Introduction
Glaucoma, an irreversible neurodegenerative disease
(1), affects ~60 million people
worldwide (2). Primary open-angle
glaucoma (POAG) and primary angle-closure glaucoma (PACG) are the
predominant types of glaucoma in various populations (2). Genetic factors have well-known
important roles in the development of glaucoma (3–7);
mutations in 7 genes (8–14) are responsible for a small portion
of glaucoma (15–17), and recent studies have disclosed a
number of new genes or loci associated with glaucoma (18–27). However, the exact genetic defects
involved remain elusive for the majority of patients.
Glaucoma is frequently observed in patients with
anterior segment dysgenesis (ASD), microcornea or microphthalmia.
Approximately 50% of patients with ASD will eventually develop
glaucoma (28). The incidence of
glaucoma is 77% in elderly patients with relative anterior
microphthalmus (cornea diameter <11 mm, axial length >20 mm)
(29). Microphthalmia, which is
always accompanied with microcornea, is considered a primary risk
factor of angle-closure glaucoma (30). Mutations in a number of genes have
been linked to ASD, microcornea and microphthalmia (31–36), and some of these were recently
reported to be responsible for primary glaucoma (37,38). Systemic analysis of these genes in
patients with primary glaucoma may provide an overview of the
contribution of their mutations to primary glaucoma.
In our previous study, whole-exome sequencing was
performed for 257 patients with primary glaucoma, where mutations
in 7 known glaucoma genes were present in 7.8% of patients
(15). In the present study,
variants from exome sequencing for 43 genes known to be associated
with ASD, microcornea or microphthalmia were selected for further
analysis. Overall, 27 potential pathogenic variants in 14 of the 43
genes were identified in 28 of 257 patients with primary glaucoma,
suggesting a possible association of these genes with primary
glaucoma.
Materials and methods
Patients
The 257 unrelated patients with primary glaucoma,
including 125 with POAG and 132 with PACG, have been described in
our previous study (15). Written
informed consent was obtained from the participants or their
guardians prior to the collection of clinical data and peripheral
venous blood samples. The study was consistent with the tenets of
the Declaration of Helsinki and was approved by the Institutional
Review Board of the Zhongshan Ophthalmic Center (Guangdong, China).
Whole-exome sequencing on genomic DNA from the patients has been
described in our previous study (15). In brief, the solution-based exome
capture system (TruSeq Exome Enrichment kits; Illumina, Inc., San
Diego, CA, USA) was applied and the average sequencing depth was
set at 125-fold.
Selection of genes for analysis
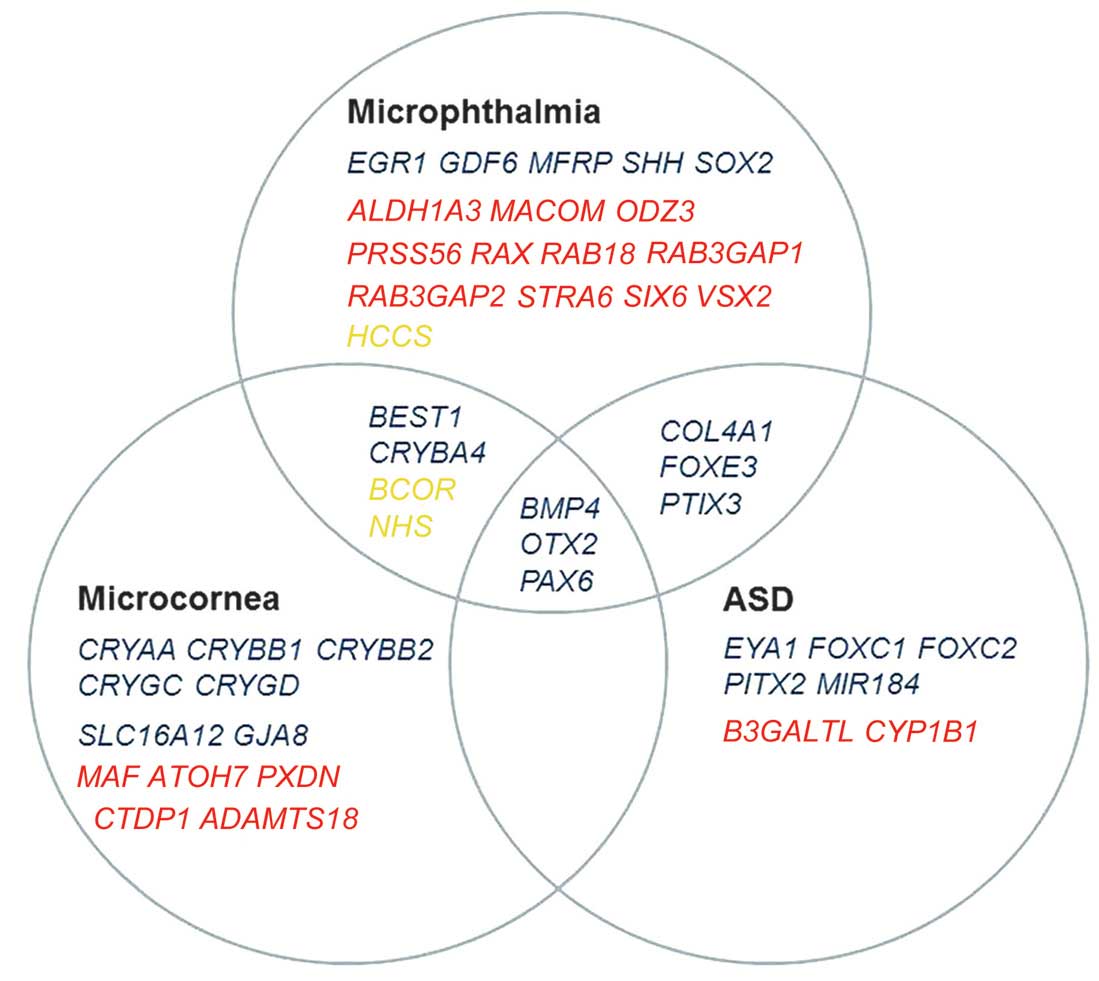
Genes associated with ASD, microcornea or
microphthalmia were selected based on the PubMed search (http://www.ncbi.nlm.nih.gov/) accessed on February 1,
2014. The classification of phenotypic spectrum of ASD was based on
a previous review (28). The
following search terms were used: [mutation AND (ASD OR
Axenfeld-Rieger Syndrome OR Peters anomaly OR Peters Plus syndrome
OR aniridia OR sclerocornea OR megalocornea OR microcornea OR
microphthalmia)] AND ('2009/02/01' [Date-Publication]: '2014/02/01'
[Date-Publication]). From all the reports identified with the
associated results, only those describing genes with mutations in
humans were selected for further analysis, which resulted in 46
candidate genes (Fig. 1). Of the
46 genes, 43 were included in the present study, while one
(CYP1B1) had been analyzed in our previous study (15) and two, PRSS56 and
MACOM, were excluded as they were not captured by the TruSeq
Exome Enrichment kit. Variants in the 43 genes were selected from
whole-exome sequencing and subsequently filtered through the
following steps: ⅰ) Inclusion criteria of variant selection:
Variants predicted to affect the coding residue or mRNA splicing;
variants with minor allele frequency <0.01 compared with the
1000 Genomes Project database accessed on September 1, 2014;
missense variants predicted to be damaging by either
PolyPhen-2(http://genetics.bwh.harvard. edu/pph2/) or SIFT
(http://sift.jcvi.org/www/SIFT_enst_submit.html)
(39,40); intronic variants predicted to
affect splicing site by BDGP (http://www.fruitfly.org/); nonsense variants,
insertions and deletions; and heterozygous variants in genes
associated with autosomal dominant diseases, compound heterozygous
or homozygous variants in genes associated with autosomal recessive
diseases, hemizygous variants in genes associated with X-linked
recessive diseases, and both hemizygous and heterozygous variants
in genes associated with X-linked dominant diseases. ⅱ) Selected
variants confirmed by Sanger sequencing were analyzed further. ⅲ)
For genes only with specific types of variants reported to be
correlated with associated eye diseases, other types of variants
were tentatively listed as less likely pathogenic variants. For
example, missense variants in NHS were listed as less likely
pathogenic variants as only truncation mutations in this gene had
been reported to be causative. ⅳ) The remaining variants were
validated based on 192 ethnicity-matched normal controls and
available family members.
Primer design
The primers used to confirm the candidate variant
were designed using the Primer3 online tool (http://primer3.ut.ee/) (41). Polymerase chain reaction was used
to amplify the fragments harboring the target variants. The
sequence of the amplicons was determined with an ABI BigDye
Terminator v3.1 Cycle Sequencing kit on an ABI3130 Genetic Analyzer
(both from Applied Biosystems, Foster City, CA, USA) as described
previously (42).
Results
Analysis of the variants
Overall, 70 candidate variants of the 43 genes were
selected from data derived from whole-exome sequencing on the 257
patients. Of the 70, 53 (75.7%) were confirmed by Sanger
sequencing, while 17 were false-positives. The compound
heterozygous variants in B3GALTL were excluded as only one
was confirmed and the other was a false-positive. Fifteen variants
in NHS, BCOR and COL4A1 were tentatively
categorized as less likely pathogenic variants as these types of
causative mutations had not been previously reported. Six of the
remaining 37 variants were excluded as they were also presented in
normal individuals. Three of the remaining variants were of
uncertain significance as they were detected in patients with
potential pathogenic mutations in known glaucoma genes. In
addition, one variant in PAX6 was excluded as it was absent
in other affected family members. Eventually, 27 potential
pathogenic mutations in 14 genes were identified (Table I). Of the 27, 20 were not present
in the 1000 Genomes Project or Exome Variant Server, while 7 were
present in the 1000 Genomes Project and Exome Variant Server with a
frequency of 2/2,184 to 1/13,006. All the 27 mutations were absent
in the 192 ethnicity-matched normal controls and were predicted to
be damaging to the encoded protein by bioinformatic analysis.
 | Table IPotential pathogenic mutations
identified in 28 unrelated Chinese patients with primary
glaucoma. |
Table I
Potential pathogenic mutations
identified in 28 unrelated Chinese patients with primary
glaucoma.
| Gene | Inh | PatientID | Diagnosis | Variations
| Online prediction
| MAF in NC | Reported or
nota | MAF in1000G or
EVS |
|---|
| Nucleotide | Amino acid | SIFT | PolyPhen-2 |
|---|
| CRYAA | AD | G636 | PACG |
c.[307C>T];[=] | p.[R103C];[=] | D | PrD | 0/384 | Novel | None |
| CRYGC | AD | G217 | PACG |
c.[110G>A];[=] | p.[R37Q];[=] | D | PrD | 0/384 | rs140859599 | 1/2184,
1/13006 |
| CRYGD | AD | G598 | PACG |
c.[19T>C];[=] | p.[Y7H];[=] | D | PrD | 0/384 | Novel | None |
| COL4A1 | AD | G353 | POAG |
c.[502G>A];[=] | p.[G168R];[=] | T | PrD | 0/384 | rs144171664 | Unknown |
| FOXC1 | AD | G378 | POAG |
c.[553_555del];[=] |
p.[185_185del];[=] | NA | NA | 0/384 | Novel | None |
| GJA8 | AD | G462 | POAG |
c.[569A>G];[=] | p.[N190S];[=] | D | PrD | 0/384 | Novel | None |
| PITX2 | AD | G654 | PACG |
c.[891C>A];[=] | p.[Q297H];[=] | T | PrD | 0/384 | Novel | None |
| SHH | AD | G408 | POAG |
c.[682G>A];[=] | p.[D228N];[=] | D | PrD | 0/384 | Novel | None |
| BMP4 | AD | G555 | PACG |
c.[450C>G];[=] | p.[N150K];[=] | T | PrD | 0/384 | Reportedb | None |
| G370 | POAG |
c.[502G>C];[=] | p.[G168R];[=] | D | PrD | 0/384 | Novel | None |
| CRYBA4 | AD | G644 | PACG |
c.[383C>T];[=] | p.[S128F];[=] | D | PrD | 0/384 | Novel | None |
| G603 | PACG |
c.[413A>G];[=] | p.[E138G];[=] | D | PrD | 0/384 | Novel | None |
| GDF6 | AD | G629 | PACG |
c.[136C>T];[=] | p.[R46C];[=] | D | B | 0/384 | Novel | None |
| G479 | POAG |
c.[1271A>G];[=] | p.[K424R];[=] | T | PrD | 0/384 | rs121909353c | 2/2184, none |
| G539 | PACG |
c.[1288A>G];[=] | p.[I430V];[=] | T | PrD | 0/384 | Novel | None |
| EYA1 | AD | G443 | POAG |
c.[35G>A];[=] | p.[R12H];[=] | T | PrD | 0/384 | rs74720958 | 1/2184, none |
| G447 | POAG |
c.[175G>A];[=] | p.[G59R];[=] | D | PrD | 0/384 | rs146216506 | Unknown,
1/13006 |
| G455 | POAG |
c.[585A>G];[=] | p.[I195M];[=] | D | B | 0/384 | Novel | None |
| G543 | PACG |
c.[679G>C];[=] | p.[A227P];[=] | T | PrD | 0/384 | Novel | None |
| BEST1 | AD | G617 | PACG |
c.[205T>C];[=] | p.[C69R];[=] | D | PrD | 0/384 | Novel | None |
| G381 | POAG |
c.[436G>T];[=] | p.[A146S];[=] | T | PrD | 0/384 | Novel | None |
| G664 | PACG |
c.[652C>A];[=] | p.[R218S];[=] | D | PrD | 0/384 | Reportedd | None |
| G38 | PACG |
c.[698C>T];[=] | p.[P233L];[=] | D | PrD | 0/384 | Reportede | None |
| G402, G587 | POAG, PACG |
c.[763C>T];[=] | p.[R255W];[=] | D | PrD | 0/384 | rs372989281f | Unknown,
1/13002 |
| G663 | PACG |
c.[910_912del];[=] |
p.[304_304del];[=] | NA | NA | 0/384 | Novel | None |
| HCCS | XL | G592 | PACG |
c.[175C>T];[0] | p.[R59C];[0] | D | PrD | 0/286g | rs200354469 | Unknown |
| G524 | PACG |
c.[572A>T];[=] | p.[E191V];[=] | T | PrD | 0/286g | Novel | None |
Associations of the mutations with
disease
Of the 27 mutations, 25 were heterozygous in 13
genes associated with autosomal dominant diseases, one was
heterozygous and one was hemizygous in HCCS associated with
X-linked dominant diseases, and none were present in the genes
associated with autosomal recessive diseases. Five of the 27
mutations have been previously reported to be pathogenic (43–47), while the remaining 22 were novel.
The 27 mutations were detected in 28 of 257 patients with glaucoma,
including 11 patients with POAG and 17 patients with PACG (Table I). The distributions of the 27
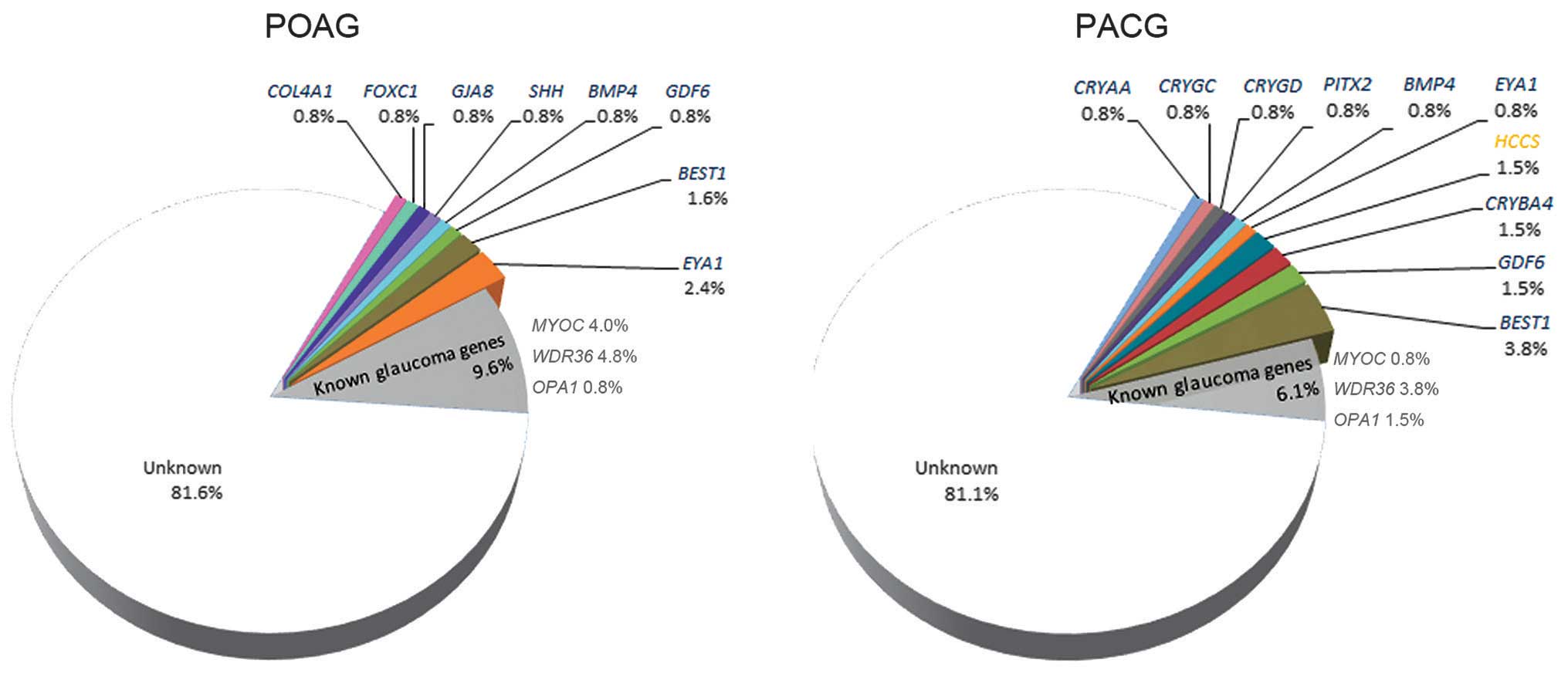
mutations in POAG and PACG are illustrated in Fig. 2. Mutations in COL4A1,
FOXC1, GJA8 and SHH were only detected in
patients with POAG, while mutations in CRYAA, CRYGC,
CRYGD, CRYBA4, PITX2 and HCCS were only
detected in patients with PACG. Mutations in BMP4,
GDF6, EYA1 and BEST1 were detected in the two
groups of patients. Of the 27 mutations, 26 were detected in 26
patients, respectively; while the remaining mutation, a previously
reported c.763C>T mutation in BEST1 (45), was detected in 1 patient with PACG
and 1 patient with POAG.
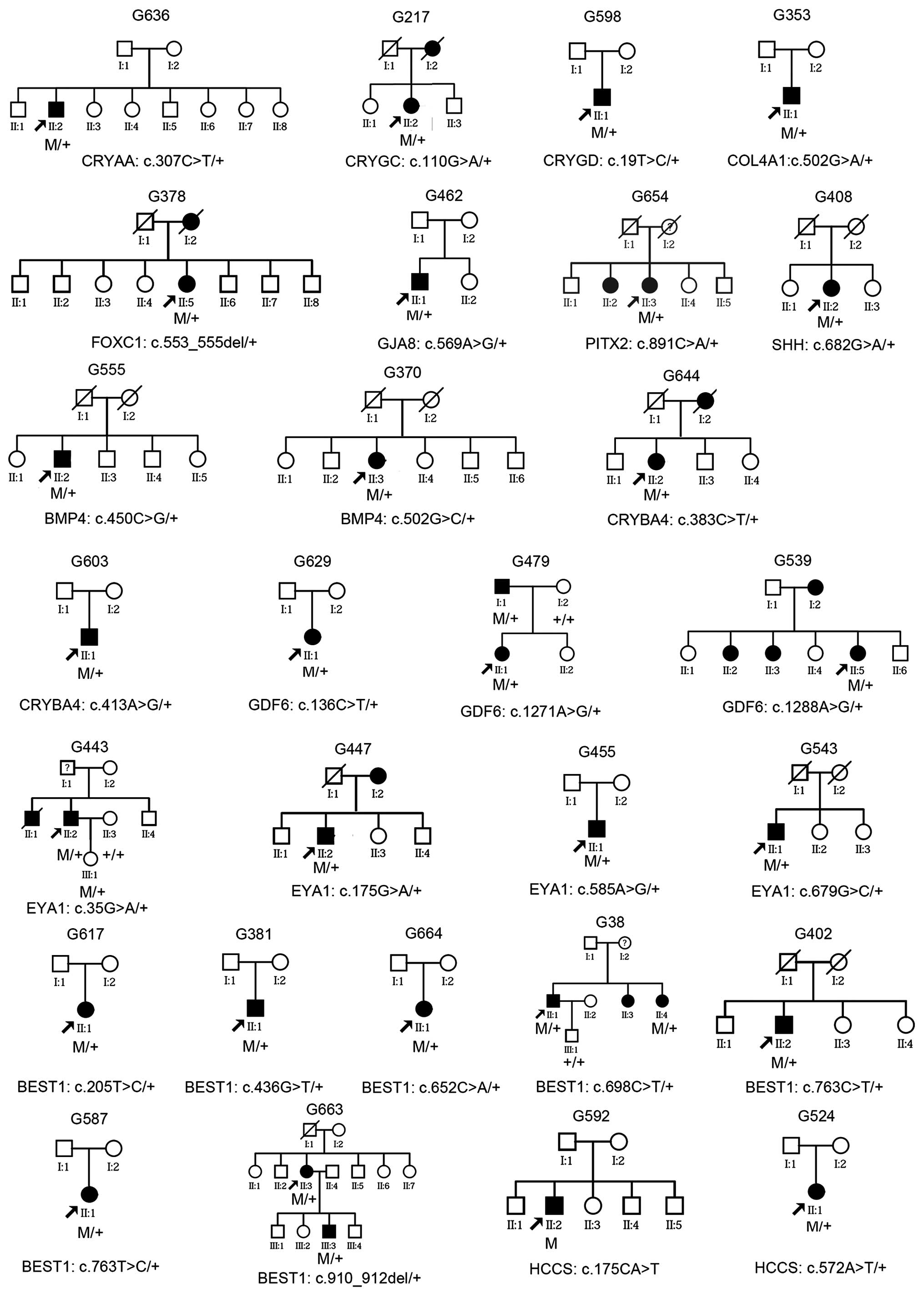
Analysis of family history
Of the 28 patients with mutations, 10 had a family
history of glaucoma suggesting an autosomal dominant trait, and the
other 18 were sporadic (Fig. 3).
Analysis of limited family members from four families showed
segregation of glaucoma with mutations in the GDF6,
EYA1 and BEST1 genes (Fig. 3). In one of the five families, the
patient (G443) and his daughter had the c.35G>A (p.R12H)
mutation in EYA1; however, the phenotype of the daughter had
signs of glaucoma risk but did not meet the diagnostic criteria:
Unilateral elevated intraocular pressure (18 mmHg for the right eye
and 23 mmHg for the left) at the age of 11 years, but had normal
visual field and retinal nerve fiber layers on optical coherence
tomography. For the 29 patients with mutations and an initial
diagnosis of primary glaucoma, other signs associated with ASD,
microcornea and microphthalmia were not observed except for a
slightly smaller corneal diameter (10–11 mm) in 3 patients
(patients G38, G479 and G587; Table
II) following careful re-examination. In addition, macular
lesion with yellow-white deposits was observed in 2 patients with
BEST1 mutation and in affected family members in the two
respective families.
| Table IIClinical data of the 28 patients with
potential pathogenic mutations. |
Table II
Clinical data of the 28 patients with
potential pathogenic mutations.
| Family ID | Diagnosis | Gene | Mutationa | Gender | Diagnosis age,
years | Cornea, mm | AL, mm | BCVA | Peak IOP, mmHg | VCDR |
|---|
| G636 | PACG | CRYAA |
c.[307C>T];[=] | M | 60 | 11.6/11.6 | 21.99/22.00 | 0.7/0.8 | 39/14 | 1.0/0.3 |
| G217 | PACG | CRYGC |
c.[110G>A];[=] | F | 49 | 11.0/11.0 | NA | 0.6/0.4 | 52/NA | 0.3/0.9 |
| G598 | PACG | CRYGD |
c.[19T>C];[=] | F | 63 | 11.8/11.3 | 22.11/22.27 | HM/1.0 | 49/20.3 | 0.9/0.9 |
| G353 | POAG | COL4A1 |
c.[502G>A];[=] | M | 60 | 12.0/11.9 | 26.45/26.42 | 1.0/1.0 | NA | 0.7/0.8 |
| G378 | POAG | FOXC1 |
c.[553_555del];[=] | F | 68 | 12.3/12.3 | 26.20/26.15 | 1.2/1.2 | 23/23 | 0.4/0.5 |
| G462 | POAG | GJA8 |
c.[569A>G];[=] | M | 29 | 11.6/11.5 | 22.25/22.38 | 1.2/HM | 24/22 | 0.6/1.0 |
| G654 | PACG | PITX2 |
c.[891C>A];[=] | F | 51 | NA | 22.50/24.02 | 0.3/FC | NAb | 0.9/0.9 |
| G408 | POAG | SHH |
c.[682G>A];[=] | F | 58 | 11.9/11.4 | 23.48/23.43 | 0.7/1.2 | NAb | 0.5/0.5 |
| G555 | PACG | BMP4 |
c.[450C>G];[=] | M | 64 | 11.0/11.0 | 22.57/23.01 | 1.0/NLP | NA/32 | 0.7/1.0 |
| G370 | POAG | BMP4 |
c.[502G>C];[=] | F | 52 | 11.3/11.2 | 23.45/23.69 | 0.9/1.0 | 33/28 | 0.8/0.8 |
| G644 | PACG | CRYBA4 |
c.[383C>T];[=] | F | 65 | 11.4/11.8 | 21.80/21.72 | 0.4/1.2 | NAb | 0.3/0.6 |
| G603 | PACG | CRYBA4 |
c.[413A>G];[=] | M | 70 | NA/11.2 | 23.70/23.67 | NLP/0.6 | 37/14 | 0.9/0.4 |
| G629 | PACG | GDF6 |
c.[136C>T];[=] | F | 53 | NA | 21.29/21.29 | 0.5/0.2 | 16/54 | 0.3/NA |
| G479 | POAG | GDF6 |
c.[1271A>G];[=] | F | 30 | 10.0/10.0 | 26.06/25.92 | 0.7/0.8 | 22/25 | 0.6/0.4 |
| G539 | PACG | GDF6 |
c.[1288A>G];[=] | F | 49 | NA | NA | 0.6/0.7 | NA | 0.3/0.3 |
| G443 | POAG | EYA1 |
c.[35G>A];[=] | M | 30 | 11.0/11.0 | 29.05/NA | 0.2/LP | 30/NA | 0.3/NA |
| G447 | POAG | EYA1 |
c.[175G>A];[=] | M | 56 | 11.8/11.3 | 23.73/23.55 | 0.6/0.7 | NAb | 0.9/0.9 |
| G455 | POAG | EYA1 |
c.[585A>G];[=] | M | 32 | 12.4/12.4 | 23.90/23.95 | 1.5/NLP | NAb | 0.9/1.0 |
| G543 | PACG | EYA1 |
c.[679G>C];[=] | M | 56 | 11.5/11.5 | 22.25/22.30 | 1.2/0.9 | 13/35 | 0.3/0.4 |
| G617 | PACG | BEST1 |
c.[205T>C];[=] | F | 72 | 11.5/11.4 | 24.48/23.91 | 0.5/0.5 | NA | 0.4/0.7 |
| G381 | POAG | BEST1 |
c.[436G>T];[=] | M | 34 | 12.2/11.6 | 25.36/25.09 | 1.5/FC | 48/55 | 0.9/1.0 |
| G664 | PACG | BEST1 |
c.[652C>A];[=] | F | 47 | 11.6/12.1 | 21.24/21.33 | 1.0/1.2 | NAb | 0.4/0.4 |
| G38 | PACG | BEST1 |
c.[698C>T];[=] | M | 20 | 10.5/10.0 | 21.38/21.38 | 0.5/0.2 | 27/32 | 0.5/0.7 |
| G402 | POAG | BEST1 |
c.[763C>T];[=] | M | 56 | 12.3/12.4 | 25.23/25.17 | 0.4/0.6 | 40/40 | 0.9/0.9 |
| G587 | PACG | BEST1 |
c.[763C>T];[=] | F | 68 | 11.2/10.6 | 22.17/21.91 | 1.0/0.2 | 17/39 | 0.5/0.9 |
| G663 | PACG | BEST1 |
c.[910_912del];[=] | F | 44 | NA/12.4 | 21.20/21.42 | 0.05/0.05 | 51/33 | 1.0/1.0 |
| G592 | PACG | HCCS |
c.[175C>T];[0] | M | 47 | NA | 22.48/22.44 | 0.5/0.8 | 36/23 | 0.9/0.5 |
| G524 | PACG | HCCS |
c.[572A>T];[=] | F | 80 | 11.0/11.0 | 22.62/22.75 | 0.1/0.3 | 50/9.5 | 0.5/0.3 |
Discussion
In the present study, 27 potential pathogenic
mutations in 14 genes have been identified in 28 of 257 patients
with primary glaucoma based on analysis of exome sequencing results
for 43 genes associated with ASD, microcornea or microphthalmia.
The 27 mutations were confirmed by Sanger sequencing and were
predicted as damaging by bioinformatic analysis. Five of the 27
mutations have been previously reported to be correlated with
different forms of associated ocular diseases (43–47) and the remaining 22 are novel. All
the mutations were absent in normal controls and the majority of
them were not present in existing human genome variant databases.
Analysis of family members from five families suggests a
segregation of primary glaucoma with mutations. These lines of
evidence suggest that the mutations in these genes are likely to
have roles in the development of primary glaucoma.
Glaucoma, secondary to ASD, microcornea or
microphthalmia, has been described in patients with mutations in
one of the following genes: BEST1 (48), BMP4 (49), COL4A1 (50), FOXC1 (51), FOXE3 (52), PAX6 (53), PITX2 (54,55), PXDN (56), PRSS56 (38), SIX6 (37) and VSX2 (57). The association of mutations in
these genes with primary glaucoma has not been previously studied,
except for a recent study in which rare and common variants in
SIX6 have been demonstrated as a risk factor for POAG
(37). Such variants in other
associated genes may also be risk factors for primary glaucoma. The
identification of 27 rare damaging variants in 14 associated genes
in 28 of the 257 patients in the present study further supports the
potential involvement of these genes in primary glaucoma. By
contrast, certain patients with variants in these genes may have
minor or subtle changes in anterior segment, as seen in 3 (G38,
G479 and G587) of the 28 patients with a relatively smaller corneal
diameter. These changes may possibly be neglected or undetected,
and therefore, the patients with such changes may mimic primary
glaucoma. In either case, variants in these genes are possibly risk
factors for primary and secondary glaucoma.
The present preliminary study provides a brief
overview of variants in the 43 genes associated with ASD,
microcornea and microphthalmia in patients with primary glaucoma.
The identification of 27 potential pathogenic variants in genes
associated with ASD, microcornea and microphthalmia in 28 of 257
patients with primary glaucoma suggests potential risk factors in
the development of primary glaucoma. Further studies are expected
to enrich the understanding between variants in these genes and
primary glaucoma.
Acknowledgments
The authors would like to thank the patients and
their families for their participation. The present study was
supported by the National Natural Science Foundation of China
(grant no. U1201221), Natural Science Foundation of Guangdong
(grant no. S2013030012978), Guangdong Department of Science &
Technology Transla tional Medicine Center (grant no.
2011A080300002), and the Fundamental Research Funds of the State
Key Laboratory of Ophthalmology.
References
|
1
|
Foster PJ, Buhrmann R, Quigley HA and
Johnson GJ: The definition and classification of glaucoma in
prevalence surveys. Br J Ophthalmol. 86:238–242. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cook C and Foster P: Epidemiology of
glaucoma: What's new? Can J Ophthalmol. 47:223–226. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Quigley HA: Glaucoma. Lancet.
377:1367–1377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Janssen SF, Gorgels TG, Ramdas WD, Klaver
CC, van Duijn CM, Jansonius NM and Bergen AA: The vast complexity
of primary open angle glaucoma: Disease genes, risks, molecular
mechanisms and pathobiology. Prog Retin Eye Res. 37:31–67. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ojha P, Wiggs JL and Pasquale LR: The
genetics of intraocular pressure. Semin Ophthalmol. 28:301–305.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wiggs JL: Genetic etiologies of glaucoma.
Arch Ophthalmol. 125:30–37. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rao KN, Nagireddy S and Chakrabarti S:
Complex genetic mechanisms in glaucoma: An overview. Indian J
Ophthalmol. 59(Suppl): S31–S42. 2011. View Article : Google Scholar :
|
|
8
|
Monemi S, Spaeth G, DaSilva A, et al:
Identification of a novel adult-onset primary open-angle glaucoma
(POAG) gene on 5q22.1. Hum Mol Genet. 14:725–733. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stone EM, Fingert JH, Alward WL, et al:
Identification of a gene that causes primary open angle glaucoma.
Science. 275:668–670. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rezaie T, Child A, Hitchings R, et al:
Adult-onset primary open- angle glaucoma caused by mutations in
optineurin. Science. 295:1077–1079. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pasutto F, Matsumoto T, Mardin CY, et al:
Heterozygous NTF4 mutations impairing neurotrophin-4 signaling in
patients with primary open-angle glaucoma. Am J Hum Genet.
85:447–456. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Melki R, Colomb E, Lefort N, Brézin AP and
Garchon HJ: CYP1B1 mutations in French patients with early-onset
primary open-angle glaucoma. J Med Genet. 41:647–651. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ali M, McKibbin M, Booth A, et al: Null
mutations in LTBP2 cause primary congenital glaucoma. Am J Hum
Genet. 84:664–671. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aung T, Ocaka L, Ebenezer ND, et al: A
major marker for normal tension glaucoma: Association with
polymorphisms in the OPA1 gene. Hum Genet. 110:52–56. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang X, Li M, Guo X, Li S, Xiao X, Jia X,
Liu X and Zhang Q: Mutation analysis of seven known
glaucoma-associated genes in Chinese patients with glaucoma. Invest
Ophthalmol Vis Sci. 55:3594–3602. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fingert JH: Primary open-angle glaucoma
genes. Eye (Lond). 25:587–595. 2011. View Article : Google Scholar
|
|
17
|
Sripriya S, Uthra S, Sangeetha R, George
RJ, Hemamalini A, Paul PG, Amali J, Vijaya L and Kumaramanickavel
G: Low frequency of myocilin mutations in Indian primary open-angle
glaucoma patients. Clin Genet. 65:333–337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hysi PG, Cheng CY, Springelkamp H, et al:
BMES GWAS Group; NEIGHBORHOOD Consortium; Wellcome Trust Case
Control Consortium 2: Genome-wide analysis of multi-ancestry
cohorts identifies new loci influencing intraocular pressure and
susceptibility to glaucoma. Nat Genet. 46:1126–1130. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gharahkhani P, Burdon KP, Fogarty R, et al
Wellcome Trust Case Control Consortium 2; NEIGHBORHOOD Consortium:
Common variants near ABCA1, AFAP1 and GMDS confer risk of primary
open-angle glaucoma. Nat Genet. 46:1120–1125. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wiggs JL, Yaspan BL, Hauser MA, et al:
Common variants at 9p21 and 8q22 are associated with increased
susceptibility to optic nerve degeneration in glaucoma. PLoS Genet.
8:e10026542012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rao KN, Kaur I, Parikh RS, Mandal AK,
Chandrasekhar G, Thomas R and Chakrabarti S: Variations in NTF4,
VAV2, and VAV3 genes are not involved with primary open-angle and
primary angle-closure glaucomas in an indian population. Invest
Ophthalmol Vis Sci. 51:4937–4941. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ozel AB, Moroi SE, Reed DM, et al NEIGHBOR
Consortium: Genome-wide association study and meta-analysis of
intraocular pressure. Hum Genet. 133:41–57. 2014. View Article : Google Scholar :
|
|
23
|
Dietz JA, Maes ME, Huang S, Yandell BS,
Schlamp CL, Montgomery AD, Allingham RR, Hauser MA and Nickells RW:
Spink2 modulates apoptotic susceptibility and is a candidate gene
in the Rgcs1 QTL that affects retinal ganglion cell death after
optic nerve damage. PLoS One. 9:e935642014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen Y, Lin Y, Vithana EN, et al: Common
variants near ABCA1 and in PMM2 are associated with primary
open-angle glaucoma. Nat Genet. 46:1115–1119. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen Y, Chen X, Wang L, Hughes G, Qian S
and Sun X: Extended association study of PLEKHA7 and COL11A1 with
primary angle closure glaucoma in a Han Chinese population. Invest
Ophthalmol Vis Sci. 55:3797–3802. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ritch R, Darbro B, Menon G, et al: TBK1
gene duplication and normal-tension glaucoma. JAMA Ophthalmol.
132:544–548. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Awadalla MS, Fingert JH, Roos BE, et al:
Copy number variations of TBK1 in Australian patients with primary
open-angle glaucoma. Am J Ophthalmol. 159:124–130.e1. 2015.
View Article : Google Scholar
|
|
28
|
Ito YA and Walter MA: Genomics and
anterior segment dysgenesis: A review. Clin Experiment Ophthalmol.
42:13–24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Auffarth GU, Blum M, Faller U, Tetz MR and
Völcker HE: Relative anterior microphthalmos: Morphometric analysis
and its implications for cataract surgery. Ophthalmology.
107:1555–1560. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nishina S, Kurosaka D, Nishida Y, Kondo H,
Kobayashi Y and Azuma N: Survey of microphthalmia in Japan. Jpn J
Ophthalmol. 56:198–202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reis LM and Semina EV: Genetics of
anterior segment dysgenesis disorders. Curr Opin Ophthalmol.
22:314–324. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weh E, Reis LM, Happ HC, Levin AV, Wheeler
PG, David KL, Carney E, Angle B, Hauser N and Semina EV: Whole
exome sequence analysis of Peters anomaly. Hum Genet.
133:1497–1511. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jordan T, Hanson I, Zaletayev D, Hodgson
S, Prosser J, Seawright A, Hastie N and van Heyningen V: The human
PAX6 gene is mutated in two patients with aniridia. Nat Genet.
1:328–332. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang P, Sun W, Li S, Xiao X, Guo X and
Zhang Q: PAX6 mutations identified in 4 of 35 families with
microcornea. Invest Ophthalmol Vis Sci. 53:6338–6342. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chang TC, Summers CG, Schimmenti LA and
Grajewski AL: Axenfeld-Rieger syndrome: New perspectives. Br J
Ophthalmol. 96:318–322. 2012. View Article : Google Scholar
|
|
36
|
Lehmann OJ, Tuft S, Brice G, et al: Novel
anterior segment phenotypes resulting from forkhead gene
alterations: Evidence for cross-species conservation of function.
Invest Ophthalmol Vis Sci. 44:2627–2633. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Carnes MU, Liu YP, Allingham RR, et al
NEIGHBORHOOD Consortium Investigators: Discovery and functional
annotation of SIX6 variants in primary open-angle glaucoma. PLoS
Genet. 10:e10043722014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jiang D, Yang Z, Li S, Xiao X, Jia X, Wang
P, Guo X, Liu X and Zhang Q: Evaluation of PRSS56 in Chinese
subjects with high hyperopia or primary angle-closure glaucoma. Mol
Vis. 19:2217–2226. 2013.PubMed/NCBI
|
|
39
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rozen S and Skaletsky H: Primer3 on the
WWW for general users and for biologist programmers. Methods Mol
Biol. 132:365–386. 2000.
|
|
42
|
Chen Y, Zhang Q, Shen T, et al:
Comprehensive mutation analysis by whole-exome sequencing in 41
Chinese families with Leber congenital amaurosis. Invest Ophthalmol
Vis Sci. 54:4351–4357. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Asai-Coakwell M, French CR, Ye M, et al:
Incomplete penetrance and phenotypic variability characterize
Gdf6-attributable oculoskeletal phenotypes. Hum Mol Genet.
18:1110–1121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kinnick TR, Mullins RF, Dev S, et al:
Autosomal recessive vitelliform macular dystrophy in a large cohort
of vitelliform macular dystrophy patients. Retina. 31:581–595.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wong RL, Hou P, Choy KW, Chiang SW, Tam
PO, Li H, Chan WM, Lam DS, Pang CP and Lai TY: Novel and homozygous
BEST1 mutations in Chinese patients with Best vitelliform macular
dystrophy. Retina. 30:820–827. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Weber S, Taylor JC, Winyard P, et al: SIX2
and BMP4 mutations associate with anomalous kidney development. J
Am Soc Nephrol. 19:891–903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Marquardt A, Stöhr H, Passmore LA, Krämer
F, Rivera A and Weber BH: Mutations in a novel gene, VMD2, encoding
a protein of unknown properties cause juvenile-onset vitelliform
macular dystrophy (Best's disease). Hum Mol Genet. 7:1517–1525.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Vincent A, McAlister C, Vandenhoven C and
Héon E: BEST1- related autosomal dominant
vitreoretinochoroidopathy: A degenerative disease with a range of
developmental ocular anomalies. Eye (Lond). 25:113–118. 2011.
View Article : Google Scholar
|
|
49
|
Bakrania P, Efthymiou M, Klein JC, et al:
Mutations in BMP4 cause eye, brain, and digit developmental
anomalies: Overlap between the BMP4 and hedgehog signaling
pathways. Am J Hum Genet. 82:304–319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kuo DS, Labelle-Dumais C and Gould DB:
COL4A1 and COL4A2 mutations and disease: Insights into pathogenic
mechanisms and potential therapeutic targets. Hum Mol Genet.
21:R97–R110. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lehmann OJ, Ebenezer ND, Jordan T, et al:
Chromosomal duplication involving the forkhead transcription factor
gene FOXC1 causes iris hypoplasia and glaucoma. Am J Hum Genet.
67:1129–1135. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Iseri SU, Osborne RJ, Farrall M, et al:
Seeing clearly: The dominant and recessive nature of FOXE3 in eye
developmental anomalies. Hum Mutat. 30:1378–1386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wolf MT, Lorenz B, Winterpacht A,
Drechsler M, Schumacher V, Royer-Pokora B, Blankenagel A, Zabel B
and Wildhardt G: Ten novel mutations found in Aniridia. Hum Mutat.
12:304–313. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Semina EV, Reiter R, Leysens NJ, et al:
Cloning and characterization of a novel bicoid-related homeobox
transcription factor gene, RIEG, involved in Rieger syndrome. Nat
Genet. 14:392–399. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Reis LM, Tyler RC, Volkmann Kloss BA, et
al: PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur
J Hum Genet. 20:1224–1233. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Khan K, Rudkin A, Parry DA, et al:
Homozygous mutations in PXDN cause congenital cataract, corneal
opacity, and developmental glaucoma. Am J Hum Genet. 89:464–473.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Aung T, Lim MC, Wong TT, Thalamuthu A,
Yong VH, Venkataraman D, Venkatraman A, Chew PT and Vithana EN:
Molecular analysis of CHX10 and MFRP in Chinese subjects with
primary angle closure glaucoma and short axial length eyes. Mol
Vis. 14:1313–1318. 2008.PubMed/NCBI
|