Introduction
Acute lung injury (ALI), a clinical syndrome caused
by a variety of non-cardiogenic factors, is characterized by
increased capillary permeability, pulmonary edema, atelectasis and
refractory hypoxemia and is induced by injury to pulmonary
capillary endothelial cells and alveolar epithelial cells; ALI has
the potential to develop into acute respiratory distress syndrome
(ARDS), which is associated with a mortality rate of 40–60%
worldwide (1). ALI that manifests
clinically as ARDS is a major cause of multiple organ dysfunction
syndrome (MODS) (1). Inflammatory
mediators, such as interleukin (IL)-1β and IL-8, play key roles in
the pathogenesis of ARDS, which is the primary cause of mortality
in patients with these conditions.
Pre-B cell colony-enhancing factor (PBEF) is a
highly conserved 52-kDa protein (2). It was originally cloned from a
complementary DNA (cDNA) library of activated human peripheral
blood mononuclear cells (PBMCs) and was identified as a secreted
protein that enhances the effects of stem cell factor and IL-7 on
pre-B cell colony formation. It is now evident that PBEF is a
multifunctional protein, having nicotinamide
phosphoribosyltransferase, adipokine and cytokine activities.
Experimental and clinical analyses on the inflammatory aspects of
PBEF conducted by Moschen et al (2) demonstrated that PBEF exhibited
inflammatory and immune stimulatory activity, and that it regulated
the expression of several inflammatory factors, such as tumor
necrosis factor (TNF)-α, IL-1β and IL-6. Ye et al (3) found that the level of PBEF in
bronchoalveolar lavage fluid and blood plasma of animal and
patients with ALI was significantly increased, suggesting that PBEF
may be an indicator of lung injury. Bajwa et al (4) confirmed that the PBEF gene
polymorphism was closely related to the prognosis of ARDS. Taken
together, these data indicate that PBEF plays an important role in
ALI. Ye et al (5)
demonstrated that PBEF played an important role in the loss of
barrier function in pulmonary vascular endothelial cells. Liu et
al (6) found that IL-1β
promoted the expression of PBEF in pulmonary vascular endothelial
cells, and that the secretion of inflammatory factors [IL-8, IL-16
and chemokine (C-C motif) receptor 3 (CCR3)] was involved. However,
the exact mechanisms of PBEF-related pulmonary vascular endothelial
cell injury remain unclear. Studies have proven that
mitogen-activated protein kinases (MAPKs) are involved in the
pathological process of lung injury (2,7,8);
however, the association between the signal transduction pathways
of the PBEF pro-inflammatory response in ALI and MAPK pathways
remains unclear.
Researchers have indicated that aquaporins (AQPs)
play an important role in the pathogenesis of ALI (9,10).
Studies have focused on vascular endothelial permeability; however,
little information on the effects of the fluid-transport function
of AQPs in the formation of pneumonedema is available. Furthermore,
the expression of AQPs in lung tissue in ALI remains a
controversial topic. Certain studies have demonstrated a decrease
in AQP expression in patients suffering from ALI (11,12), whereas in another study, Lai et
al (13) demonstrated that
inflammatory factors promoted the expression of AQP1 in
vitro. In the present study, PBEF was upregulated using an
overexpression plasmid, and TNF-α and siRNA were also used to
investigate the association of PBEF with inflammatory factors and
AQP activity. The mechanisms of ALI were further examined with the
use of MAPK inhibitors. Our study of the pathological mechanisms of
ALI provides information which may prove to be useful in the
prevention and treatment of ALI.
Materials and methods
Cell line and cell culture
Human pulmonary microvascular endothelial cells
(HPMECs) were purchased from the American Type Culture Collection
(ATCC, Manassas, VA, USA) and cultured in DMEM medium supplemented
with 10% fetal bovine serum (FBS; both from Gibco, Paisley, UK),
100 U/ml penicillin and 50 U/ml streptomycin (both from
Sigma-Aldrich China, Inc., Shanghai, China) at 37°C in an incubator
with 5% CO2.
Construction of the PBEF-overexpressing
plasmid
The PBEF-overexpressing plasmid was constructed by
inserting the cDNA clone of PBEF reverse transcriptase from the
HPMECs into the pEGFP-N1 vector [from Takara Biotechnology (Dalian)
Co., Ltd., Dalian, China]. Total RNA was extracted using TRIzol
reagent (Shanghai Invitrogen Biotechnology Co., Ltd., Shanghai,
China) to prepare the cDNA clone of PBEF. Reverse transcription
polymerase chain reaction (RT-PCR) was performed to retrieve the
full-length mouse PBEF cDNA using cDNA and a reverse transcription
kit (Promega Biotech Co., Ltd., Beijing, China). The PCR conditions
were as follows: a pre-denaturation step at 94°C for 5 min,
followed by 94°C for 30 sec, 58°C for 30 sec, 72°C for 90 sec for
30 cycles, and finally a complete cycle at 72°C for 10 min. The
amplified products were analyzed by electro phoresis, and the
specific 1,473-bp band of the amplified cDNA fragment was confirmed
and collected. The cloned PBEF cDNA was inserted into the pEGFP-N1
vector with XhoI and BamHI [Takara Biotechnology
(Dalian) Co., Ltd.], sequenced and verified. The primers used for
PBEF amplification were: PBEF-F, 5′-CCGCTCGAGATGAATCCTGCGGCAGAAGC-3′
and PBEF-R, 5′-CGGGATCCCGATGATGTGCTGCTTCCAGTTC-3′.
Underlined primers indicate the restriction enzyme site.
Design and synthesis of siRNA
The PBEF-targeting siRNA was designed and
synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China). The
siRNA sequences are presented in Table I.
 | Table IsiRNA sequences. |
Table I
siRNA sequences.
| Name | Sense/antisense siRNA
(5′→3′) |
|---|
| PBEF-homo-514 |
GGCCAAAUAUUUGUUAGAATT
UUCUAACAAAUAUUUGGCCTT |
| PBEF-homo-1056 |
GGGAUGGAGUAGAUAUUAATT
UUAAUAUCUACUCCAUCCCTT |
| PBEF-homo-1279 |
GGGCCGAUUAUCUUUACAUTT
AUGUAAAGAUAAUCGGCCCTT |
| Negative
control |
UUCUCCGAACGUGUCACGUTT
ACGUGACACGUUCGGAGAATT |
Cell transfection and induction of PBEF
expression by TNF-α
Cell transfection was carried out using
Lipofectamine 2000 according to the manufacturer's instructions
(Shanghai Invitrogen Biotechnology Co., Ltd.). Briefly, the cells
were seeded in 6-well plates at a concentration of 1×105
cells/ml (1 ml/well). When the cells reached 70% confluency 24 h
after seeding, the plasmid (pEGFP-N1-PBEF) and PBEF siRNA were
transfected into the cells at final concentrations of 4
µg/ml and 50 nM, respectively. The medium was changed 4–6 h
after transfection. TNF-α (Peprotech, Inc., Rocky Hill, NJ, USA)
was added at a concentration of 5.75 nM, as previously described
(14), and was re-added every 24
h. The effects of the induction of PBEF expression were detected by
reverse transcription-quantitative PCR (RT-qPCR) and western blot
anlaysis after 72 h.
Determination of cell apoptosis
Cell apoptosis was evaluated by co-staining of the
cells with Annexin V-FITC and propidium iodide (PI; BD Pharmingen,
San Diego, CA, USA). Briefly, the cells were collected 72 h after
transfection and resuspended in 0.5 ml binding buffer and stained
with 5 µl Annexin V-FITC and 5 µl PI, and the
solution was then incubated for 15 min at room temperature in the
dark. Thereafter, the cells were immediately analyzed on a Coulter
Epics XL Flow Cytometer (Beckman Coulter, Inc., Brea, CA, USA). The
fluorochrome was excited at 488 nm by an argon ion laser, and
Annexin V and PI emissions were monitored at 525 and 630 nm,
respectively. In each analysis, 10,000 events were recorded. The
dual parametric dot plots were used to calculate the percentage of
non-apoptotic viable cells in the lower left quadrant (Annexin
V-negative/PI-negative), early apoptotic cells in the lower right
quadrant (Annexin V-positive/PI-negative), late apoptotic cells in
the upper right quadrant (Annexin V-positive/PI-positive) and
necrotic cells in the upper left quadrant (Annexin
V-negative/PI-positive).
Blocking MAPK and AKT signaling
pathways
SB 203580 (a p38 inhibitor), PD 98059 [an
extracellular signal-regulated kinase (ERK)1/2 inhibitor], c-Jun
N-terminal kinase (JNK) inhibitor II and LY 294002 [a
phosphoinositide 3-kinase (PI3K) inhibitor] were all purchased from
Calbiochem (Merck Millipore, Shanghai, China). The cells were
exposed to the inhibitors at a final concentration of 20 µM
for 1 h prior to transfection and induction of epression. DMSO
(Sigma-Aldrich China, Inc.) was used as a negative control, as
previously described (15).
RT-qPCR
Total RNA was extracted using TRIzol reagent
(Shanghai Invitrogen Biotechnology Co., Ltd.) after the cells had
been collected and quantitified. This was followed by reverse
transcription with 1 µg RNA. Quantitative (real-time) PCR
was performed using the SYBR-Green dye method (SYBR-Green PCR
Master Mix; Toyobo Co., Ltd., Osaka, Japan) under the follows
conditions: 95°C for 5 min, 95°C for 30 sec, 55°C for 30 sec, 72°C
for 30 sec, for 40 cycles. The reaction mixture (35 µl)
contained 100 ng cDNA (20 µl). The primers used are
presented in Table II. The
experiments were repeated 3 times and analyzed by comparing the
2−ΔΔCt values.
| Table IIPrimers used for quantitative
PCR. |
Table II
Primers used for quantitative
PCR.
| Primer name | Primer sequences
(5′→3′) | Product size
(bp) | GenBank accession
no. |
|---|
| PBEF-F |
TGCTACAGAAGTTGACAAGAGATC | 102 | U02020 |
| PBEF-R |
CCCTGCTGGCGTCCTATG | | |
| IL-1β-F |
TGGCTTATTACAGTGGCAATG | 134 | NM_000576 |
| IL-1β-R |
GTGGTGGTCGGAGATTCG | | |
| IL-6-F |
TAGGACTGGAGATGTCTGAG | 104 | NM_000600 |
| IL-6-R |
GTGGAGAAGGAGTTCATAGC | | |
| IL-8-F |
CACAAACTTTCAGAGACAGCAGAG | 85 | NM_000584 |
| IL-8-R |
ACACAGTGAGATGGTTCCTTCC | | |
| AQP1-F |
ACTCATCTACGACTTCATCC | 100 | NM_001185060 |
| AQP1-R |
GGCATCCAGGTCATACTC | | |
| GAPDH-F |
TCTCTGCTCCTCCTGTTC | 97 | NM_002046 |
| GAPDH-F |
ACTCCGACCTTCACCTTC | | |
Western blot anlaysis
Total cellular proteins were extracted by incubating
the cells in lysis buffer obtained from Cell Signaling Technology,
Inc. (Beverly, MA, USA). The protein concentrations of the cell
lysates were determined using a bicinchoninic acid assay kit
(Pierce Biotechnology, Inc., Rockford, IL, USA) according to the
manufacturer's instructions. For SDS-PAGE, 10% gels were used, and
equal amount of proteins were loaded per lane. Following
electrophoresis, the separated proteins were transferred onto
nitrocellulose membranes (Pierce Biotechnology, Inc.) and blocked
with 5% non-fat milk in TBST buffer for 1 h. Subsequently, the
membranes were incubated with primary antibodies [anti-PBEF
(ab45890), anti-IL-1β (ab9722), anti-IL-6 (ab6672), anti-IL-8
(ab7747), anti-AQP1 (ab15080) and anti-GAPDH (ab181602) antibodies;
Abcam, Shanghai, China] at a 1:1,000 dilution in 5% non-fat milk
overnight at 4°C followed by secondary antibodies [goat anti-rabbit
IgG H&L (HRP); ab97051; Abcam] at a 1:2,000 dilution for 1 h at
room temperature. Protein bands were visualized on X-ray film using
an enhanced chemiluminescence system (Pierce Biotechnology,
Inc.).
Statistical analysis
Experiments were carried out at least in triplicate,
and the results are expressed as the means ± SD. Statistical
analysis was performed using the SPSS statistical package (SPSS
17.0 for Windows; SPSS, Inc., Chicago, IL, USA). The differences
between 2 groups were analyzed using a two-tailed Student's t-test,
and those between 3 or more groups were analyzed by one-way
analysis of variance (ANOVA) multiple comparisons. A P-value
<0.05 was considered to indicate a statistically significant
difference.
Results
Regulation of the intracellular
expression level of PBEF
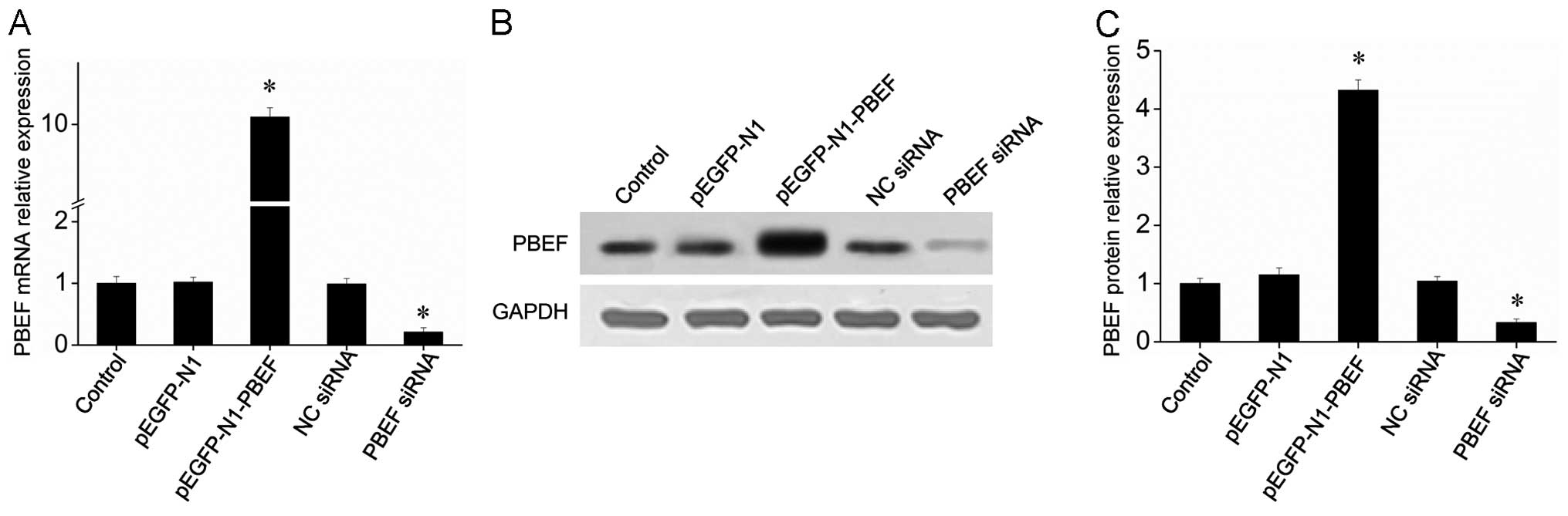
RT-qPCR was employed to evaluate the inhibitory
effects of siRNA, and the results revealed that siRNA
PBEF-homo-1279 inhibited PBEF gene expression by up to 85% at a
concentration of 50 nM (data not shown). The results of RT-qPCR and
western blot anlaysis revealed that the PBEF-targeting siRNA
significantly inhibited the mRNA and protein expression of PBEF
(Fig. 1). Moreover, transfection
of the cells with the PBEF-overexpressing plasmid (pEGFP-N1-PBEF)
increased the expression level of PBEF to a level 10-fold greater
than that of the control group (Fig.
1). These results indicated the powerful effects of the
PBEF-overexpressing plasmid and PBEF-targeting siRNA on the
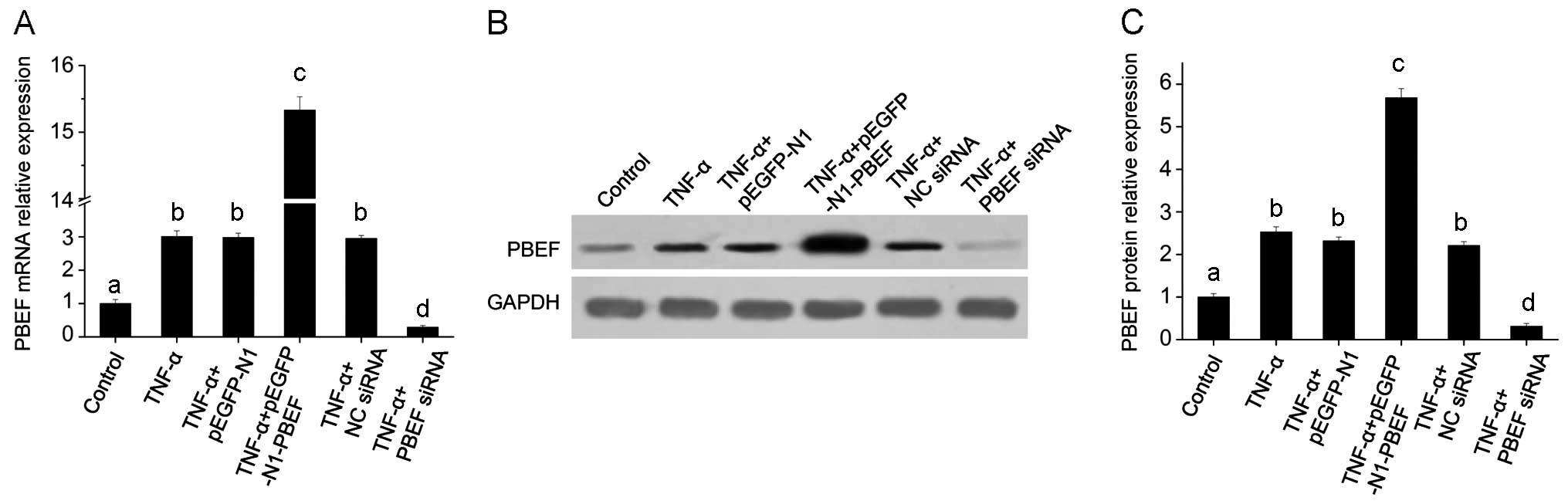
expression of PBEF. The effects of TNF-α on the overexpression and
knockdown of PBEF were also investigated, and the results revealed
that the expression of PBEF increased significantly with the
presence of TNF-α, which indicated that TNF-α promoted the
expression of PBEF in the HPMECs (Fig. 2). The effects of TNF-α on the
expression of PBEF were further enhanced by combining TNF-α and
transfection with the PBEF-overexpressing plasmid. However, the
TNF-α-induced overexpression of PBEF was blocked by PBEF siRNA
(Fig. 2). Taken together, our
results suggest that TNF-α has an enhancing effect when combined
with the PBEF-overexpressing plasmid and an antagonistic effect
when combined with PBEF siRNA.
Overexpression of PBEF promotes HPMEC
apoptosis
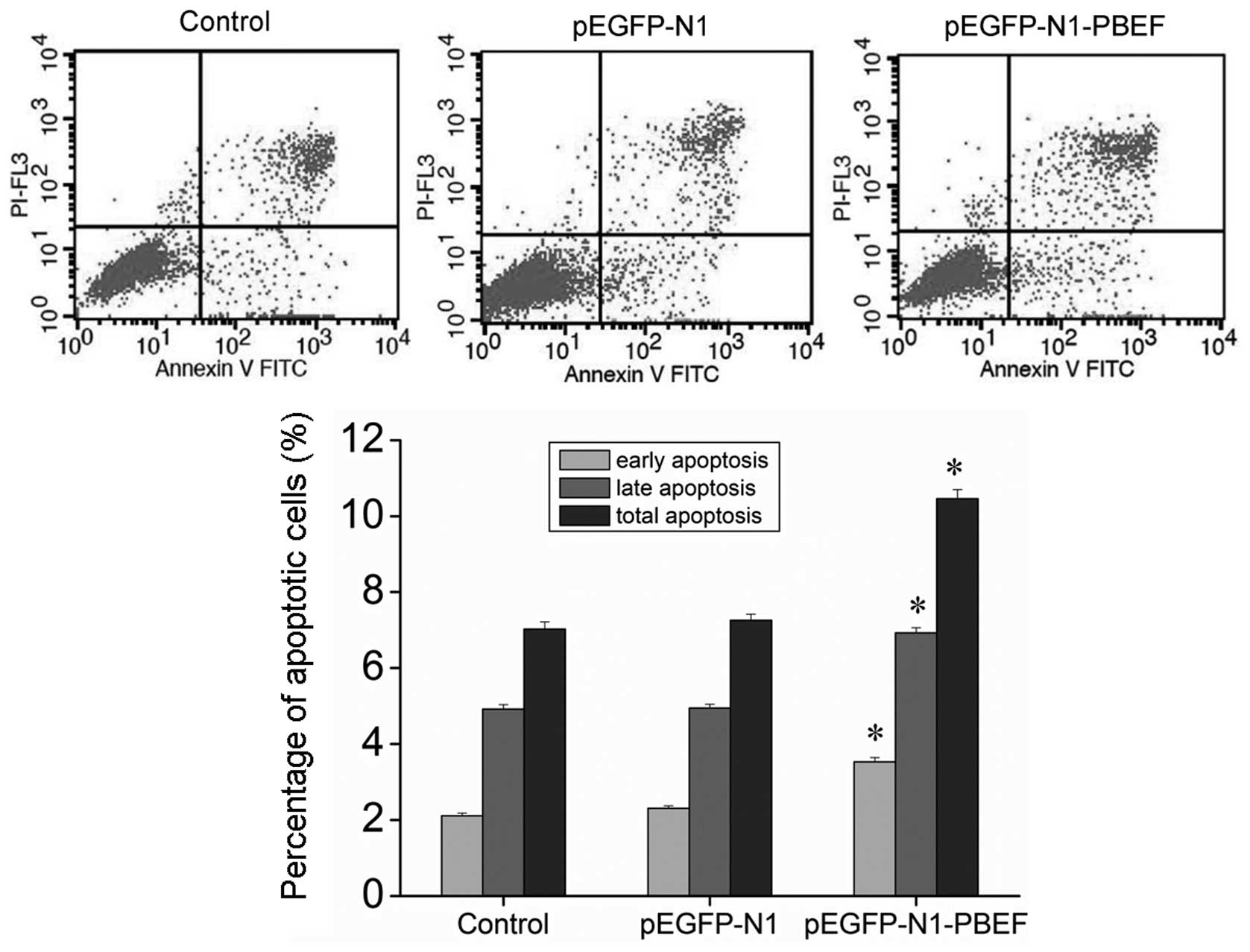
To examine the effects of PBEF overexpression on
HPMEC apoptosis, flow cytometric analysis was employed on the cells
co-stained with Annexin V-FITC and PI. The results revealed that
the overexpression of PBEF significantly promoted both the early-
and late-phase apoptosis of the HPMECs (Fig. 3). These results suggest that an
increase in the PBEF expression level causes apoptotic cell death,
and this in turn, may lead to the development of ALI.
PBEF regulates the expression of
inflammatory factors and AQPs
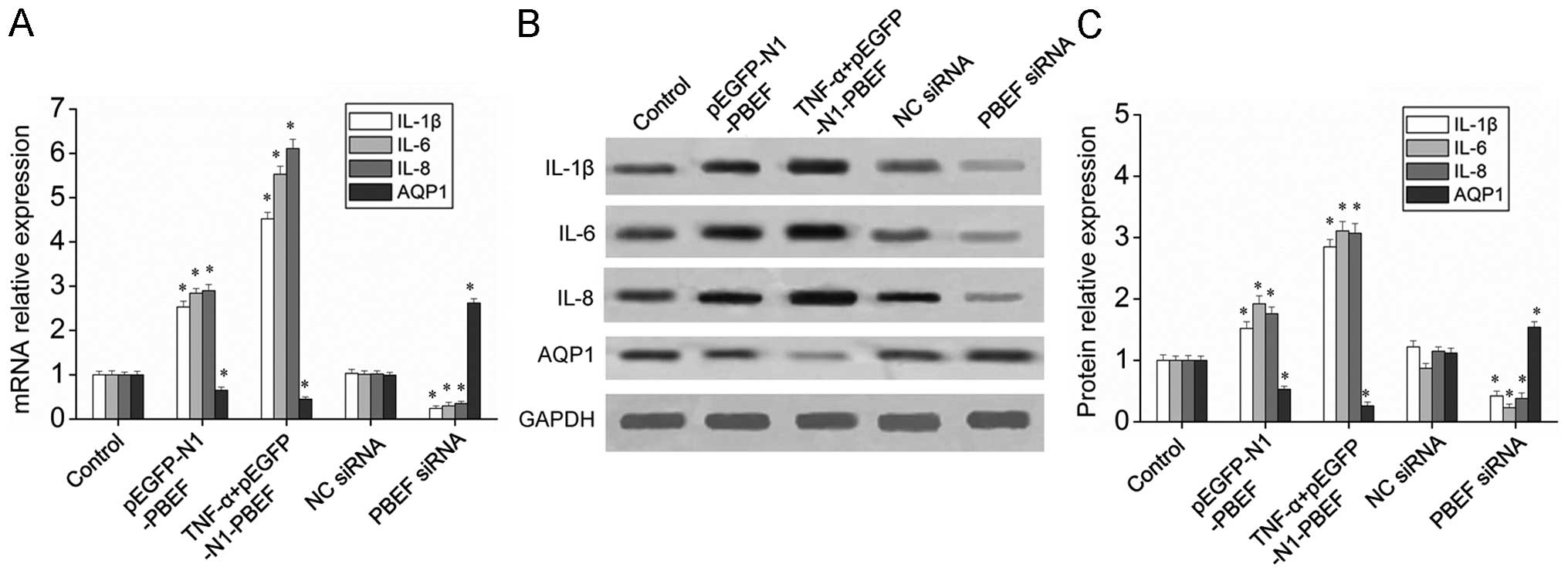
To examine the effects of PBEF expression on
inflammatory factors and AQPs, RT-qPCR and western blot analysis
were employed to measure the mRNA and protein levels of PBEF in the
HPMECs. The results indicated that PBEF overexpression
significantly increased the mRNA and protein expression of IL-1β,
IL-6 and IL-8, and this was further enhanced by the addition of
TNF-α (Fig. 4). The mRNA and
protein expression of AQP1 was inhibited by the overexpression of
PBEF and this effect was enhanced by TNF-α (Fig. 4). Furthermore, the inhibition of
PBEF using siRNA downregulated the mRNA and protein levels of
IL-1β, IL-6 and IL-8 and increased the mRNA and protein level of
AQP1. Taken together, these results indicate that PBEF enhances the
mRNA and protein expression of IL-1β, IL-6 and IL-8, and inhibits
the mRNA and protein expression of AQP1.
PBEF regulates the expression of
inflammatory factors and AQPs through the MAPK pathways
The regulatory effects of MAPKs on inflammatory
factors and AQPs have previously been demonstrated (16). Therefore, in this study,
experiments were also carried out to investigate the association
between the PBEF-regulated expression of inflammatory factors and
AQPs, and the activation of the MAPK and PI3K pathways. Inhibitors
of several signaling pathways (p38, ERK, JNK and PI3K) at
non-cytotoxic concentrations (20 µM) were introduced into
PBEF-overexpressing HPMECs that were treated or not with TNF-α. The
mRNA and protein levels of the inflammatory factors and AQP1 were
measured by RT-qPCR and western blot analysis. The results
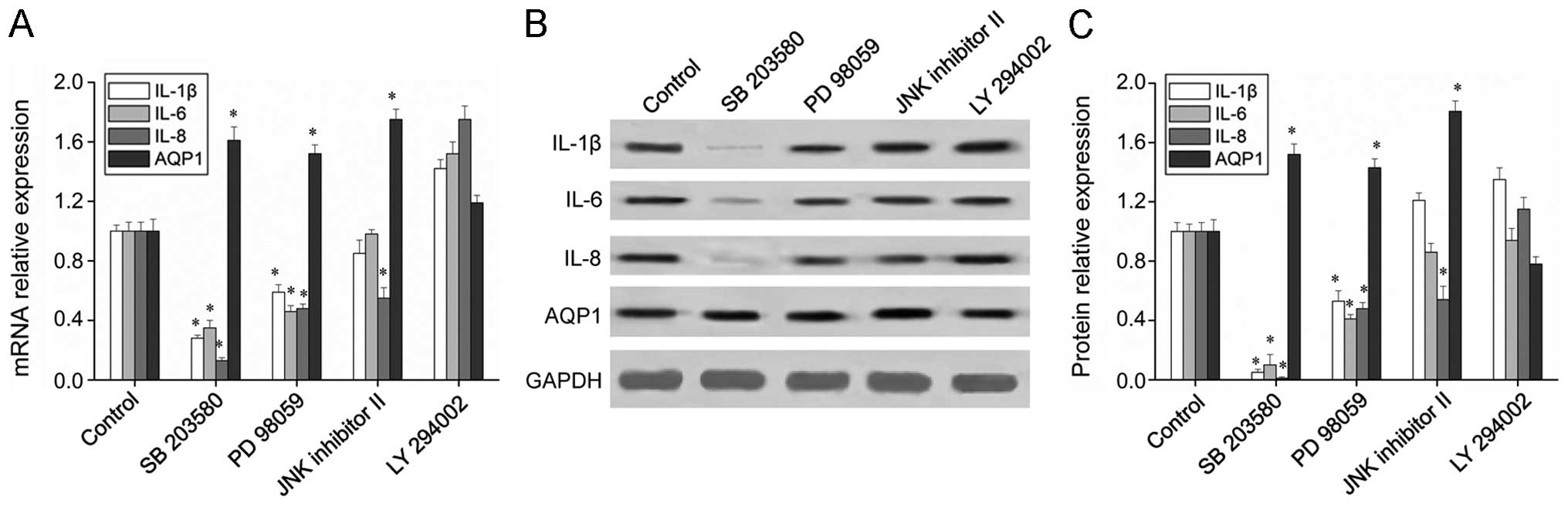
(Fig. 5) revealed that the
expression of the inflammatory factors, IL-1β, IL-6 and IL-8, was
significantly downregulated in the presence of the p38 inhibitor
(SB 203580), as well as in the presence of the ERK inhibitor (PD
98059). However, the JNK inhibitor (JNK inhibitor II) decreased
only the expression level of IL-8, and no significant changes were
observed in the expression of inflammatory factors in the presence
of the PI3K inhibitor (LY 294002). These results indicate that PBEF
regulates the expression of inflammatory factors and that the MAPK
pathways, particularly p38 MAPK, are involved. The expression of
AQP1 was upregulated significantly by the p38, ERK and JNK
inhibitors, indicating that the regulatory effects of PBEF on AQP1
are dependent on the MAPK pathways. Taken together, our results
suggest that PBEF regulates the expression of inflammatory factors
and AQPs through the MAPK pathways.
Discussion
ARDS is still one of the main causes of death of
patients in the intensive care unit (ICU), with a mortality rate of
40–60% (1); it is proving
difficult to reduce the incidence and mortality rate of ARDS. The
prevention of ALI is the main measure which must be taken in order
to reduce the incidence of ARDS. There have been many important
developments in the study of the pathogenesis, treatment strategies
and other aspects of ALI (17,18). Studies have emphasized the
important role which PBEF plays in the development of ALI. For
instance, Kamp et al (19)
proved that PBEF is a candidate gene which is related to ALI
through genome-wide association studies (GWAS). The present study
focused on investigating the important role of PBEF in ALI and its
association with inflammatory signaling pathways and pulmonary
water transport. Our results revealed that TNF-α promoted the
expression of PBEF in HPMECs, which was consistent with the results
of Hector et al (14).
Moreover, Wang et al (20)
previously reported that TNF-α enhanced the hyperbaric
oxygen-induced visfatin expression through the JNK pathway in human
coronary arterial endothelial cells.
It has been demonstrated that endothelial cell
injury is an important marker of ALI, and that the degree of injury
is closely related to the prognosis of ALI patients (21). Therefore, the study of the link
between HPMEC dysfunction and ALI has attracted increasing
attention. In the present study, the influence of PBEF
overexpression on HPMEC apoptosis was detected by flow cytometry,
and the results revealed that PBEF overexpression promoted HPMEC
apoptosis, which was consistent with the findings of the study by
Martin et al (22) and de
Souza et al (23). In
addition, Martin et al (22) found that ALI was related to an
increase in cell apoptosis in lung tissue, a delay in inflammatory
cell apoptosis, and the exacerbation of lung injury. However,
during the healing process, the apoptosis of proliferous
endothelial cells and fibroblasts occurred, and the lung tissue was
reconstructed. de Souza et al (23) found that the increase in
endothelial/epithelial cell apoptosis and the decrease in
inflammatory cell apoptosis were closely related to the occurrence
of ALI. Gao et al (24)
also demonstrated that PBEF silencing using siRNA inhibited the
expression of inflammatory cytokines and decreased the apoptosis
mediated by FasL in HPMECs.
It has also been demonstrated that PBEF plays an
important role in inflammatory signaling pathways and endothelial
cell permeability; however, the exact mechanisms remain unclear
(25). In the present study, we
demonstrated that PBEF promoted the expression levels of the
inflammatory factors, IL-1β, IL-6 and IL-8, which resulted in the
enhancement of the inflammatory response. Liu et al
(6) and Li et al (26) proved that PBEF promoted the
expression of IL-8 in human pulmonary alveolar epithelial cells.
Ognjanovic et al (27)
indicated that recombinant PBEF increased the expression of IL-6
and IL-8 in amniotic epithelial cells. PBEF has also been found to
induce the mRNA and protein expression of IL-1, IL-6, IL-10 and
TNF-α in PBMCs and that of IL-1β, IL-6 and TNF-α in CD14 monocytes
(2). Therefore, PBEF and some
corresponding inflammatory factors may have mutually promoting
effects and may exhibit a ʽwaterfall-like' chain reaction in
HPMECs. Our results confirmed that PBEF inhibited the expression of
AQP1 in HPMECs. AQP1 is a crucial molecule in maintaining the
balance of water movement between blood vessels and interstitial
fluid (9). In a previous study
using an animal model of lipopolysaccharide-induced ALI, the
expression of AQP1 was decreased, and pneumonedema with ALI
aggravation were observed (12).
In a murine model of pneumonedema induced by viral infection, it
was found that the expression of AQP1 and AQP5 in the mouse lungs
was decreased, which then recovered when inflammation became
slighter (11).
The mechanisms of the PBEF-regulated inflammatory
response remain elusive. In the present study, we found that PBEF
regulated the expression of inflammatory factors mainly through the
MAPK pathways, which was consistent with the conclusion drawn by
Moschen et al (2).
However, they also found that PBEF regulated the expression of
IL-10 and TNF-α through the PI3K pathway. Liu et al
(7) demonstrated that the
visfatin-induced monocyte chemoattractant protein (MCP)-1 and IL-6
production involved the p38 MAPK, PI3K and ERK1/2 pathways in human
umbilical vein endothelial cells. They also found that PBEF induced
the expression of IL-8, IL-16 and CCR3 through the p38 and JNK
pathways in pulmonary epithelial cells. Taken together, these
results suggest that p38 MAPK is the main pathway through which
PBEF regulates inflammatory factors.
The molecular mechanisms through which PBEF affects
the expression of AQP1 have yet to be clarified. In this study, we
found that the expression of AQP1 increased after blocking p38, ERK
and JNK, which indicated that PBEF regulates the expression of AQP1
mainly through the MAPK pathways. Therefore, it may be inferred
that, in the pathological process, the increase in PBEF expression
inhibits AQP1 expression and causes pneumonedema, mainly through
the MAPK pathways. It was also confirmed in this study that the
MAPK pathways have a close association with AQP1. Similarly,
Umenishi et al (28)
demonstrated that the activation of the ERK, p38 and JNK pathways,
as well as the hypertonicity response element in the AQP1 promoter,
was involved in hypertonicity-induced AQP1 expression in mouse
innermedullary collecting duct (mIMCD)-3 cells. MEK/ERK mediated
the UVB-induced AQP1 downregulation and water-permeability
impairment in human retinal pigment epithelial cells, which was
also confirmed by Jiang et al (29). Furthermore, Shankardas et
al (30) found that AQP1
played an important role in human corneal endothelial cell (HCEC)
proliferation and migration through the ERK signaling pathway.
In conclusion, our study demonstrated that PBEF
regulated the pathological process of ALI by promoting HPMEC
apoptosis and regulating the expression of inflammatory factors and
AQP1 through the MAPK and PI3K pathways. These results provide
valuable experimental evidence which may be used in future studies
on the biological function, molecular mechanisms and clinical
application potential of PBEF in ALl pathogenesis.
Acknowledgments
This study was supported by the Hunan Provincial
Natural Science Foundation of China (2015JJ4056), Hunan Provincial
Science and Technology Plan Project of China (2013FJ4106) and the
High Technology Industry Department, Development and Reform
Commission of Hunan Province, China (2012–1493).
References
|
1
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moschen AR, Kaser A, Enrich B, Mosheimer
B, Theurl M, Niederegger H and Tilg H: Visfatin, an adipocytokine
with proinflammatory and immunomodulating properties. J Immunol.
178:1748–1758. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ye SQ, Simon BA, Maloney JP,
Zambelli-Weiner A, Gao L, Grant A, Easley RB, McVerry BJ, Tuder RM,
Standiford T, et al: Pre-B-cell colony-enhancing factor as a
potential novel biomarker in acute lung injury. Am J Respir Crit
Care Med. 171:361–370. 2005. View Article : Google Scholar
|
|
4
|
Bajwa EK, Yu CL, Gong MN, Thompson BT and
Christiani DC: Pre-B-cell colony-enhancing factor gene
polymorphisms and risk of acute respiratory distress syndrome. Crit
Care Med. 35:1290–1295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ye SQ, Zhang LQ, Adyshev D, Usatyuk PV,
Garcia AN, Lavoie TL, Verin AD, Natarajan V and Garcia JG:
Pre-B-cell-colony-enhancing factor is critically involved in
thrombin-induced lung endothelial cell barrier dysregulation.
Microvasc Res. 70:142–151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu P, Li H, Cepeda J, Zhang LQ, Cui X,
Garcia JG and Ye SQ: Critical role of PBEF expression in pulmonary
cell inflammation and permeability. Cell Biol Int. 33:19–30. 2009.
View Article : Google Scholar
|
|
7
|
Liu SW, Qiao SB, Yuan JS and Liu DQ:
Visfatin stimulates production of monocyte chemotactic protein-1
and interleukin-6 in human vein umbilical endothelial cells. Horm
Metab Res. 41:281–286. 2009. View Article : Google Scholar
|
|
8
|
Liu P, Li H, Cepeda J, Xia Y, Kempf JA, Ye
H, Zhang LQ and Ye SQ: Regulation of inflammatory cytokine
expression in pulmonary epithelial cells by pre-B-cell
colony-enhancing factor via a nonenzymatic and AP-1-dependent
mechanism. J Biol Chem. 284:27344–27351. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Verkman AS: Role of aquaporins in lung
liquid physiology. Respir Physiol Neurobiol. 159:324–330. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li J, Xu M, Fan Q, Xie X, Zhang Y, Mu D,
Zhao P, Zhang B, Cao F, Wang Y, et al: Tanshinone IIA ameliorates
seawater exposure-induced lung injury by inhibiting aquaporins
(AQP) 1 and AQP5 expression in lung. Respir Physiol Neurobiol.
176:39–49. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Towne JE, Harrod KS, Krane CM and Menon
AG: Decreased expression of aquaporin (AQP)1 and AQP5 in mouse lung
after acute viral infection. Am J Respir Cell Mol Biol. 22:34–44.
2000. View Article : Google Scholar
|
|
12
|
Su X, Song Y, Jiang J and Bai C: The role
of aquaporin-1 (AQP1) expression in a murine model of
lipopolysaccharide-induced acute lung injury. Respir Physiol
Neurobiol. 142:1–11. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lai KN, Leung JC, Metz CN, Lai FM, Bucala
R and Lan HY: Role for macrophage migration inhibitory factor in
acute respiratory distress syndrome. J Pathol. 199:496–508. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hector J, Schwarzloh B, Goehring J, Strate
TG, Hess UF, Deuretzbacher G, Hansen-Algenstaedt N, Beil FU and
Algenstaedt P: TNF-alpha alters visfatin and adiponectin levels in
human fat. Horm Metab Res. 39:250–255. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fujita-Yoshigaki J, Matsuki-Fukushima M
and Sugiya H: Inhibition of Src and p38 MAP kinases suppresses the
change of claudin expression induced on dedifferentiation of
primary cultured parotid acinar cells. Am J Physiol Cell Physiol.
294:C774–C785. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schindler JF, Monahan JB and Smith WG: p38
pathway kinases as anti-inflammatory drug targets. J Dent Res.
86:800–811. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Raghavendran K, Pryhuber GS, Chess PR,
Davidson BA, Knight PR and Notter RH: Pharmacotherapy of acute lung
injury and acute respiratory distress syndrome. Curr Med Chem.
15:1911–1924. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Licker M, Fauconnet P, Villiger Y and
Tschopp JM: Acute lung injury and outcomes after thoracic surgery.
Curr Opin Anaesthesiol. 22:61–67. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kamp R, Sun X and Garcia JG: Making
genomics functional: deciphering the genetics of acute lung injury.
Proc Am Thorac Soc. 5:348–353. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang BW, Lin CM, Wu GJ and Shyu KG: Tumor
necrosis factor-α enhances hyperbaric oxygen-induced visfatin
expression via JNK pathway in human coronary arterial endothelial
cells. J Biomed Sci. 18:272011. View Article : Google Scholar
|
|
21
|
Maniatis NA, Kotanidou A, Catravas JD and
Orfanos SE: Endothelial pathomechanisms in acute lung injury.
Vascul Pharmacol. 49:119–133. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martin TR, Nakamura M and Matute-Bello G:
The role of apoptosis in acute lung injury. Crit Care Med. 31(4
Suppl): S184–S188. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
de Souza PM and Lindsay MA: Apoptosis as a
therapeutic target for the treatment of lung disease. Curr Opin
Pharmacol. 5:232–237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao W, Mao Q, Feng AW, Sun HM, Sun WK, Lu
X, Su X and Shi Y: Inhibition of pre-B cell colony-enhancing factor
attenuates inflammation and apoptosis induced by pandemic H1N1 2009
in lung endothelium. Respir Physiol Neurobiol. 178:235–241. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pilz S, Mangge H, Obermayer-Pietsch B and
März W: Visfatin/pre-B-cell colony-enhancing factor: a protein with
various suggested functions. J Endocrinol Invest. 30:138–144. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li H, Liu P, Cepeda J, Fang D, Easley RB,
Simon BA, Zhang LQ and Ye SQ: Augmentation of pulmonary epithelial
cell IL-8 expression and permeability by pre-B-cell colony
enhancing factor. J Inflamm (Lond). 5:152008. View Article : Google Scholar :
|
|
27
|
Ognjanovic S and Bryant-Greenwood GD:
Pre-B-cell colony-enhancing factor, a novel cytokine of human fetal
membranes. Am J Obstet Gynecol. 187:1051–1058. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Umenishi F and Schrier RW:
Hypertonicity-induced aquaporin-1 (AQP1) expression is mediated by
the activation of MAPK pathways and hypertonicity-responsive
element in the AQP1 gene. J Biol Chem. 278:15765–15770. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang Q, Cao C, Lu S, Kivlin R, Wallin B,
Chu W, Bi Z, Wang X and Wan Y: MEK/ERK pathway mediates UVB-induced
AQP1 downregulation and water permeability impairment in human
retinal pigment epithelial cells. Int J Mol Med. 23:771–777. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shankardas J, Patil RV and Vishwanatha JK:
Effect of down-regulation of aquaporins in human corneal
endothelial and epithelial cell lines. Mol Vis. 16:1538–1548.
2010.PubMed/NCBI
|