Introduction
Chronic obstructive pulmonary disease (COPD) is
characterized by chronic inflammation and oxidative damage that
results in airflow limitation; these changes are not fully
reversible and the disease is associated with progressive
development. Emphysema is the main pathological change and the
major factor responsible for the decline in lung function in COPD.
In recent years, COPD has become a major global public health
concern and is associated with high morbidity and mortality
(1). Currently, smoking is widely
recognized as a major risk factor for COPD. Cigarette smoke
contains several harmful chemicals that may lead to inflammatory
responses and oxidative stress in airway epithelial cells,
fibroblasts and alveolar epithelial cells. Inflammatory and
alveolar epithelial cells release reactive oxygen species (ROS),
which stimulate inflammatory cells to release inflammatory
mediators. The interactions between these mediators promote the
development of COPD (1–4).
Inflammatory cells, such as neutrophils, alveolar
macrophages and T lymphocytes are activated by stimuli in
vivo and release a variety of inflammatory mediators, such as
leukotriene B4 (LTB4), tumor necrosis factor (TNF)-α, interleukin
(IL)-17 and chemokines, such as IL-8 [also known as chemokine
(C-X-C motif) ligand 8 (CXCL8)] (1–5).
These mediators can damage the lung structure, promote neutrophil
inflammation and play an important role in the inflammation and
pathogenesis of COPD. IL-8, macrophage inflammatory protein-2α
[MIP-2α, also known as chemokine (C-X-C motif) ligand 2 (CXCL2)]
and monocyte chemotactic protein-1 [MCP-1, also known as chemokine
(C-C motif) ligand 2 (CCL2)] are chemokines, that recruit
neutrophils and monocytes to sites of inflammation (6–10).
Thus, these inflammatory mediators play an important role in the
development of emphysema.
Heme oxygenase-1 (HO-1) is a rate-limiting enzyme
involved in mammalian heme biosynthesis. HO-1 breaks down heme to
form equimolar amounts of biliverdin, free iron and carbon monoxide
(CO) and it is rapidly activated by its physiological substrate,
heme, as well as inflammation and oxidative stress-related stimuli,
nitric oxide (NO), heat shock and other factors. HO-1 and its
products exert potent anti-inflammatory, antioxidant,
anti-apoptotic and anti-proliferative effects under several and
pathologies, such as asthma, organ transplantation, acute lung
injury (11–17).
Studies have found that HO-1 plays a role in the
development of COPD characterized by chronic inflammation and
oxidative damage and the overexpression of HO-1 mediated by
adenovirus in the lungs attenuates the development of
elastase-induced pulmonary emphysema in mice (18–27). We thus hypothesized that the
upregulation of HO-1 expression induced by hemin may prevent the
development of emphysema by inhibiting inflammation and the
associated damage induced by oxidative stress.
Materials and methods
Animal model
Healthy male Wistar rats (4 weeks of age, weighing
100–120 g) were obtained from the Experimental Animal Center,
Chinese Academy of Military Science. The present study was approved
by the Institutional Review Board for Biomedical Research of Shanxi
Medical University, Taiyuan, China. The rats were allowed to
acclimatize for 2 weeks and had free access to food and water. The
rats were then randomly divided into 5 groups (n=10/group) as
follows: i) the control group (C): normal saline (NS) + sham smoke;
ii) the smoke-exposed group (SM): NS + smoke; iii) the hemin group
(H): hemin + smoke; iv) the SnPP group (S): SnPP + smoke; and v)
the H+S group (HS): hemin + SnPP + smoke. For 20 weeks, the rats
were treated with 0.3 ml NS or 0.3 ml protoporphyrin IX
[tin-protoporphyrin IX (SnPP), an inhibitor of HO-1 activity or
ferriprotoporphyrin IX chloride (hemin), an inducer of HO-1] once
daily for the first 3 days of each week and exposed to smoke (or
sham smoke) once daily from the 2nd to the 6th day of each week.
After 20 weeks, the animals were sacrificed, as previously
described (28).
The smoking regimen was 16 cigarettes each day, each
for 10 min, with an interval of 5 min between cigarettes. The rats
were exposed to 24 puffs of cigarette smoke from 2 cigarettes in a
smoke exposure device (PAB-S200; Beijing Biolaunching Technologies
Co., Ltd., Beijing, China). The Monkey King brand of cigarettes
were commercial filter cigarettes produced by the Baoji Cigarette
Factory, Baoji, China and had a cigarette standard tar volume of 11
mg.
Hemin or SnPP (Frontier Scientific, Inc., Logan, UT,
USA) were dissolved in 1 M NaOH and adjusted to pH 7.3–7.5 with HCl
and then diluted to the final concentration with NS. The rats were
subcutaneously injected with hemin (20 µmol/kg) or SnPP (20
µmol/kg), or hemin (20 µmol/kg) + SnPP (20
µmol/kg) or NS. The dose of hemin used in the present study
was decided upon following testing in preliminary experiments (data
not shown) and examing doses used in a previous study (41). In a previous study (41) the dose of hemin used was 75
µmol/kg. However, we found that this dose was too high for
long-term use, and from our preliminary experiments, we found that
the dose of 20 µmol/kg hemin was the opitam one for
long-term use.
Collection of bronchoalveolar lavage and
other specimens
The animals were anesthetized and 8-10 ml blood
specimens were drawn from the abdominal aorta. The trachea was
cannulated, the right lung was ligated and the left lung was
intubated and injected thrice with 2 ml pre-cooled saline (4°C).
Bronchoalveolar lavage fluid (BALF) was recovered and centrifuged.
The supernatant was stored at −80°C and the cells were allowed to
settle, stained with Wright's dye (dye includes methylene blue and
eosin Y dissolved in methyl alcohol; all obtained from Beyotime
Institute of Biotechnology, Shanghai, China) and counted using a
standard technique. The right upper lung tissue was removed, fixed
with formalin, embedded in paraffin, sectioned and stained with
hematoxylin and eosin [(H&E), H&E staining kit (C0105;
Beyotime Institute of Biotechnology, Shanghai, China)]. The
pathological changes of the lung tissues were then observed at ×10
and ×40 magnification under a light microscope. The lung, BALF
supernatant and serum were stored at −80°C for other
measurements.
Cell count in BALF
Following the recovery of BALF and placed in a
hemocytometer. The number of cells counted was the sum of all cells
across 4 squares in one chamber. The number of cells was calculated
using the following formula: total number of cells/ml = cells in 4
squares/4×104). The remaining BALF was centrifuged at
400 × g at 4°C for 5 min. The supernatant was saved and the
precipitated cells were smeared and stained with Wright's staining
solution. The total number of cells, macrophages and neutrophils
was then counted in 5 high-power (×40 magnification) microscopic
fields, and we then calculated the mean percentage of macrophages
(or neutrophils) in the total cells and the numbers/ml = the mean
percentage × the total number of cells/ml.
Morphometric evaluation of emphysema
Alveolar airspace enlargement was assessed according
to the mean linear intercept (MLI), mean alveolar number (MAN) and
mean alveolar area (MAA) by 2 independent individuals in a blinded
manner, as previously described (29). MLI involved centering a 'cross' on
the middle of each field, counting the number of alveolar septa
(Ns) through the crosshairs and measuring the total length of the
crosshairs (L), MLI = L/Ns. MAN was calculated by counting the
number of alveoli (Na) within each field divided by the area of the
field (TA), MAN = Na/TA. MAA involved counting the number of
alveoli (Na) within each field, measuring the area of each
field(TA) and H&E staining (lung parenchyma) of the area (PA),
MAA = (TA-PA)/Na.
Preparation of 10% lung tissue
homogenates
The rat lung tissue (100 mg) was accurately weighed,
cut into small pieces and placed into 9 volumes of normal saline by
weight (g)/volume (ml) = 1:9, and homogenized using an ultrasonic
processor (CP-750, Cole-Parmer North America, Vernon Hills, IL,
USA). The supernatant was isolated by centrifugation at 3,000 rpm
for 10 min and stored at −80°C for later use in other analyses.
Measurement of cytokine levels
In accordance with the instructions provided with
the ELISA kit (Ebioscience, San Diego, CA, USA), the levels of
cytokines (TNF-α and IL-8 in serum and BALF, IL-17, IL-10, MIP-2α
and MCP-1 in serum, BALF and lung tissue supernatant) were
measured. The OD values were measured using a microplate reader
(Gemini EM, Molecular Devices, Sunnyvale, CA, USA) at 450 nm.
Standard curves were created and the concentrations were calculated
according to the relevant formulas.
Immunohistochemistry (IHC)
Tissue blocks were formalin-fixed,
paraffin-embedded, sectioned, de-paraffinized in 3 changes of
xylene (5 min each), hydrated in 2 changes of 100% ethanol (10 min
each) and subsequently hydrated in 2 changes of 95% ethanol (10 min
each). Antigen unmasking was performed in a pressure cooker with 10
mM sodium citrate buffer, pH 6.0. Endogenous peroxidase activity
was quenched by placing the sections in a 1% hydrogen peroxide
solution in methanol for 30 min. The tissue sections were then
washed and immunostaining was performed using a commercially
available SABC (streptavidin biotinylated complex) kit (SA1025,
Boster, Wuhan, China) according to the manufacturer's instructions.
The tissue sections were stained with HO-1 antibody (ab13248;
Abcam, Cambridge, MA, USA). Images of the stained sections were
digitally captured at ×40 magnification using a Nanozoomer system
(Olympus American Inc., Center Valley, PA, USA) and the average
gray value of staining was measured on each image by one of the
investigators in a blinded manner. The average gray value was
negatively-related to the depth of protein staining.
Western blot analysis
Whole lung tissue (50 mg) was accurately weighed,
cut into small pieces and placed into 500 µl RIPA lysis
buffer (Beyotime Institute of Biotechnology, Haimen, China) and
homogenized using an ultrasonic homogenizer on ice. The supernatant
was isolated by centrifugation at 12,000 rpm for 10 min and stored
at −80°C for use in western blot analysis. β-actin and HO-1 protein
expression was then measured by western blot analysis. Proteins
were separated according to their molecular weight and blotted onto
PVDF membranes. The membranes were blocked for 2 h in 5% BSA and
incubated with rabbit-anti-rat HO-1 antibody (1/1,000, H-105; Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA) followed by a
peroxidase labeled goat anti-rabbit antibody (1/2,000, BA1055;
Boster). For the protein loading control, the membrane was stripped
using a 25 mM glycine-HCl buffer containing 1% SDS and incubated
with mouse anti-rat β-actin antibody (1/1,000, BM0626; Boster)
followed by a peroxidase labeled goat-anti-mouse antibody (1/2,000,
BA1050; Boster). The bands of interest were visualized by enhanced
chemiluminescence kit (AR1111, Boster) according to the protocol in
provided with the FluorChem HD2 System (Protein Simple, San Jose,
CA, USA). The relative protein expression levels of HO-1/β-actin
were then obtained by analyzing the OD value ratios of the bands
with Image-Pro Plus 6.0 software (Media Cybernetics, Inc.,
Rockville, MD, USA).
Malondialdehyde (MDA), superoxide
dismutase (SOD) and glutathione (GSH) assays
The tissue specimens were allowed to naturally thaw
to room temperature. The OD values representing the MDA levels in
lung homogenates and serum were measured at 532 nm according to the
manufacturer's instructions provided with the TBA kit (Jiancheng
Biotech Co., Ltd., Nanjing, China). The OD values representing the
SOD levels in the lung tissue supernatant and serum were measured
at 550 nm according to the manufacturer's instructions provided
with the hydroxylamine kit (Jiancheng Biotech Co., Ltd). The OD
values representing the GSH levels in the lung tissue supernatant
and serum were measured at 420 nm according to the manufacturer's
instructions provided with the reduced glutathione assay kit
(Jiancheng Biotech Co., Ltd). The concentrations of MDA, SOD and
GSH were then calculated according to the instructions provided
with the respective kits.
Statistical analysis
The results are expressed as the means ± SD.
Statistical analysis was performed using SPSS 17.0 software and
two-way analysis of variance (ANOVA) was used to assess the
differences between groups. A P-value <0.05 was considered to
indicate a statistically significant difference.
Results
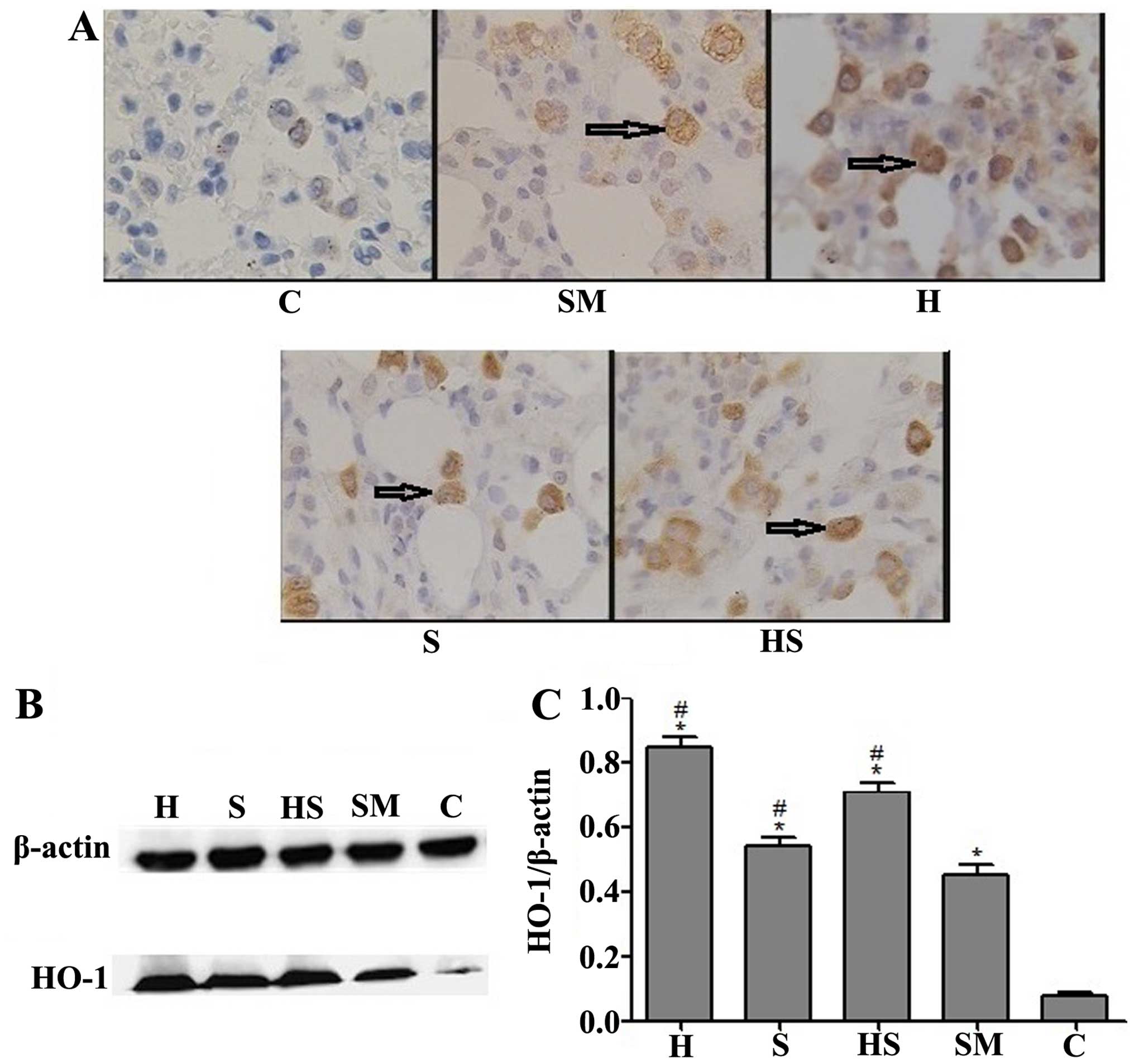
Effects of treatment with hemin and/or
SnPP and exposure to cigarette smoke on the expression levels of
HO-1
The results of IHC and western blot analysis
revealed that compared with the control (C) and smoke-exposed (SM)
groups, treatment with hemin (an inducer of HO-1), SnPP (an
inhibtor of HO-1 activity) or SnPP + hemin (H, S and HS groups)
resulted in a significant increase in HO-1 protein expression in
the lungs, particularly in alveolar macrophages. In IHC, this
increase in HO-1 expression was evidenced by an increase in the
amount of dark brown staining. Exposure to cigarette smoke (SM
group) also resulted in a small increase in HO-1 expression
(Fig. 1).
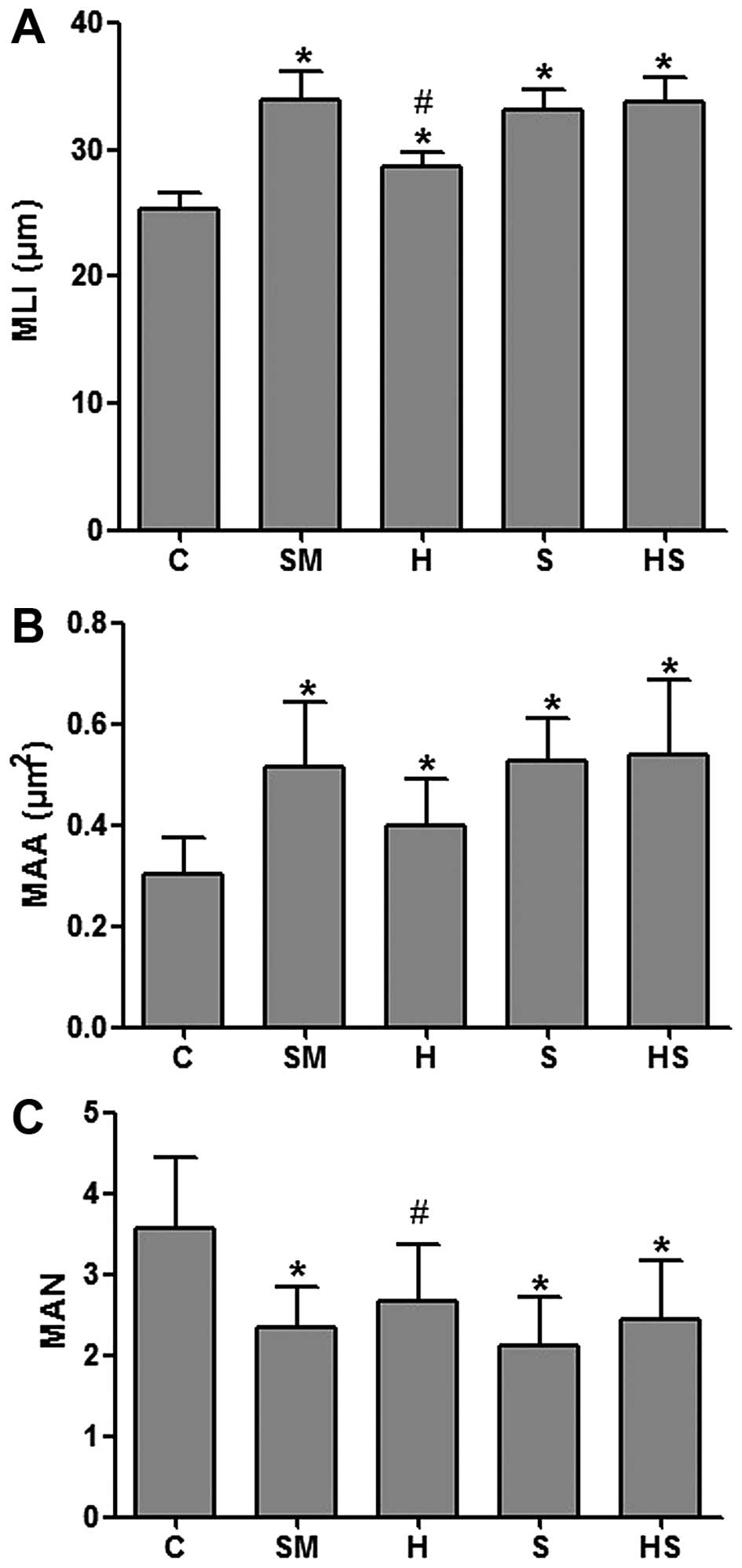
Exposure to cigarette smoke induces
pathological changes associated with emphysema and these changes
are attenuated by treatment with hemin
Cigarette smoke-induced emphysema was observed after
20 weeks of exposure to cigarette smoke, evidenced by pathological
changes associated with emphysema (alveolar airspaces expanded
universally and integrated into a larger cavity, alveolar pores
expanded and many alveolar septa became narrow and broken).
Treatment with hemin (inducer of HO-1; H group) attenuated these
pathological changes; however, treatment with SnPP (an inhibitor of
HO-1 activity; S group) did not reverse the adverse effects of
smoke (Fig. 2). The development
of emphysema was also evidenced by a significant increase in the
MLI and MAA values and a significant decrease in the MAN values in
the rats exposed to cigarette smoke (SM group) compared to the
control rats (C), indicating alveolar airspace enlargement.
Treatment with hemin partly reversed these effects, decreasing the
MLI and MAA values and increasing the MAN values (the values did
not completely return to those of the controls though). However,
treatment with SnPP increased the MLI and MAA values and decreased
the MAN values (Fig. 3). These
results demonstrate that the activation of HO-1 attenuates the
adverse effects of cigarette smoke, preventing alveolar airspace
enlargement.
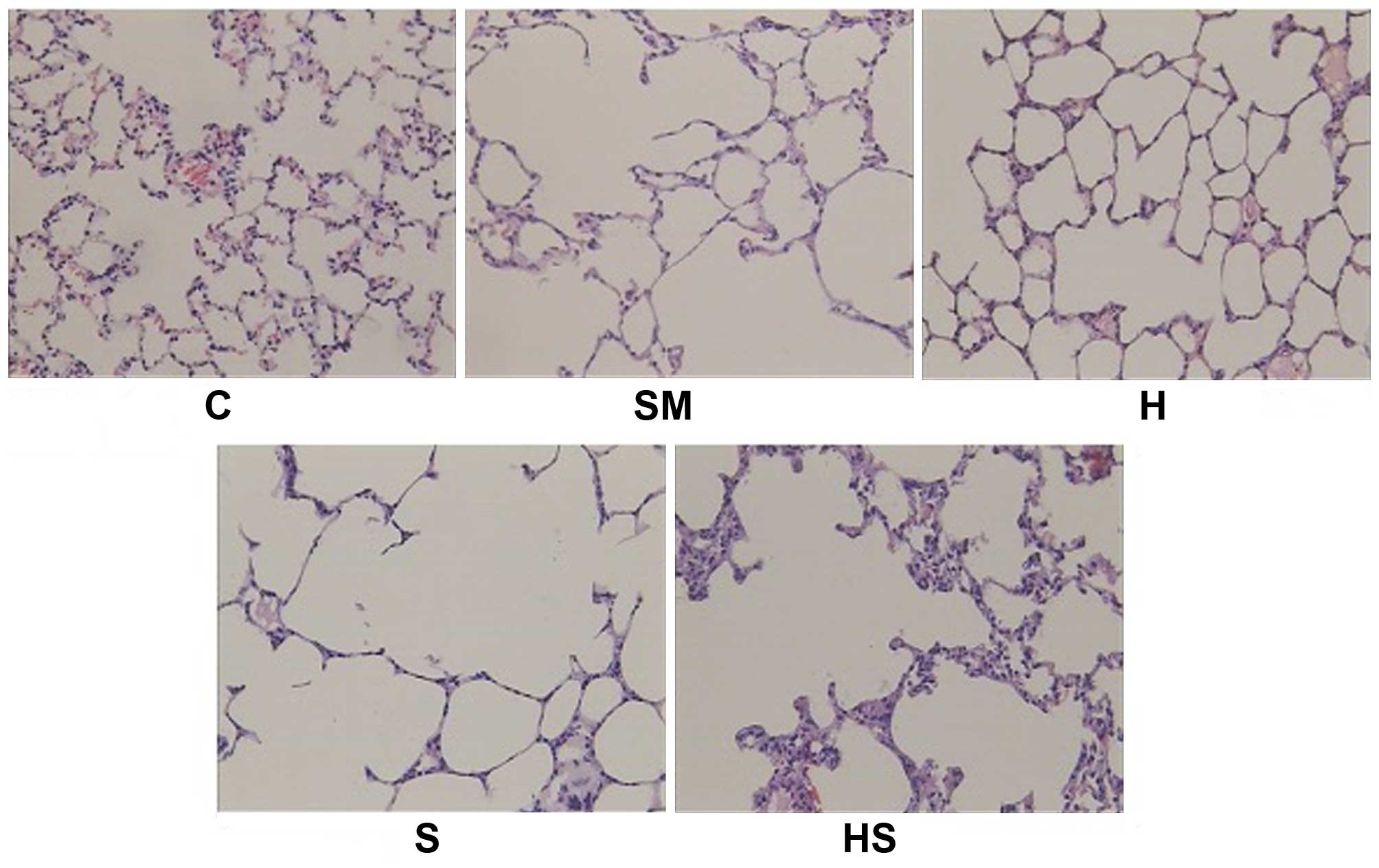 | Figure 2Pathological changes in lung tissue.
A representative pathological image is shown for each group
(H&E, ×10 magnification). Cigarette smoke exposure induced
pathological changes associated with emphysema (SM group) and hemin
treatment reduced pulmonary injury (H and HS groups). In the
control (C) group, alveolar distribution was uniform and alveolar
septa were complete. Alveolar airspaces expanded universally and
integrated into a larger cavity, alveolar pores expanded, and many
alveolar septa became narrowed and broken in the SM, S and HS
groups, whereas these changes were alleviated in the H group. C,
sham smoke; SM, cigarette smoke; H, hemin + smoke; S, SnPP + smoke;
HS, hemin + SnPP + smoke. |
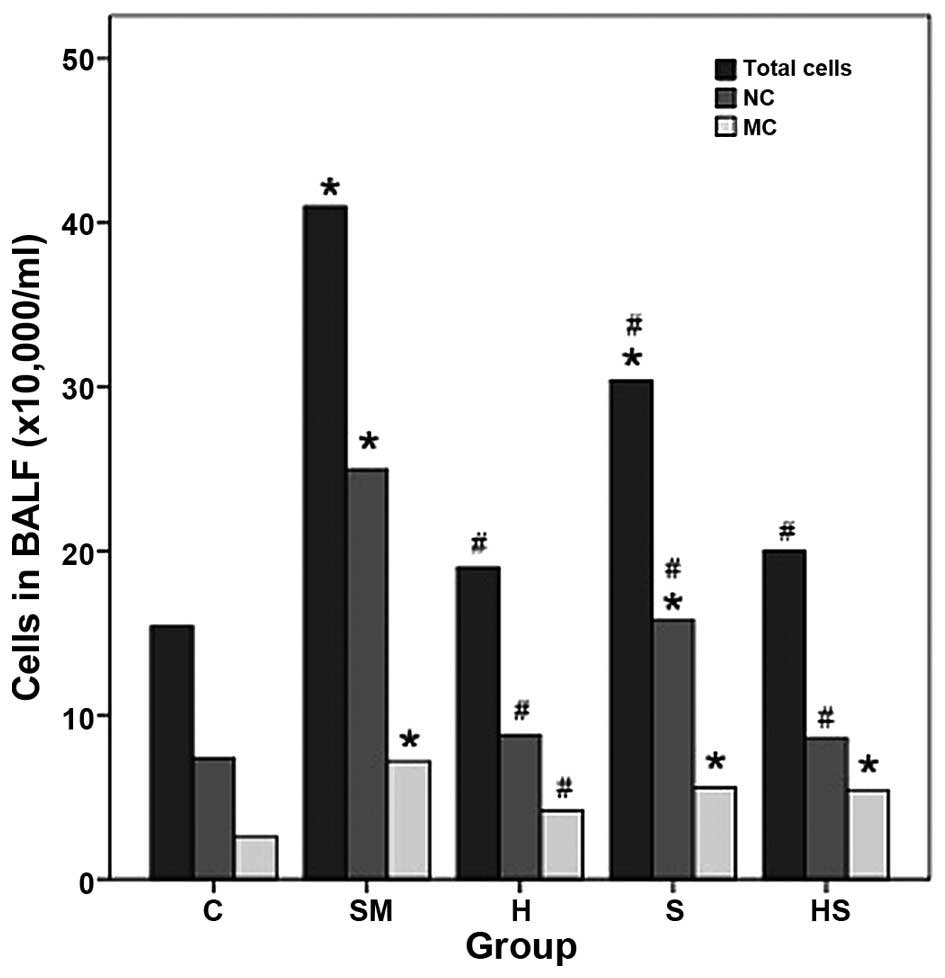
Exposure to cigarette smoke results in an
increase in inflammatory cell infiltration and elevated levels of
inflammatory mediators and these effects are partly reversed by
treatment with hemin
Exposure to cigarette smoke (SM group) induced an
increase in inflammatory cell infiltration; the total cell,
neutrophil and macrophage counts in BALF were significantly higher
in the SM group than those in the control group (C) (Fig. 4). Treatment with hemin (inducer of
HO-1; H group) reduced inflammatory cell infiltration to a certain
extent (shown by the decrease in the total cell, neutrophil and
macrophage count in BALF). However, treatment with SnPP (an
inhibitor of HO-1; S group) did not reverse the adverse effects of
smoke; the total cell and neutrophil counts significantly increased
compared to the control (C group) (Fig. 4). Exposure to cigarette smoke also
significantly increased the levels of the inflammatory mediators,
including IL-17, TNF-α, IL-8, MCP-1 and MIP-2α. The TNF-α and IL-8
levels significantly increased in the serum and BALF in the SM
group compared to the control group. Treatment with hemin
significantly reduced these effects. The IL-17 levels significantly
increased in the BALF and lung tissue following exposure to
cigarette smoke. Treatment with hemin decreased these levels in the
BALF and lung tissue. SnPP increased the IL-17 levels in the BALF
and lung tissue. The MIP-2α levels increased in the lung tissue,
serum and BALF following exposure to cigarette smoke, and treatment
with hemin decreased these levels. However, in the SnPP-treated
group, the MIP-2α levels increased. The IL-10 levels significantly
decreased in the serum of the smoke-exposed rats. Treatment with
hemin significantly increased the IL-10 levels in the serum, BALF
and lung tissue compared to the smoke-exposed rats. SnPP had no
significant effect on the IL-10 levels. The levels of MCP-1
significantly increased in the serum, BALF and lung tissue in the
SM group compared to the control group. Hemin significantly
decreased these levels compared to the SM group. Treatment with
SnPP also increased the MCP-1 levels compared to the controls. The
levels of the inflammatory mediators did not completely return to
those of the controls, indicating that hemin partly suppressed
inflammation induced by cigarette smoke (Fig. 5).
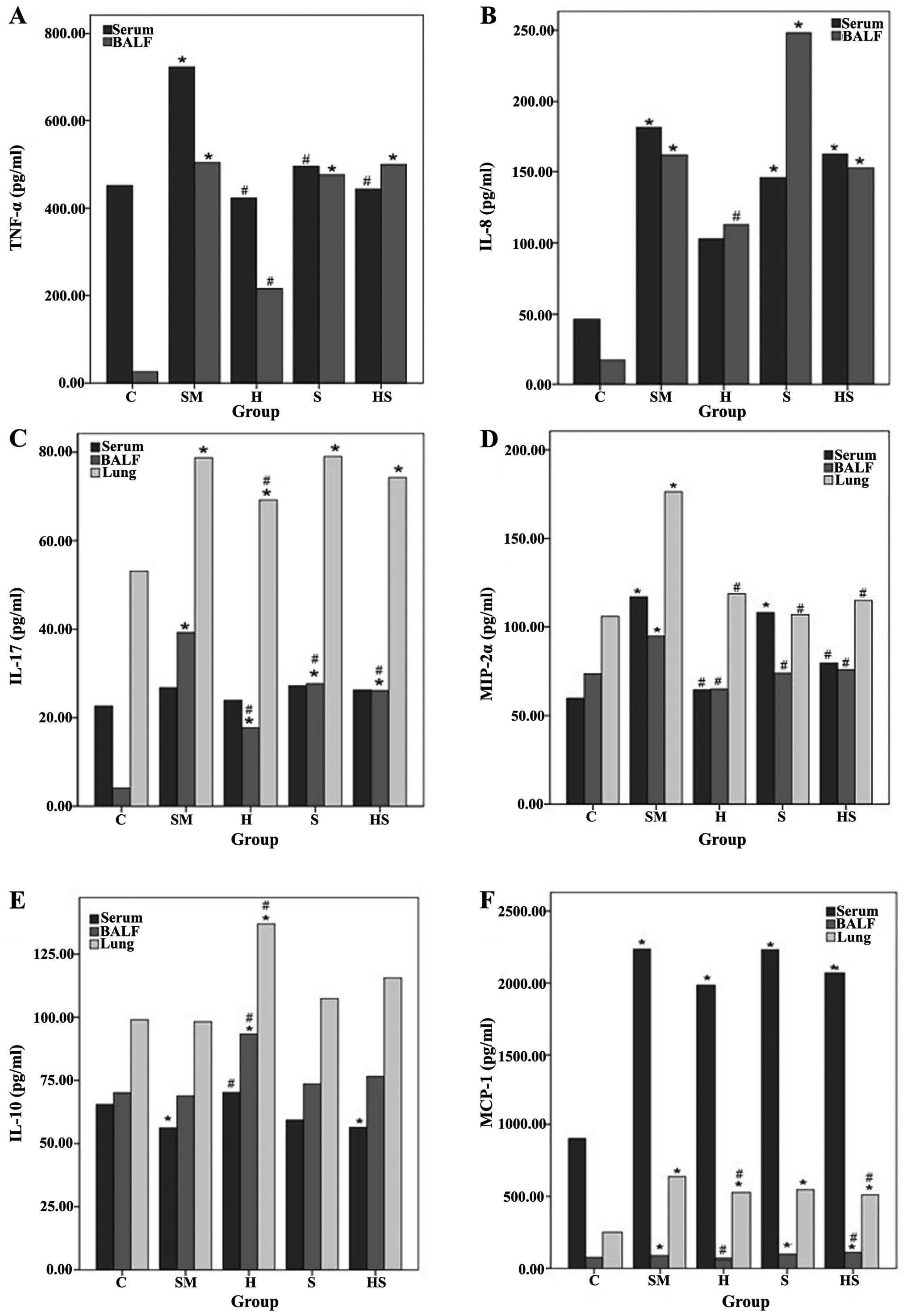 | Figure 5(A-F) Levels of inflammatory
cytokines and chemokines in serum, BALF and lung tissue. Cigarette
smoke exposure significantly increased the levels of the
inflammatory mediators, such as IL-17, TNF-α, IL-8, MCP-1 and
MIP-2α, and hemin treatment reduced the levels of these mediators,
except for IL-10. *P<0.05 compared with the control
(C) group; #P<0.05 compared with the smoke-exposed
(SM) group. BALF, bronchoalveolar lavage fluid; C, sham smoke; SM,
cigarette smoke; H, hemin + smoke; S, SnPP + smoke; HS, hemin +
SnPP + smoke. |
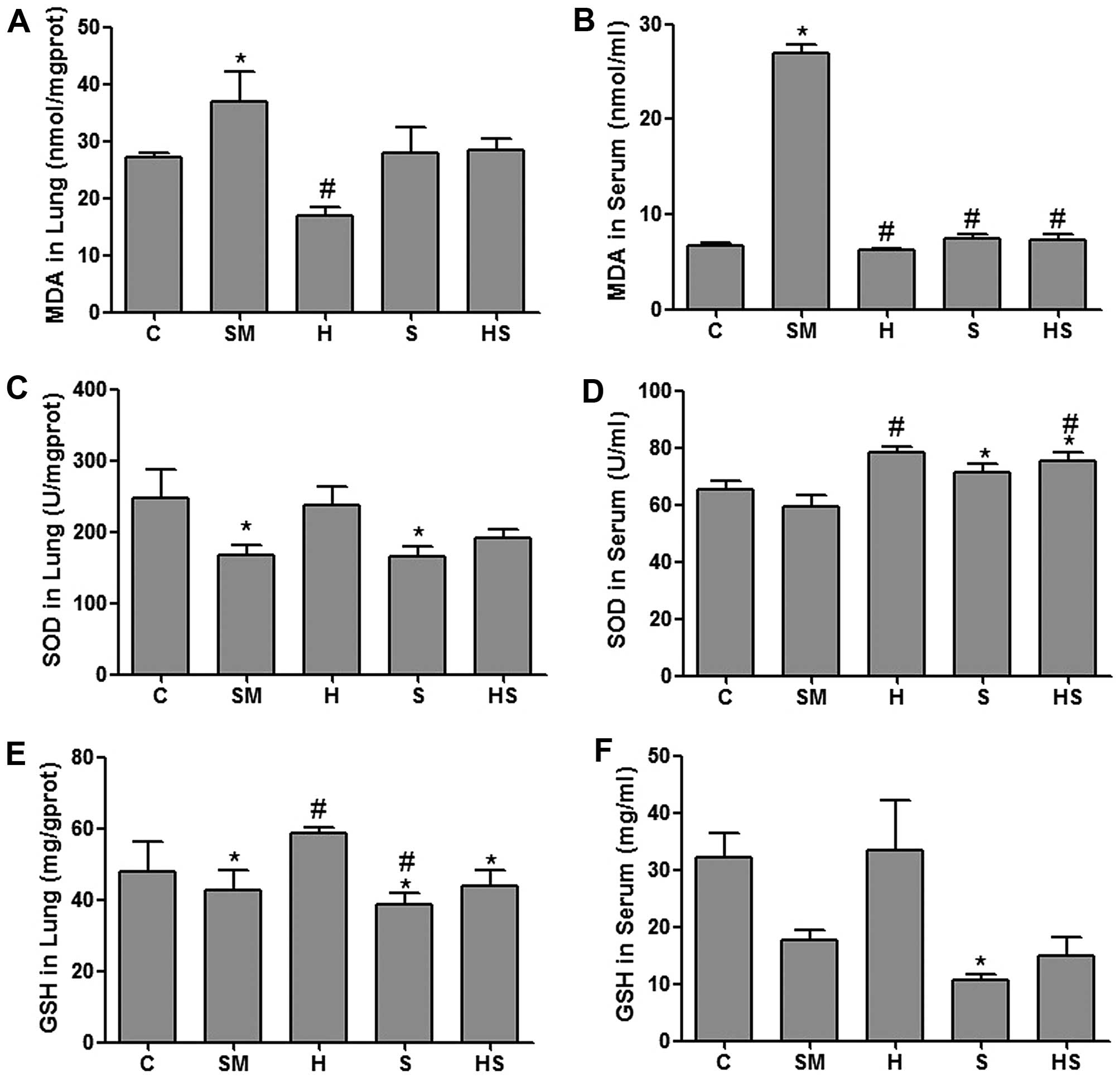
Treatment with hemin decreases the MDA,
and increases the SOD and GSH contents
Following exposure to cigarette smoke (SM group),
the MDA levels in the serum and lung tissue homogenate were higher
than those in the sham smoke (control, C) group (P<0.05), and
the SOD levels in the lung tissue in the SM group were lower than
those in the control group (P<0.05). Treatment with hemin
decreased the MDA, and increased the SOD and GSH contents. These
results indicate that HO-1 exerts antioxidant effects in rats
exposed to cigarette smoke (Fig.
6).
Discussion
In the present study, we found that long-term
treatment with hemin significantly upregulated HO-1 protein
expression, reduced the levels of inflammatory cytokines and
chemokines and the infiltration of inflammatory cells, inhibited
oxidative stress-induced damage, and protected the lungs from the
development of pathological changes associated with emphysema.
Hemin, as an inducer of HO-1, significantly
upregulated HO-1 protein expression and attenuated pathological
changes associated with emphysema in rats exposed to long-term
cigarette smoke for 20 weeks. Previous studies have found that
exposure to cigarette smoke extract (CSE) induces an increase in
the expression of HO-1 in primary human lung fibroblasts, tracheal
smooth muscle and alveolar epithelial cells (18–21). However, the increased expression
levels of HO-1 did not provide sufficient protection against the
inflammation and oxidative damage induced by CSE in order to
inhibit the development of pulmonary emphysema. In addition, it has
been demonstrated that a decreased HO-1 expression in the alveolar
macrophages of patients with severe COPD may be related to a lower
HO-1 inducibility by ROS, caused by HO-1 gene promoter
polymorphisms (22–26).
As an HO-1 inhibitor, treatment with SnPP slightly
decreased HO-1 expression compared to treatment with hemin plus
SnPP; treatment with hemin + SnPP slightly increased HO-1 protein
expression in the lungs, while HO-1 activity was inhibited, but it
did not aggravate the adverse effects of exposure to smoke or
increase the levels of inflammatory mediators induced by smoking.
Although HO-1 activity is inhibited by SnPP, the upregulation of
HO-1 expression can have anti-inflammatory and antioxidant effects
(30).
Long-term exposure to smoke leads to inflammation
and oxidative stress in the airways and lung tissue. In a previous
study, exposure to smoke for 20 weeks significantly increased the
levels of the pro-inflammatory cytokines, including TNF-α, IL-1,
IL-6, KC (CXCL1) and MCP-1 in the mouse lung tissue homogenate
(30). Another study found that 3
days of mainstream smoke exposure induced an acute inflammatory
response characterized by a neutrophilic influx, increased cytokine
secretion (KC, TNF-α, MIP-2, MIP-1α and MCP-1) in a mouse model of
COPD (31). The present study
also found that smoking significantly increased the infiltration of
inflammatory cells and the levels of cytokines, such as IL-17,
TNF-α, IL-8, MCP-1 and MIP-2α and resulted in pathological changes
associated with emphysema.
It has been found that the CD4+
IL-17+ cell number positively correlates with airflow
limitations and pathological changes and negatively correlates with
FEV(1)%p and FEV(1)/FVC in patients with COPD. Increased
CD4+ IL-17+ cell numbers are closely related
to chronic lung inflammation and may contribute to the pathogenesis
of COPD (32). The administration
of IL-17 antibodies to mice exposed to cigarette smoke has been
shown to reduce the number of neutrophils, the levels of IL-6 and
IL-8, and neutrophil airway inflammation (33,34). In a mouse model of
elastase-induced pulmonary emphysema, the number of
IL-17A+ CD4 T cells and the levels of IL-17A, IL-1β, KC
and MIP-2 increased; however, the levels of these cytokines
decreased in IL-17A-deficient mice following elastase treatment
(35). IL-17 not only promotes
the secretion of IL-8 (CXCL8) and TNF-α, but also promotes the
recruitment of neutrophils and monocytes, increases their
functionality, extends their survival time in the lungs and thus
causes lung inflammation (5,32,34). Thus, IL-17A may be a more critical
cytokine and may play an important role in the development of
emphysema and pulmonary inflammation. However, a study of patients
with COPD and smoke-induced COPD found elevated levels of IL-8 and
decreased IL-17 levels in sputum and BALF. Time course experiments
demonstrated that IL-8 levels began to increase after 8 weeks of
smoke exposure and IL-17 levels decreased after 10 weeks. Elevated
IL-8 levels in patients with COPD appeared to be primarily derived
from neutrophils and macrophages, and decreased IL-17 levels were
mainly due to the decreased number of monocytes and damaged
epithelial cells (36). These
results were inconsistent as monocytes and cells other than Th17
cells can also secrete IL-17 and a decrease in the number of these
cells may also affect the secretion of IL-17. Therefore, further
studies evaluating the distribution of lymphocytes together with
other cells are required.
Long-term treatment with hemin upregulated the
protein expression of HO-1, reduced the infiltration of neutrophils
and monocytes/macrophages, decreased the levels of pro-inflammatory
cytokines, including IL-17, TNF-α, IL-8, MCP-1 and MIP-2α, and
inhibited pathological changes in the lungs, although the changes
in the levels of these cytokines levels were not consistent. For
example, the levels of IL-17 in the serum did not differ
significantly among the experimental groups, possibly since IL-17
is mainly secreted by cells at sites of inflammation in lung
tissue, and activated cells in the blood are recruited to the
lungs. In a previous study, in a mouse model of non-eosinophilic
asthma, the induction of HO-1 exerted anti-inflammatory effects by
decreasing IL-17A levels via inhibition of Th17 cell
differentiation. The HO-1 inhibitor, SnPP, reversed these effects
and transfection with HO-1 siRNA abolished the effects of hemin on
the induction of HO-1 in vivo (37). In a murine model of dextran
sulfate sodium (DSS)-induced colitis, the upregulation of HO-1 by
hemin attenuated IL-17 and Th17-related cytokine production and
ameliorated experimental colitis (38). Thus, HO-1 may exert
anti-inflammatory effects by inhibiting IL-17 and other cytokines,
including TNF-α, IL-8, MCP-1 and MIP-2α.
IL-10 is an important anti-inflammatory cytokine
secreted by B cells, monocytes, regulatory T cells and others
(39). Studies have found that
the upregulation of HO-1 expression promotes the secretion of
IL-10, thus promoting the anti-inflammatory effects of IL-10 in
several animal disease models, such as asthma and acute lung injury
(40,41). In addition, IL-10 induces the
expression of HO-1, a downstream effector of IL-10, which plays an
anti-inflammatory role in lipopolysaccharide-induced septic shock
in mice (42,43). These results suggest that HO-1 and
IL-10 may form a positive loop to enhance the anti-inflammatory
effect. The present study also found that the levels of IL-10 in
BALF, serum and lung tissue homogenate were higher in the hemin
group than those in the smoke-exposed group, indicating that hemin
treatment exerts anti-inflammatory effects by promoting the
secretion of IL-10.
Inflammation and oxidative stress, in combination,
promote the development of emphysema. MDA is a major oxidative
stress product produced by the attack of oxygen free radicals on
polyunsaturated fatty acids and may be used as a marker to reflect
the level of oxidative lipid damage in vivo. SOD is a
indirect marker of the body's ability to eliminate oxygen free
radicals. The concentrations of MDA and SOD often complement each
other. In the present study, the MDA levels increased, whereas the
SOD and GSH levels decreased in the smoke-exposed group, while
hemin treatment decreased the MDA, and increased the SOD and GSH
levels. These results indicated that hemin treatment improved the
body's antioxidant capacity and reduced oxidative lipid damage.
The most important finding of the present study was
the protective effects of HO-1 against the development of cigarette
smoke-induced emphysema. HO-1 can alleviate lung pathological
changes, possibly due to its capability to reduce the infiltration
of inflammatory cells in lung tissue, the levels of inflammatory
mediators and associated oxidative stress damage. However, HO-1 did
not completely inhibit the inflammatory response and oxidative
stress. A previous study found that long-term HO-1 upregulation
induced by CoPP prevented smoke-induced B cell infiltrates and
increased CD4+CD25+ Tregs, but had no effect
on smoke-induced emphysema and the increase in inflammatory cells
and cytokines in a mouse model (30). This result is not consistent with
our study. This may be due to the differences in species selection
in rodent models (rat vs. others) and treatment regimens (dose,
treatment interval, or single or combinatorial pharmacological
interventions). Thus, the exact role of HO-1 in the smoke-induced
inflammatory response and oxidative stress remains to be
elucidated.
In conclusion, our findings demonstrate that
long-term HO-1 upregulation exerts a protective effect against the
development of cigarette smoke-induced emphysema. A possible
explanation for this effect of HO-1 on emphysema is its
anti-inflammatory and antioxidant effects.
Acknowledgments
The authors would like to thank Dr Hongjuan Yang, Dr
Fangrun Yuan, Dr Enguang Wang, Dr Junfang Wu and Mrs. Jiahui Zhao
for their assistance in performing the smoke exposures and
collecting experimental specimens. The present study was supported
by the grants from the Health and Family Planning Commission of
Shanxi Provice (2014039) and the Natural Science Foundation from
the Science and Technology Department of Shanxi Province (project
no. 2013011055-3).
References
|
1
|
Vestbo J, Hurd SS, Agustí AG, Jones PW,
Vogelmeier C, Anzueto A, Barnes PJ, Fabbri LM, Martinez FJ,
Nishimura M, et al: Global strategy for the diagnosis, management,
and prevention of chronic obstructive pulmonary disease: GOLD
executive summary. Am J Respir Crit Care Med. 187:347–365. 2013.
View Article : Google Scholar
|
|
2
|
Barnes PJ: Chronic obstructive pulmonary
disease. N Engl J Med. 343:269–280. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yoshida T and Tuder RM: Pathobiology of
cigarette smoke-induced chronic obstructive pulmonary disease.
Physiol Rev. 87:1047–1082. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tuder RM and Petrache I: Pathogenesis of
chronic obstructive pulmonary disease. J Clin Invest.
122:2749–2755. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang J, Chu S, Zhong X, Lao Q, He Z and
Liang Y: Increased expression of CD4+IL-17+
cells in the lung tissue of patients with stable chronic
obstructive pulmonary disease (COPD) and smokers. Int
Immunopharmacol. 15:58–66. 2013. View Article : Google Scholar
|
|
6
|
Driscoll KE: Macrophage inflammatory
proteins: biology and role in pulmonary inflammation. Exp Lung Res.
20:473–490. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Driscoll KE: TNFα and MIP-2: role in
particle-induced inflammation and regulation by oxidative stress.
Toxicol Lett. 112–113. 177–183. 2000.
|
|
8
|
Traves SL, Culpitt SV, Russell RE, Barnes
PJ and Donnelly LE: Increased levels of the chemokines GROα and
MCP-1 in sputum samples from patients with COPD. Thorax.
57:590–595. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Boer WI, Alagappan VK and Sharma HS:
Molecular mechanisms in chronic obstructive pulmonary disease:
Potential targets for therapy. Cell Biochem Biophys. 47:131–148.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
de Boer WI, Sont JK, van Schadewijk A,
Stolk J, van Krieken JH and Hiemstra PS: Monocyte chemoattractant
protein 1, interleukin 8, and chronic airways inflammation in COPD.
J Pathol. 190:619–626. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Abraham NG: Therapeutic applications of
human heme oxygenase gene transfer and gene therapy. Curr Pharm
Des. 9:2513–2524. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abraham NG and Kappas A: Pharmacological
and clinical aspects of heme oxygenase. Pharmacol Rev. 60:79–127.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chauveau C, Bouchet D, Roussel JC, Mathieu
P, Braudeau C, Renaudin K, Tesson L, Soulillou JP, Iyer S and
Buelow R: Gene transfer of heme oxygenase-1 and carbon monoxide
delivery inhibit chronic rejection. Am J Transplant. 2:581–592.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Araujo JA, Meng L, Tward AD, Hancock WW,
Zhai Y, Lee A, Ishikawa K, Iyer S, Buelow R, Busuttil RW, et al:
Systemic rather than local heme oxygenase-1 overexpression improves
cardiac allograft outcomes in a new transgenic mouse. J Immunol.
171:1572–1580. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ryter SW and Choi AM: Heme oxygenase-1:
Redox regulation of a stress protein in lung and cell culture
models. Antioxid Redox Signal. 7:80–91. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jia YX, Sekizawa K, Okinaga S, Lie R and
Sasaki H: Role of heme oxygenase in pulmonary response to antigen
challenge in sensitized rats in vivo. Int Arch Allergy Immunol.
120:141–145. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang XH, Wang K, Zhang F, Li XC, Li J, De
W, Guo J, Qian XF and Fan Y: Heme oxygenase-1 alleviates
ischemia/reperfusion injury in aged liver. World J Gastroenterol.
11:690–694. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baglole CJ, Sime PJ and Phipps RP:
Cigarette smoke-induced expression of heme oxygenase-1 in human
lung fibroblasts is regulated by intracellular glutathione. Am J
Physiol Lung Cell Mol Physiol. 295:L624–L636. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng SE, Lee IT, Lin CC, Kou YR and Yang
CM: Cigarette smoke particle-phase extract induces HO-1 expression
in human tracheal smooth muscle cells: role of the c-Src/NADPH
oxidase/MAPK/Nrf2 signaling pathway. Free Radic Biol Med.
48:1410–1422. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fukano Y, Oishi M, Chibana F, Numazawa S
and Yoshida T: Analysis of the expression of heme oxygenase-1 gene
in human alveolar epithelial cells exposed to cigarette smoke
condensate. J Toxicol Sci. 31:99–109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maestrelli P, Messlemani E AH, De Fina O,
Nowicki Y, Saetta M, Mapp C and Fabbri LM: Increased expression of
heme oxygenase (HO)-1 in alveolar spaces and HO-2 in alveolar walls
of smokers. Am J Respir Crit Care Med. 164:1508–1513. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsoumakidou M, Tzanakis N, Chrysofakis G
and Siafakas NM: Nitrosative stress, heme oxygenase-1 expression
and airway inflammation during severe exacerbations of COPD. Chest.
127:1911–1918. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maestrelli P, Páska C, Saetta M, Turato G,
Nowicki Y, Monti S, Formichi B, Miniati M and Fabbri LM: Decreased
haem oxygenase-1 and increased inducible nitric oxide synthase in
the lung of severe COPD patients. Eur Respir J. 21:971–976. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Slebos DJ, Kerstjens HA, Rutgers SR,
Kauffman HF, Choi AM and Postma DS: Haem oxygenase-1 expression is
diminished in alveolar macrophages of patients with COPD. Eur
Respir J. 23:652–653. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Exner M, Minar E, Wagner O and Schillinger
M: The role of heme oxygenase-1 promoter polymorphisms in human
disease. Free Radic Biol Med. 37:1097–1104. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lakhdar R, Denden S, Kassab A, Leban N,
Knani J, Lefranc G, Miled A, Chibani JB and Khelil AH: Update in
chronic obstructive pulmonary disease: Role of antioxidant and
metabolizing gene polymorphisms. Exp Lung Res. 37:364–375. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shinohara T, Kaneko T, Nagashima Y, Ueda
A, Tagawa A and Ishigatsubo Y: Adenovirus-mediated transfer and
overexpression of heme oxygenase 1 cDNA in lungs attenuates
elastase-induced pulmonary emphysema in mice. Hum Gene Ther.
16:318–327. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li JQ, Wen Y, Zhao H, Liu ZL, Song MJ, Xu
YJ and Zhang ZX: The effects of bilirubin concentration on laminin
and epidermal growth factor expression in lung tissue and type II
pneumocytes in smoking rats model. Zhonghua Nei Ke Za Zhi.
44:129–132. 2005.In Chinese. PubMed/NCBI
|
|
29
|
Thurlbeck WM: Measurement of pulmonary
emphysema. Am Rev Respir Dis. 95:752–764. 1967.PubMed/NCBI
|
|
30
|
Brandsma CA, Hylkema MN, van der Strate
BW, Slebos DJ, Luinge MA, Geerlings M, Timens W, Postma DS and
Kerstjens HA: Heme oxygenase-1 prevents smoke induced B-cell
infiltrates: a role for regulatory T cells? Respir Res. 9:172008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
John G, Kohse K, Orasche J, Reda A,
Schnelle-Kreis J, Zimmermann R, Schmid O, Eickelberg O and Yildirim
AÖ: The composition of cigarette smoke determines inflammatory cell
recruitment to the lung in COPD mouse models. Clin Sci (Lond).
126:207–221. 2014. View Article : Google Scholar
|
|
32
|
Vargas-Rojas MI, Ramírez-Venegas A,
Limón-Camacho L, Ochoa L, Hernández-Zenteno R and Sansores RH:
Increase of Th17 cells in peripheral blood of patients with chronic
obstructive pulmonary disease. Respir Med. 105:1648–1654. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shen N, Wang J, Zhao M, Pei F and He B:
Anti-interleukin-17 antibodies attenuate airway inflammation in
tobacco-smoke-exposed mice. Inhal Toxicol. 23:212–218. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Linden A and Adachi M: Neutrophilic airway
inflammation and IL-17. Allergy. 57:769–775. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kurimoto E, Miyahara N, Kanehiro A, Waseda
K, Taniguchi A, Ikeda G, Koga H, Nishimori H, Tanimoto Y, Kataoka
M, et al: IL-17A is essential to the development of
elastase-induced pulmonary inflammation and emphysema in mice.
Respir Res. 14:52013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang X, Zheng H, Zhang H, Ma W, Wang F,
Liu C and He S: Increased interleukin (IL)-8 and decreased IL-17
production in chronic obstructive pulmonary disease (COPD) provoked
by cigarette smoke. Cytokine. 56:717–725. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang Y, Zhang L, Wu J, Di C and Xia Z:
Heme oxygenase-1 exerts a protective role in ovalbumin-induced
neutrophilic airway inflammation by inhibiting Th17 cell-mediated
immune response. J Biol Chem. 288:34612–34626. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhong W, Xia Z, Hinrichs D, Rosenbaum JT,
Wegmann KW, Meyrowitz J and Zhang Z: Hemin exerts multiple
protective mechanisms and attenuates dextran sulfate sodium-induced
colitis. J Pediatr Gastroenterol Nutr. 50:132–139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Taylor A, Verhagen J, Blaser K, Akdis M
and Akdis CA: Mechanisms of immune suppression by interleukin-10
and transforming growth factor-beta: the role of T regulatory
cells. Immunology. 117:433–442. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Inoue S, Suzuki M, Nagashima Y, Suzuki S,
Hashiba T, Tsuburai T, Ikehara K, Matsuse T and Ishigatsubo Y:
Transfer of heme oxygenase 1 cDNA by a replication-deficient
adenovirus enhances interleukin 10 production from alveolar
macrophages that attenuates lipopolysaccharide-induced acute lung
injury in mice. Hum Gene Ther. 12:967–979. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xia ZW, Xu LQ, Zhong WW, Wei JJ, Li NL,
Shao J, Li YZ, Yu SC and Zhang ZL: Heme oxygenase-1 attenuates
ovalbumin-induced airway inflammation by up-regulation of foxp3
T-regulatory cells, interleukin-10, and membrane-bound transforming
growth factor-1. Am J Pathol. 171:1904–1914. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee TS and Chau LY: Heme oxygenase-1
mediates the anti-inflammatory effect of interleukin-10 in mice.
Nat Med. 8:240–246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Drechsler Y, Dolganiuc A, Norkina O,
Romics L, Li W, Kodys K, Bach FH, Mandrekar P and Szabo G: Heme
oxygenase-1 mediates the anti-inflammatory effects of acute alcohol
on IL-10 induction involving p38 MAPK activation in monocytes. J
Immunol. 177:2592–2600. 2006. View Article : Google Scholar : PubMed/NCBI
|