAS1 were not altered (Fig. 4A). We then treated the A549 and
MCF-7 cells with the HAS inhibitor, 4-MU, and measured the mRNA
expression levels of HAS2 and HAS3. The results revealed that the
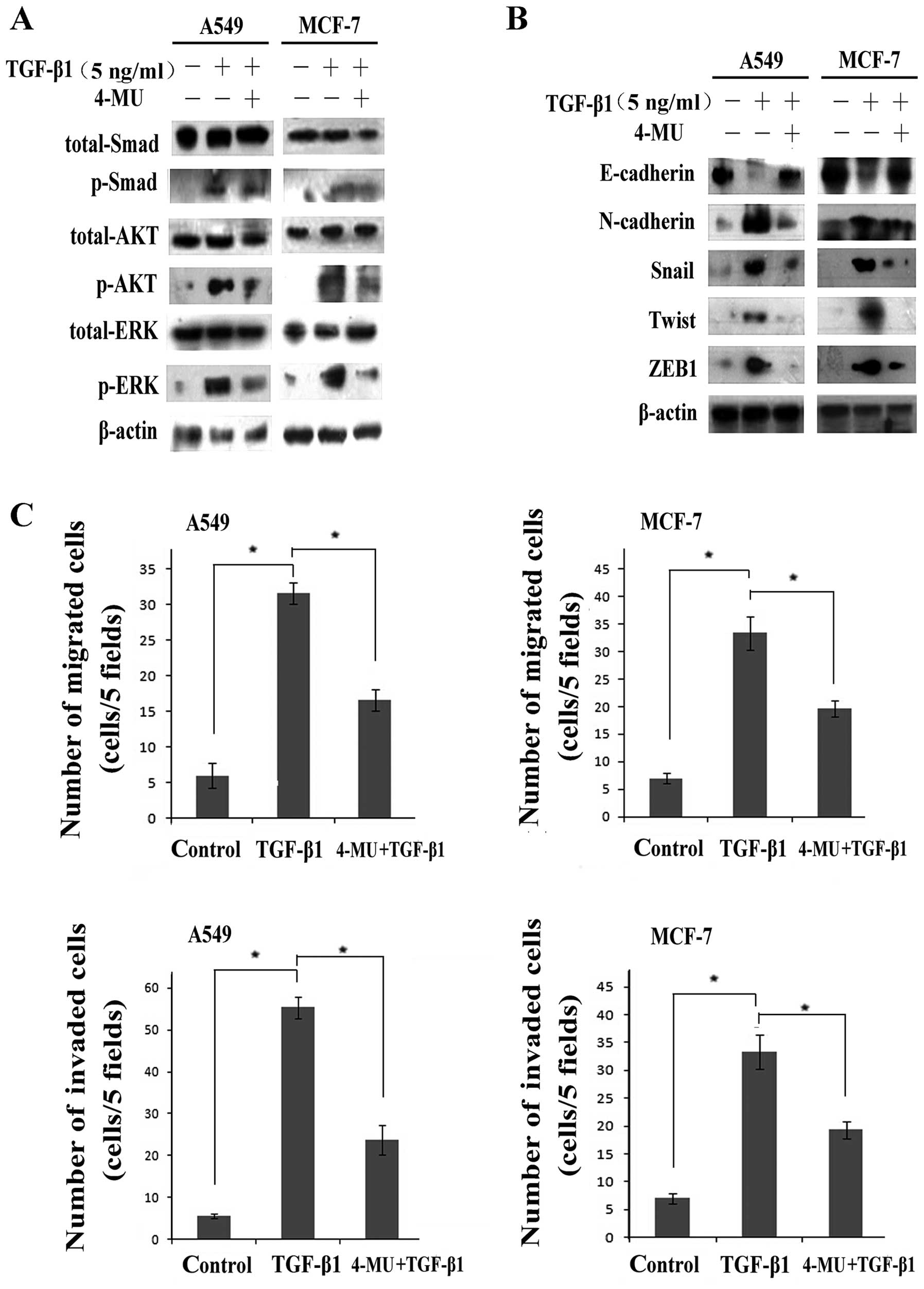
expression levels were significantly downregulated (Fig. 4A). Western blot analysis (Fig. 4A) and immunofluorescence staining
(Fig. 4B) also demonstrated that
4-MU suppressed the expression of CD44 and EGFR, decreasing their
expression to levels similar to those of the cells not stimulated
with TGF-β1 and disrupted the CD44-EGFR co-localization induced by
TGF-β1 (Fig. 4A and B).
Additionally, 4-MU inhibited the interaction between CD44 and EGFR,
even in the presence of TGF-β1 (Fig.
4C).
We further investigated the effects of 4-MU on
pathway activation. Pathways downstream of EGF/EGFR were shown to
be inactivated, the levels of p-ERK and p-AKT were signifi-cantly
decreased, whereas the expression levels of total ERK and AKT
proteins remained unaltered (Fig.
5A). Moreover, no significant effect on Smad2/3 phosphorylation
was observed following treastment with 4-MU, which suggesting
suggests that 4-MU suppressed the EGFR pathway rather than the
TGF-β1 pathway. In other words, HA contributed to the regulation of
TGF-β1-induced EGF/EGFR signaling.
In addition, we detected the expression of a set of
EMT markers, including E-cadherin, N-cadherin, Snail, Twist and
ZEB1, as well as the cell migration and invasion ability. Western
blot assays revealed that the cells treated with 4-MU had higher
expression levels of E-cadherin, and lower expression levels of
N-cadherin, Snail, Twist and ZEB1 compared with the cells treated
with TGF-β1 alone (Fig. 5B),
indicating that treatment with 4-MU reversed EMT. Moreover, the
results form Transwell migration assay indicated that the number of
migrated/invaded cells was markedly decreased following treatment
with 4-MU even after stimulation with TGF-β1 (Fig. 5C).
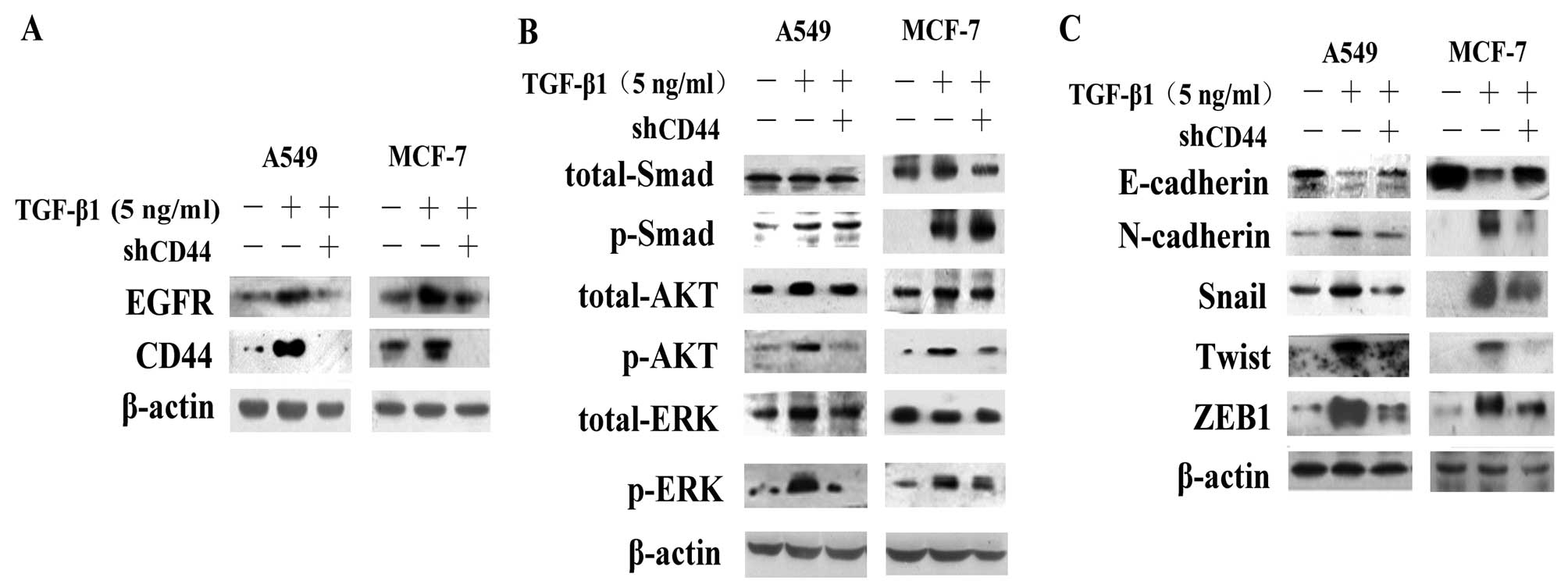
shRNA-CD44 blocks the activation of the
AKT and ERK pathways and causes the reversal of EMT
We then examined whether the disruption of CD44
expression effectively suppresses the expression of EGFR.
Transfection of the cells with shRNA-CD44 resulted in a decrease in
CD44 and EGFR expression compared to the controls and the
TGF-β1-treated group (Fig. 6A).
We found that the knockdown of CD44 also caused a marked decrease
in the levels of p-ERK and p-AKT, whereas it had no effect on the
levels of p-Smad (Fig. 6B),
demonstrating the effects of CD44 on TGF-β1-induced EGF/EGFR
signaling. Alterations in the expression of EMT-associated proteins
indicated that interference with the expression of CD44 reversed
EMT (Fig. 6C). Transfection of
the cells with shRNA-CD44 increased E-cadherin expression and
decreased N-cadherin, Snail, Twist and ZEB1 expression. Cell
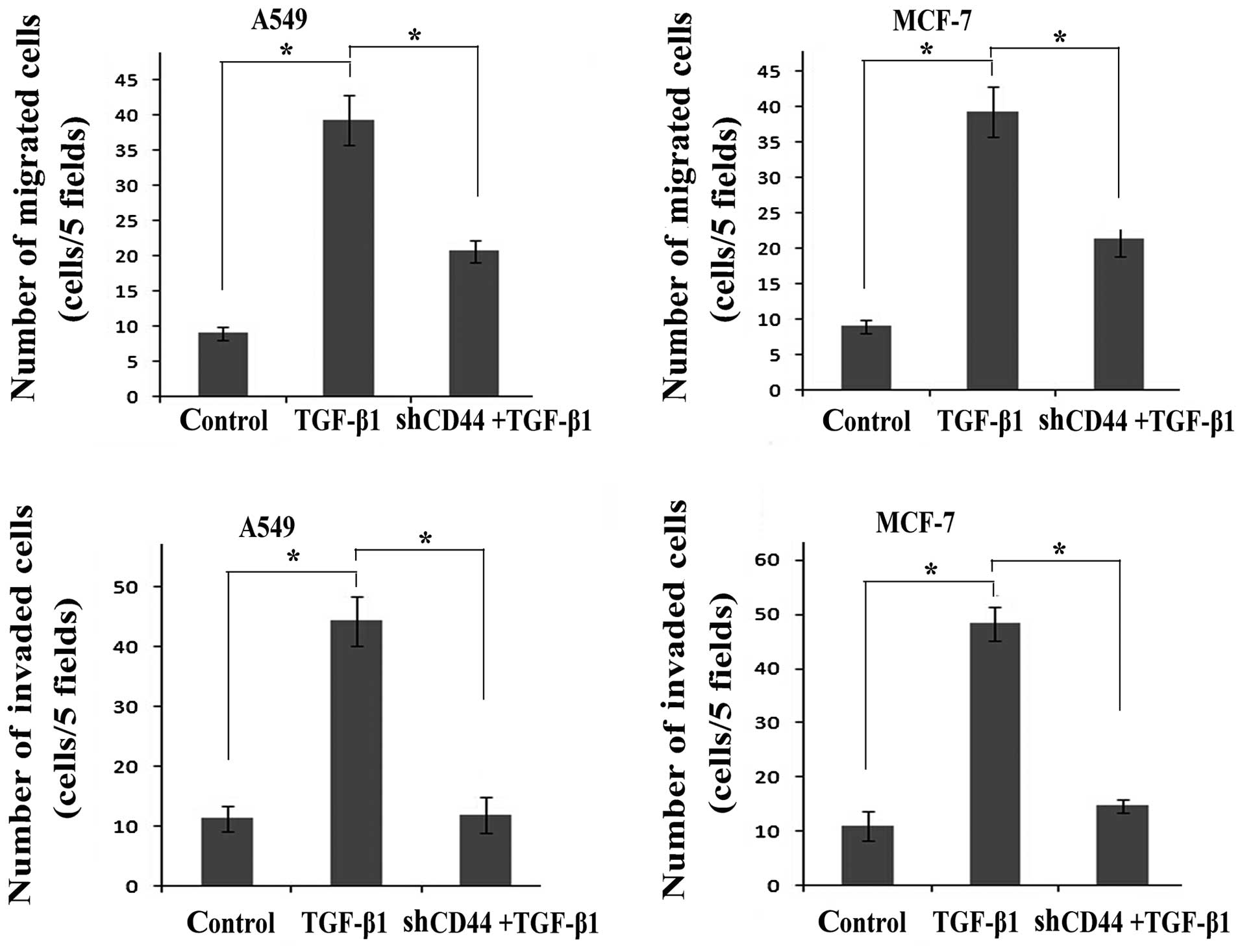
migration/invasion assays revealed that transfection with
shRNA-CD44 partially suppressed cell migration and invasion during
the EMT process induced by TGF-β1 (Fig. 7).
In conclusion, the inhibition of CD44 by shRNA-CD44
following stimulation with TGF-β1 abolished the expression of CD44
and EGFR, deactivated the EGF/EGFR signaling pathways and inhibited
the process of EMT, suggesting that there is a link between TGF-β1
and CD44/EGFR in the process of EMT.
Discussion
EMT enables epithelial cells to lose cell-cell
adhesions and acquire a mesenchymal phenotype, exhibiting enhanced
migratory and tumor-initiating capabilitie. Thus, EMT is considered
a crucial factor in cancer metastasis and progression (24). EMT is characterized by a loss of
epithelial markers (E-cadherin), while gaining mesenchymal markers
(vimentin and N-cadherin) and the upregulation of associated
transcriptional regulators (Snail, Twist and ZEB1) (25). The process of EMT involves a
variety of regulatory mechanisms supported by diverse extracellular
signal-derived activities and gene regulation. Extracellular clues
include the extracellular matrix components, TGF-β1 and EGF. TGF-β1
signaling occurs throuhg the phosphorylation of Smad2 and Smad3,
and this complex is associated with Smad4 translocation to the
nucleus, resulting in the transcriptional activation of downstream
targets (26). The EGF/EGFR
system regulates numerous biological processes by activating
multiple downstream signaling pathways, including the
mitogen-activated protein kinase (MEK)-ERK pathway and the
phosphoinositide-3 kinase (PI3K)/AKT pathways (27–29).
Kang et al (30) demonstrated that the crosstalk
between TGF-β1-mediated Smad and EGFR signaling induced
trans-membrane 4 L6 family member 5 (TM4SF5) expression and led to
the acquisition of mesenchymal cell characteristics (30). Ouyang et al (16) reported that miR-10b was engaged in
EGF-TGF-β1 crosstalk and enhanced the expression of EMT-promoting
genes in pancreatic ductal adenocarcinoma. We observed that either
TGF-β1 or EGF alone were able to induce EMT and that stimulation
with TGF-β1 was more potent than stimulation with EGF in the
regulation of EMT, as measured through a change in the behavioral
phenotype and the expression of EMT-associated molecules, further
suggesting that TGF-β1 is an important inducer of EMT.
Additionally, TGF-β1 increased the expression of EGFR, as well as
that of p-AKT and p-ERK, downstream molecules of the EGF/EGFR
pathway. There was considerable evidence to support the existence
of the transactivation of EGF signaling by TGF-β1 during the
process of EMT.
HA, an ubiquitous extracellular and cell
surface-associated component of the extracellular matrix, has been
shown to negatively correlate with clinical outcomes in several
types of cancer, such as breast, colon and prostate cancer
(31). HA binds to and signals
through the cell surface receptor, CD44, as has been confirmed by
previous studies (32,33). The majority of malignant tumors
contain elevated levels of both HA and CD44, and it has been
demonstrated that HA-activated CD44 promotes the proliferation,
invasion, metastasis and stemness of cancer cells (34). Various growth factors and
cytokines, such as TGF-β1, have been shown to modulate HA
production in tumor cells (17).
EGFR acts on CD44 to stimulate Ras-mediated signaling in an
HA-dependent manner (35–36). In the present study, we
investigated whether HA/CD44 plays an important role in the
TGF-β1-induced EGF signaling transactivation and EMT in cancer
cells. Our results revealed that TGF-β1 upregulated the expression
of HAS2 and HAS3, which increased the expression of CD44, and
subsequently promoted CD44/EGFR co-localization, and activated the
downstream AKT and ERK pathways. Our results were similar to those
of a physiological study, in which the investigators found that the
HAS2-dependent production of HA facilitates TGF-β1-dependent
fibroblast differentiation by promoting the interaction between
CD44 and EGFR held within membrane-bound lipid rafts (37).
In this study, to determine the importance of
HA/CD44 in TGF-β1-induced EGF signaling activation and EMT in
cancer cells, we used the HAS inhibitor, 4-MU, to block HAS
expression and shRNA targeting CD44 to knockdown CD44 expression.
We observed that they both 4-MU and shRNA-CD44 abolished
TGF-β1-induced CD44/EGFR expression. Co-IP and immunofluorescence
staining revealed that the inhibition of HAS disrupted CD44 and
EGFR co-localization. In addition, the downregulation of HAS and
CD44 blocked the TGF-β1-induced activation of the EGFR pathway,
presented as a decrease in the phosphorylation levels of AKT and
ERK, and reversed EMT. This indicates that HA/CD44 mediates EGF
signaling transactivation by TGF-β1 and is vital to TGF-β1-induced
EMT. Moreover, we obtained consistent results from two types of
cancer cells, suggesting that this mechanism may be common to
different types of cancer.
In conclusion, the findings of the current study
demonstrated that stimulation with TGF-β1 was more potent than EGF
in regulating EMT-associated protein expression and enhancing cell
migration and invasion. More importantly, the data presented in
this study indicated that TGF-β1 upregulated HAS expression,
promoted CD44/EGFR expression and co-localization, and subsequently
activated the AKT and ERK signaling pathways. Furthermore, the
inhibition of HAS by 4-MU and that of CD44 by shRNA abolished
TGF-β1-induced CD44/EGFR expression, inhibited the activation of
the AKT and ERK pathways and caused the reversal of EMT. The
aforementioned results suggest that HA/CD44 contributes to the
transactivation of EGF signaling through TGF-β1 in EMT during
cancer progression. Studies designed to elucidate the effects of
TGF-β1-induced HA/CD44 expression on other EMT-associated signaling
pathways, such as platelet-derived growth factor (PDGF)/signal
transducers and activators of transcription (STATs) are currently
in progress. The detailed analysis of HA/CD44 is important for the
effective targeting of EMT and may offer new options for novel
anticancer therapeutic strategies.
Acknowledgments
The authors would like to thank Dr Fei Zhang and
members of our laboratory for their technical assistance and
helpful comments. The present study was supported by grants from
the National Natural Science Foundation of China (81001186,
81201725 and 81402420) and the Tianjin Municipal Science and the
Technology Commission Research Fund (10JCYBJC14100, 13JCYBJC21800
and 15JCQNJC12400).
References
|
1
|
Wang Y and Shang Y: Epigenetic control of
epithelial-to-mesenchymal transition and cancer metastasis. Exp
Cell Res. 319:160–169. 2013. View Article : Google Scholar
|
|
2
|
Frisch SM, Schaller M and Cieply B:
Mechanisms that link the oncogenic epithelial-mesenchymal
transition to suppression of anoikis. J Cell Sci. 126:21–29. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Peng Z, Wang CX, Fang EH, Wang GB and Tong
Q: Role of epithelial-mesenchymal transition in gastric cancer
initiation and progression. World J Gastroenterol. 20:5403–5410.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Quan J, Elhousiny M, Johnson NW and Gao J:
Transforming growth factor-β1 treatment of oral cancer induces
epithelial-mesenchymal transition and promotes bone invasion via
enhanced activity of osteoclasts. Clin Exp Metastasis. 30:659–670.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sengupta S, Jana S, Biswas S, Mandal PK
and Bhattacharyya A: Cooperative involvement of NFAT and SnoN
mediates transforming growth factor-β (TGF-β) induced EMT in
metastatic breast cancer (MDA-MB 231) cells. Clin Exp Metastasis.
30:1019–1031. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Argast GM, Krueger JS, Thomson S,
Sujka-Kwok I, Carey K, Silva S, O’Connor M, Mercado P, Mulford IJ,
Young GD, et al: Inducible expression of TGFβ, snail and Zeb1
recapitulates EMT in vitro and in vivo in a NSCLC model. Clin Exp
Metastasis. 28:593–614. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Toda S, Matsumura S, Fujitani N, Nishimura
T, Yonemitsu N and Sugihara H: Transforming growth factor-β1
induces a mesenchyme-like cell shape without epithelial
polarization in thyrocytes and inhibits thyroid folliculogenesis in
collagen gel culture. Endocrinology. 138:5561–5575. 1997.PubMed/NCBI
|
|
8
|
Richter P, Umbreit C, Franz M and Berndt
A, Grimm S, Uecker A, Böhmer FD, Kosmehl H and Berndt A: EGF/TGFβ1
co-stimulation of oral squamous cell carcinoma cells causes an
epithelial-mesenchymal transition cell phenotype expressing laminin
332. J Oral Pathol Med. 40:46–54. 2011. View Article : Google Scholar
|
|
9
|
Liu Z, Du R, Long J, Dong A, Fan J, Guo K
and Xu Y: JDP2 inhibits the epithelial-to-mesenchymal transition in
pancreatic cancer BxPC3 cells. Tumour Biol. 33:1527–1534. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen J, Wang T, Zhou YC, Gao F, Zhang ZH,
Xu H, Wang SL and Shen LZ: Aquaporin 3 promotes
epithelial-mesenchymal transition in gastric cancer. J Exp Clin
Cancer Res. 33:382014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li L, Han R, Xiao H, Lin C, Wang Y, Liu H,
Li K, Chen H, Sun F, Yang Z, et al: Metformin sensitizes
EGFR-TKI-resistant human lung cancer cells in vitro and in vivo
through inhibition of IL-6 signaling and EMT reversal. Clin Cancer
Res. 20:2714–2726. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Davis FM, Peters AA, Grice DM, Cabot PJ,
Parat MO, Roberts-Thomson SJ and Monteith GR: Non-stimulated,
agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468
breast cancer cells and the effect of EGF-induced EMT on calcium
entry. PLoS One. 7:e369232012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Berndt A, Büttner R, Gühne S, Gleinig A,
Richter P, Chen Y, Franz M and Liebmann C: Effects of activated
fibroblasts on phenotype modulation, EGFR signalling and cell cycle
regulation in OSCC cells. Exp Cell Res. 322:402–414. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ren S, Su C, Wang Z, et al: Epithelial
phenotype as a predictive marker for response to EGFR-TKIs in
non-small cell lung cancer patients with wild-type EGFR. Int J
Cancer. 135:2962–2971. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tian YC, Chen YC, Chang CT, Hung CC, Wu
MS, Phillips A and Yang CW: Epidermal growth factor and
transforming growth factor-beta1 enhance HK-2 cell migration
through a synergistic increase of matrix metalloproteinase and
sustained activation of ERK signaling pathway. Exp Cell Res.
313:2367–2377. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ouyang H, Gore J, Deitz S and Korc M:
microRNA-10b enhances pancreatic cancer cell invasion by
suppressing TIP30 expression and promoting EGF and TGF-β actions.
Oncogene. 33:4664–4674. 2014. View Article : Google Scholar :
|
|
17
|
Chow G, Tauler J and Mulshine JL:
Cytokines and growth factors stimulate hyaluronan production: role
of hyaluronan in epithelial to mesenchymal-like transition in
non-small cell lung cancer. J Biomed Biotechnol. 2010:4854682010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Porsch H, Bernert B, Mehić M, Theocharis
AD, Heldin CH and Heldin P: Efficient TGFβ-induced
epithelial-mesenchymal transition depends on hyaluronan synthase
HAS2. Oncogene. 32:4355–4365. 2013. View Article : Google Scholar :
|
|
19
|
Hiscox S, Baruha B, Smith C, Bellerby R,
Goddard L, Jordan N, Poghosyan Z, Nicholson RI, Barrett-Lee P and
Gee J: Overexpression of CD44 accompanies acquired tamoxifen
resistance in MCF7 cells and augments their sensitivity to the
stromal factors, heregulin and hyaluronan. BMC Cancer. 12:4582012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goodison S, Urquidi V and Tarin D: CD44
cell adhesion molecules. Mol Pathol. 52:189–196. 1999. View Article : Google Scholar
|
|
21
|
Hiraga T, Ito S and Nakamura H: Cancer
stem-like cell marker CD44 promotes bone metastases by enhancing
tumorigenicity, cell motility, and hyaluronan production. Cancer
Res. 73:4112–4122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Midgley AC, Bowen T, Phillips AO and
Steadman R: MicroRNA-7 inhibition rescues age-associated loss of
EGF receptor and hyaluronan (HA)-dependent differentiation in
fibroblasts. Aging Cell. 13:235–244. 2013. View Article : Google Scholar
|
|
23
|
Williams K, Motiani K, Giridhar PV and
Kasper S: CD44 integrates signaling in normal stem cell, cancer
stem cell and (pre) metastatic niches. Exp Biol Med (Maywood).
238:324–338. 2013. View Article : Google Scholar
|
|
24
|
Xu Z, Jiang Y, Steed H, Davidge S and Fu
Y: TGFβ and EGF synergistically induce a more invasive phenotype of
epithelial ovarian cancer cells. Biochem Biophys Res Commun.
401:376–381. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wendt MK, Smith JA and Schiemann WP:
Transforming growth factor-β-induced epithelial-mesenchymal
transition facilitates epidermal growth factor-dependent breast
cancer progression. Oncogene. 29:6485–6498. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ohshio Y, Teramoto K, Hashimoto M,
Kitamura S, Hanaoka J and Kontani K: Inhibition of transforming
growth factor-β release from tumor cells reduces their motility
associated with epithelial-mesenchymal transition. Oncol Rep.
30:1000–1006. 2013.PubMed/NCBI
|
|
27
|
Elloul S, Kedrin D, Knoblauch NW, Beck AH
and Toker A: The adherens junction protein afadin is an AKT
substrate that regulates breast cancer cell migration. Mol Cancer
Res. 12:464–476. 2014. View Article : Google Scholar :
|
|
28
|
Voon DC, Wang H, Koo JK, Chai JH, Hor YT,
Tan TZ, Chu YS, Mori S and Ito Y: EMT-induced stemness and
tumorigenicity are fueled by the EGFR/Ras pathway. PLoS One.
8:e704272013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Buonato JM and Lazzara MJ: ERK1/2 blockade
prevents epithelial-mesenchymal transition in lung cancer cells and
promotes their sensitivity to EGFR inhibition. Cancer Res.
74:309–319. 2014. View Article : Google Scholar :
|
|
30
|
Kang M, Choi S, Jeong SJ, Lee SA, Kwak TK,
Kim H, Jung O, Lee MS, Ko Y, Ryu J, et al: Cross-talk between TGFβ1
and EGFR signalling pathways induces TM4SF5 expression and
epithelial-mesenchymal transition. Biochem J. 443:691–700. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Heffler M, Golubovskaya VM, Conroy J, Liu
S, Wang D, Cance WG and Dunn KB: FAK and HAS inhibition
synergistically decrease colon cancer cell viability and affect
expression of critical genes. Anticancer Agents Med Chem.
13:584–594. 2013. View Article : Google Scholar :
|
|
32
|
Su CY, Li YS, Han Y, Zhou SJ and Liu ZD:
Correlation between expression of cell adhesion molecules CD44 v6
and E-cadherin and lymphatic metastasis in non- small cell lung
cancer. Asian Pac J Cancer Prev. 15:2221–2224. 2014. View Article : Google Scholar
|
|
33
|
Cheng C, Yaffe MB and Sharp PA: A positive
feedback loop couples Ras activation and CD44 alternative splicing.
Genes Dev. 20:1715–1720. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Raso-Barnett L, Banky B, Barbai T, Becsagh
P, Timar J and Raso E: Demonstration of a melanoma-specific CD44
alternative splicing pattern that remains qualitatively stable, but
shows quantitative changes during tumour progression. PLoS One.
8:e538832013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Perez A, Neskey DM, Wen J, Pereira L,
Reategui EP, Goodwin WJ, Carraway KL and Franzmann EJ: CD44
interacts with EGFR and promotes head and neck squamous cell
carcinoma initiation and progression. Oral Oncol. 49:306–313. 2013.
View Article : Google Scholar :
|
|
36
|
Grass GD, Tolliver LB, Bratoeva M and
Toole BP: CD147, CD44, and the epidermal growth factor receptor
(EGFR) signaling pathway cooperate to regulate breast epithelial
cell invasiveness. J Biol Chem. 288:26089–26104. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Midgley AC, Rogers M, Hallett MB, Clayton
A, Bowen T, Phillips AO and Steadman R: Transforming growth
factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast
differentiation is mediated by hyaluronan (HA)-facilitated
epidermal growth factor receptor (EGFR) and CD44 co-localization in
lipid rafts. J Biol Chem. 288:14824–14838. 2013. View Article : Google Scholar : PubMed/NCBI
|