Introduction
Coronary heart disease (CHD) is the leading cause of
morbidity and mortality worldwide, accounting for an estimated 7.3
million deaths in 2008 according to the World Health Organization
(1). Myocardial ischemia, which
is commonly observed when arteries supplying the heart become
occluded, results when inadequate blood perfusion affects the
cardiac tissues. To minimize cardiac damage, ischemic tissue must
be sufficiently reperfused. However, reperfusion has the potential
to exacerbate severe tissue injury, a process termed 'reperfusion
injury'. The main pathological manifestation of CHD is myocardial
damage due to ischemia/reperfusion (I/R) injury (2). Clinically, myocardial I/R injury
occurs during or following coronary angioplasty, thrombolytic
therapy, coronary revascularization and heart transplantation
(3). Myocardial I/R injury may
lead to the expansion of the myocardial infarct area, cardiac
arrhythmias, contractile dysfunction and even sudden death
(4).
Myocardial I/R injury has been studied for over 50
years. Jennings et al first described myocardial I/R injury
using a canine heart coronary artery ligation model in 1960
(5). The authors observed that
reperfusion accelerated the development of myocardial necrosis, and
the degree of myocardial necrosis following I/R for 30–60 min was
similar to that observed 24 h after coronary occlusion. To date,
various studies have shown that I/R injury is associated with
calcium overload (6–8), the production of free radicals
(9,10) and mitochondrial alterations
(10,11). Cytosolic calcium overload is now
well known as an essential pathophysiological mechanism which is
involved in reperfusion injury, although the source and origin of
the calcium remains to be determined (12).
Calcium transport in cells has been reported to be
governed by non-selective cation channels referred to as 'transient
receptor potential (TRP) channels', which are highly permeable to
calcium. At the present time, >30 mammalian TRP channels have
been identified, cloned and characterized. These channels are
grouped into the following 6 subfamilies based on their amino acid
sequence homology: i) TRPC, ii) TRPM, iii) TRPV, iv) TRPA, v) TRPML
and vi) TRPP (13). TRPC proteins
are widely expressed in cardiac, pulmonary and vascular tissues and
partially regulate cellular Ca2+ flux either by acting
as Ca2+ entry channels or by altering membrane potential
(14,15). As mentioned above, I/R injury is
associated with calcium overload. As a calcium channel, we wished
to determine whether TRPC participates in the development of I/R
injury. Certain studies have previously examined this idea. The
study by Zhanget al demonstrated that interleukin (IL)-17A
contributes to brain I/R injury via the calpain-transient receptor
potential cation channel, subfamily C, member 6 (TRPC6) pathway in
mice (16). Similarly, it has
been demonstrated that the activation of TRPC6 channels is
essential for lung I/R-induced edema in mice (17). However, whether TRPC6 is
associated with myocardial I/R injury remains unknown. Thus, in the
present study, we aimed to investigate this matter.
Nuclear factor-κB (NF-κB) is one of the key factors
regulating cell gene transcription. Known as a nuclear
transcription factor, NF-κB is involved in the regulation of the
expression of several genes. As a key regulator of cardiac genes,
NF-κB regulates the expression of multiple downstream signal
transduction cascades in a variety of physiological and
pathophysiological states (18).
Studies have shown that NF-κB is associated with myocardial I/R
injury (19). The nuclear
translocation of NF-κB upregulates TRPC6 expression and enhances
Ca2+ influx (20,21). The c-Jun N-terminal kinase (JNK)
signaling channel, as a member of the mitogen-activated protein
kinase (MAPK) family, plays an important role in the cellular
stress response. Also known as the stress-activated protein kinase
(SAPK), research has indicated that JNK is closely related to
myocardial I/R injury (22,23). In a model of myocardial I/R
injury, Shimizu et al found that myocardial ischemia first
activated MAPK activity, and then activated the transcription
factors, activator protein-1 (AF-1) and NF-κB (24). We thus wished to determine whether
TRPC6 participates in myocardial I/R injury, and whether this is
associated with the activation of the JNK signaling pathway through
the translocation of NF-κB. In this study, we conducted a
preliminary investigation into this matter.
Danshensu (DSS) [(R)-3-(3,
4-dihydroxyphenyl)-2-hydroxypropanoic acid; CID 11600642] is a
water-soluble component of phenolic acid from the widely used
Chinese herb, Danshen. Previous studies have confirmed that DSS has
biological activities in that it improves microcirculation and
exerts cardiovascular protective effects. For example, DSS has been
shown to restore endothelium-dependent vasorelaxation via the
prostacyclin pathway by increasing cyclooxygenase (COX)-2 gene
expression and prostacyclin production (25), and thus it suppresses the
formation of reactive oxygen species (ROS) and protects the
myocardium against ischemia (26), protecting endothelial cells
against injury induced by inflammation and inhibiting apoptosis
(27,28), as well as inhibiting cardiac
fibrosis through the negative regulation of ROS-/p38 MAPK signaling
(29).
It has previously been suggested that DSS exerts a
number of protective effects associated with NF-κB and JNK. DSS has
also been shown to reduce lipopolysaccharide (LPS)-induced
inflammatory responses in murine RAW264 macrophages by decreasing
the nuclear translocation of NF-κB p65 (30). It has also been shown to partly
inhibit the expression of the receptor for advanced glycation
endproducts (RAGE), phosphorylated (p-)p38 and COX-2, and inhibit
NF-κB activation in diabetic mice (31). Other studies have confirmed that
DSS exerts obvious inhibitory effects on JNK and NF-κB p65
expression in hepatic stellate cells (HSCs) stimulated with IL-1β
and may thus be used to inhibit hepatic fibrosis (32).
It has been suggested in previous research that DSS
protects against myocardial I/R injury (26). However, whether DSS is associated
with TRPC6 remains unknown. Therefore, in this study, we examined
the association between the protective effects of DSS and TRPC6
following myocardial I/R injury in vitro and the
intermediary roles of JNK and NF-κB.
Materials and methods
H9c2 cell culture and treatment
Rat H9c2 cells provided by the Cell Bank of Type
Culture Collection of Chinese Academy of Sciences (Shanghai, China)
were cultured in DMEM supplemented with 10% FBS at 37°C in a
humidified incubator with 5% CO2. The cells were fed
every 3 days and subcultured upon reaching 90% confluence. The
cells were plated at an appropriate density according to each
experimental design. To induce I/R injury, the H9c2 cells were
cultured in hypoxic solution in a hypoxic incubator (95%
N2, 5% CO2) for 2 h. The hypoxic solution was
subsequently replaced with reoxygenation solution, and the cells
were cultured in a high oxygen incubator (95% O2, 5%
CO2) for various periods of time. The inhibitors,
SP600125 (10 μM) and ammonium pyrrolidine dithiocarbamate
(PDTC, 10 μM) (Sigma, St. Louis, MO, USA) were administered
30 min prior to the induction of I/R injury. DSS (Shanxi, China)
(98% purity) was provided by Xi'an Hongxing Biotechnology Co., Ltd,
Xi'an, China. DSS was added to double distilled water and prepared
in 5, 25, 50 and 100 mg/l concentrations, separately.
In this study, the H9c2 cells were divided into 6
groups as follows: normal cultured H9c2 cells (Con), 2 h of
ischemia (I), 2 h of ischemia and then 0.5, 1, 2 and 3 h of
reperfusion (I/R 0.5 h, I/R 1 h, I/R 2 h and I/R 3 h). For DSS
treatment the cells were divided into 7 groups as follows: 1) the
untreated control (Con) group; ii) the ischemia (I) group; iii) the
cells subjected to 2 h of ischemia and 3 h of reperfusion (I/R 3 h)
group; iv) cells exposed to I/R 3 h and treated with 5 mg/l DSS
(D5+I/R 3 h) group; v) cells exposed to I/R 3 h and treated with 25
mg/l DSS (D25+I/R 3 h) group; vi) cells exposed to I/R 3 h and
treated with 50 mg/l DSS (D50+I/R 3 h) group; and vii) cells
exposed to I/R 3 h and treated with 100 mg/l DSS (D100+I/R 3 h)
group.
Apoptosis assay
An Annexin V-FITC Apoptosis Detection kit (BD
Biosciences) was used to evaluate H9c2 cell injury. The H9c2 cells
were harvested and washed twice with cold phosphate-buffered saline
(PBS), and then centrifuged at 1,000 × g for 5 min. Subsequently,
the cell pellets were resuspended in 200 μl binding buffer
with 10 μl Annexin V (1 μg/ml), and then incubated
for 15 min in the dark at room temperature. Following the addition
of 300 μl binding buffer with 5 μl propidium iodide
(PI; 50 μg/ml), the cells were analyzed by flow cytometry
with a BD FACSCalibur. The positive staining of Annexin V indicated
early apoptotic cells, and double staining of Annexin V and PI
indicated late apoptotic cells. The statistical data are presented
as the percentage of apoptotic cells.
Western blot analysis
At various time points during the experiment, the
cells were harvested and lysed in cell lysis buffer (Beyotime
Institute of Biotechnology, Shanghai, China) at 4°C. Protein
samples were separated by 8–10% SDS-PAGE, transferred onto
nitrocellulose membranes (Millipore, Billerica, MA, USA) and
blocked in 5% non-fat milk in TBST (150 mM NaCl, 50 mM Tris pH 7.5,
0.1% Tween-20) for 1 h at room temperature. The membranes were
incubated with one of the following primary antibodies at the
appropriate concentrations: i) rabbit anti-TRPC6 (T6442; Sigma),
ii) mouse anti-JNK (sc-827) and anti-p-JNK (sc-12882) (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), iii) mouse anti-NF-κB
p65 (SAB4502609; Sigma), iv) mouse anti-β actin (A2228; Sigma), or
v) mouse anti-lamin B (SAB1306342-40TST; Sigma) overnight at 4°C.
Finally, the membranes were incubated with horseradish
peroxidase-conjugated anti-rabbit or -mouse IgG (both from Santa
Cruz Biotechnology, Inc.) for 1 h at room temperature. An enhanced
chemiluminescence reagent (Santa Cruz Biotechnology, Inc.) was used
to detect the bound antibodies, and the blots were developed with a
Supersignal chemiluminescence detection kit. Relative protein
levels were normalized to those of either β-actin or lamin B.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted using the TRIzol RNA kit
(Sigma) according to the manufacturer's instructions. cDNA was
prepared from total RNA using the RevertAid™ First Strand cDNA
Synthesis kit (Fermentas, Vilnius, Lithuania). The reverse
transcription reaction was performed under the following
conditions: i) 37°C for 15 min, ii) 85°C for 5 sec, and iii) 42°C
for 2 min. PCR was performed using a primer specific for either JNK
or β-actin. The following primer pairs were used for PCR: i) JNK
sense, 5′-GGUUAUGUACGGAUUGUGGtt-3′ and antisense,
5′-CCACAAUCCGUACAUAACCtt-3′ and ii) β-actin sense,
5′-GGGAAATCGTGCGTGACATTAAGG-3′ and antisense,
5′-CAGGAAGGAAGGCTGGAAGAGTG-3′.
qPCR was run on a 7300 PCR system (Applied
Biosystems, Foster City, CA, USA) using a SYBR® Premix
Ex Taq™ kit (Takara Bio, Inc., Shiga, Japan). Each reaction
consisted of a 20-μl sample containing 2 μl cDNA, 10
μl 2X SYBR Green mixtures, 2 μl of each primer and 4
μl of RNase-free water. The following cycling conditions
were used: i) 1 cycle of denaturation at 95°C for 5 min, ii) 60°C
for 30 sec, and iii) 40 two-segment cycles from amplification (95°C
for 5 sec and 60°C for 30 sec). Calculations were made using the
∆∆Ct method.
Knockdown of JNK by siRNA
Specific siRNA targeting JNK and negative control
siRNA were purchased from the Ambion (Austin, TX, USA). Cell
transfection was performed according to the manufacturer's
instructions (Ambion). First, the cells were trypsinized and
diluted to 1×105 cells/ml medium. The transfection
reagent (Lipofectamine™ 2000, 11668-027; Ambion) and siRNA were
diluted separately in serum-free mediuma, mixed and incubated at
room temperature for 10 min to allow the siRNA/1ipid complex to
form. The siRNA/1ipid complex was then added to each well at a
final siRNA concentration of 60 pmol/well. At 48 or 72 h following
transfection, the cells were harvested for RT-qPCR or western blot
analysis to determine the JNK mRNA and protein levels. The H9c2
cells were subjected to I/R injury following transfection with the
JNK siRNA for 48 h. si-con represented the H9c2 cells that were
transfected with the negative control siRNA, and OR represented the
oligofectamine reagent group without siRNA.
Calcium flux assay
The H9c2 cells were trypsinized and diluted to
1×105 cells/ml medium. Subsequently, the cells were
collected and loaded with 5 μM of the fluo-3AM calcium
indicator (Invitrogen, Carlsbad, CA, USA) in Hank's balanced salt
solution (HBSS) for 30 min at 37°C. After the baseline ofcytosolic
free Ca2+ ([Ca2+]i) was recorded,
the receptor-operated channels were activated by the addition of
100 μM 1-oleoyl-2-acetyl-sn-glycerol (OAG) (Sigma) for 1
min, followed by changing the extracellular buffer to 2 mM
Ca2+ (CaCl2). The
[Ca2+]i level was detected at 5 time points
at 1-min intervals using an FLX 800 spectrophotofluorometer
(BioTek, Winooski, VT, USA) with a filter for 480 nm excitation and
510 nm emission wavelengths.
Statistical analysis
The data are presented as the means ± SD of 5
independent experiments performed in quintuplicate. Differences
were evaluated using one-way ANOVA. Statistical analyses were
performed using SPSS version 10.0. Values of p<0.05 were
considered to indicate statistically significant differences.
Results
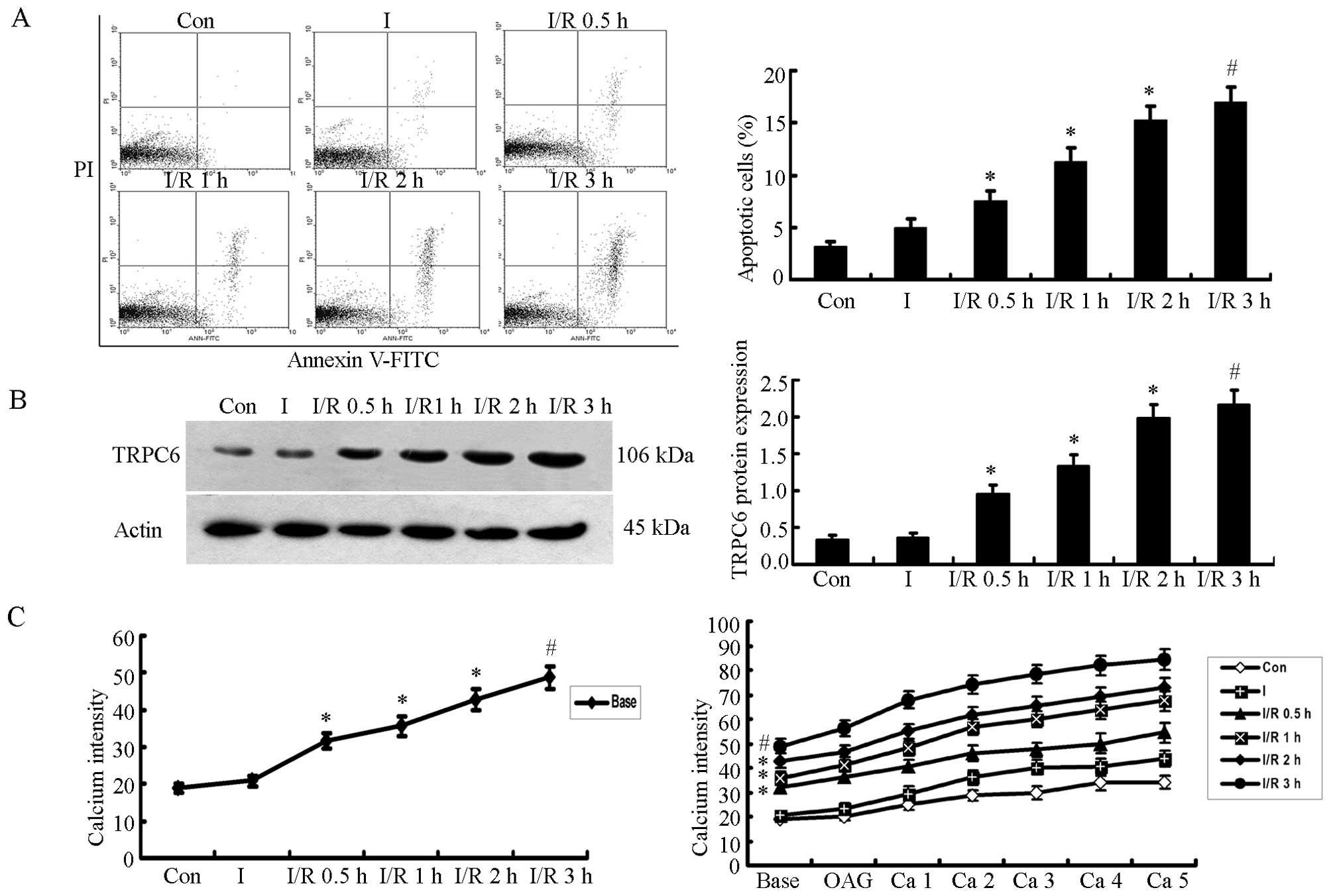
I/R injury increases the apoptosis of
H9c2 cells
In this study, the H9c2 cells were divided into 6
groups as follows: Con, I, I/R 0.5 h, I/R 1 h, I/R 2 h and I/R 3 h.
The results revealed that there was no significant difference in
the apoptotic rates between the cells in the I group and the Con
group, although the apoptotic rate was slightly increased in the I
group. However, when the ischemic cells underwent reperfusion, cell
apoptosis was not terminated, but rather increased in a
time-dependent manner, with a noticeable difference (P<0.05,
n=5). The difference between the cells exposed to 2 h of ischemia
followed by 3 h of reperfusion (I/R 3 h) and those in the I group
was the most significant (P<0.01, n=5; Fig. 1A).
TRPC6 protein expression increases over
the course of I/R
Western blot analysis was used to measure the TRPC6
protein expression levels in H9c2 cells; β-actin was used as a
protein standard. The results revealed that TRPC6 protein
expression increased in a time-dependent manner over the
reperfusion time following 2 h of ischemia (P<0.05, n=5). The
cells in the I/R 3 h group exhibited the most significant
difference compared with the cells in the I group (P<0.01, n=5;
Fig. 1B).
The [Ca2+]i level
increases following reperfusion
The [Ca2+]i level was measured
using the fluorescence indicator, fluo-3AM, which reflects the
TRPC6 channel activity under basal and OAG/CaCl2
stimulation conditions. A significant increase in the basal
[Ca2+]i level was detected in the cells in
the I/R 0.5, 1, 2 and 3 h groups compared with the cells in the the
Con or I groups (P<0.05), and the difference between the I/R 3 h
group and the I group was most significant (P<0.01, n=5).
Furthermore, following the activation of TRPC6 by OAG and
CaCl2, the [Ca2+]i level was
slightly higher at each time point (Ca 1–5) than the basal level.
This was similar to the basal [Ca2+]i level,
and the level between the Con group vs. the I group (P<0.05),
and the level between the I/R 3 h group vs. the Con or I group
(P<0.01; Fig. 1C).
In summary, the apoptosis of the H9c2 cells, the
protein expression of TRPC6, and the [Ca2+]i
levels increased significantly over the course of I/R, with I/R 3 h
being the most significant time point.
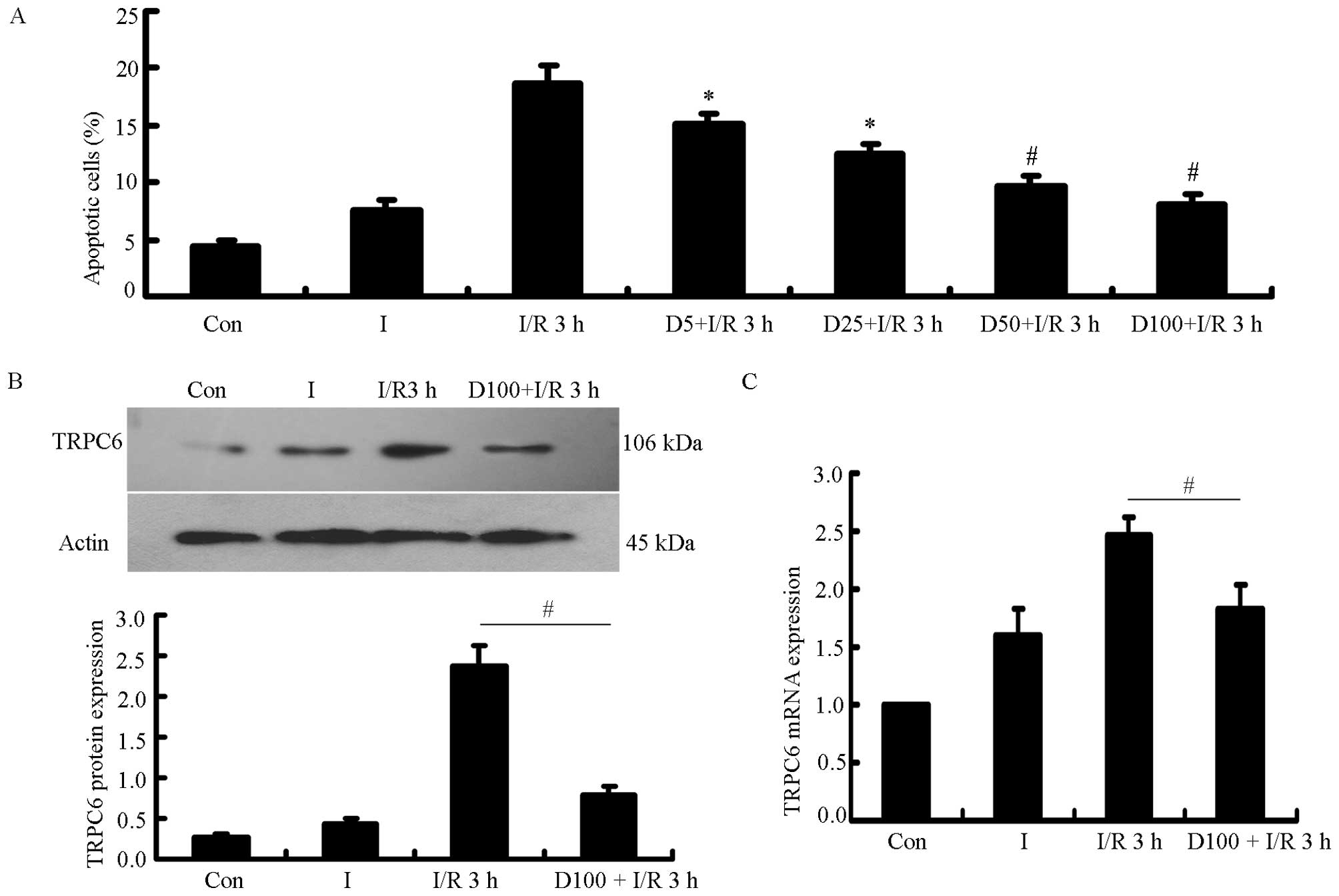
Cell apoptotic rate, and TRPC6 protein
and mRNA expression are reduced when the H9c2 cells are treated
with DSS prior to the induction of I/R injury
The H9c2 cells were treated with various
concentrations of DSS (5, 25, 50 and 100 mg/l) for 2 h prior to
being subjected to 2 h of ischemia and 3 h of reperfusion. The
apoptotic rate of the H9c2 cells was detected by flow cytometry.
There was a significant difference between the 7 groups, and the
rate of apoptosis decreased in a dose-dependent manner in the cells
treated with DSS, with the most significant difference being
observed between the cells in the I/R 3 h group (P<0.05,
P<0.01, n=5) and the cells treated with 100 mg DSS (Fig. 2A). The TRPC6 protein and mRNA
levels both decreased significantly when the H9c2 cells were
treated with 100 mg/l DSS prior to being subjected to I/R injury,
compared with the cells exposed to I/R 3 h not treated with DSS
(P<0.01, n=5; Fig. 2B and
C).
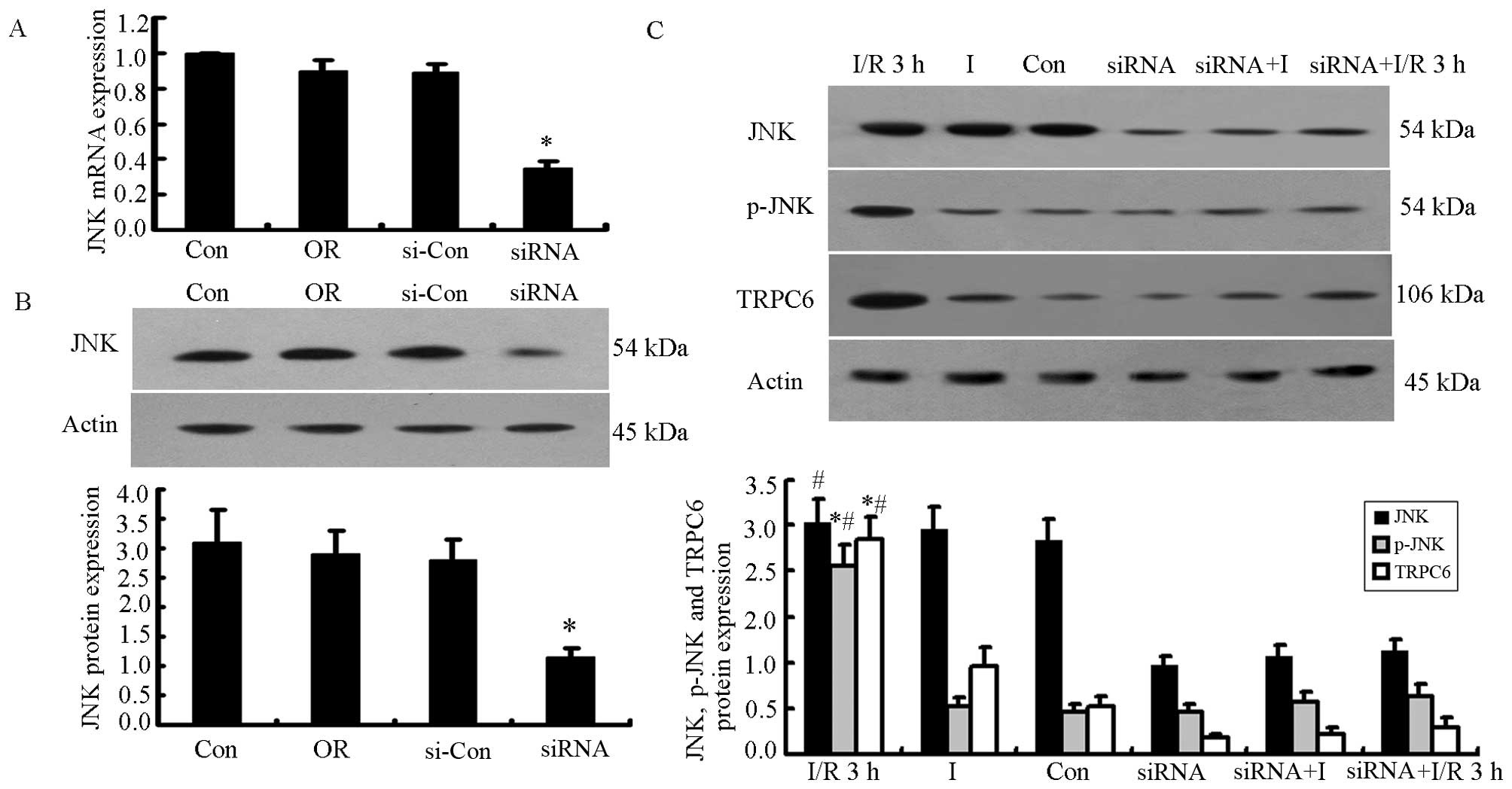
Knockdown of JNK prior to the induction
of I/R injury decreases TRPC6 protein expression, cell apoptosis
and the [Ca2+]i level in H9c2 cells
We used siRNA to selectively inhibit JNK protein
expression. The JNK mRNA level decreased significantly in the siRNA
group 48 h following transfection (P<0.05, n=5; Fig. 3A). The JNK protein level decreased
by 71.20% at 72 h following transfection (0.87±0.07 in the siRNA
group compared to 3.03±0.35 in the control group, P<0.05, n=5).
The difference in JNK expression between the OR and si-con groups
and the Con group was not significant (Fig. 3B).
Subsequently, we analyzed the changes in the JNK,
p-JNK and TRPC6 protein levels, cell apoptosis, and the
[Ca2+]i level in the H9c2 cells undergoing
I/R 3 h with or without JNK siRNA transfection. p-JNK and TRPC6
protein expression increased significantly after the H9c2 cells
were subjected to I/R injury (P<0.05 vs. Con or I group),
whereas there was no significant difference observed in JNK protein
expression between the Con and I groups. The H9c2 cells were
subjected to I/R injury following transfection with JNK siRNA, and
we found that JNK, p-JNK and TRPC6 protein expression decreased
significantly after the knockdown of JNK (P<0.05 vs. siRNA
group, siRNA+I or siRNA+I/R 3 h group; n=5; Fig. 3C).
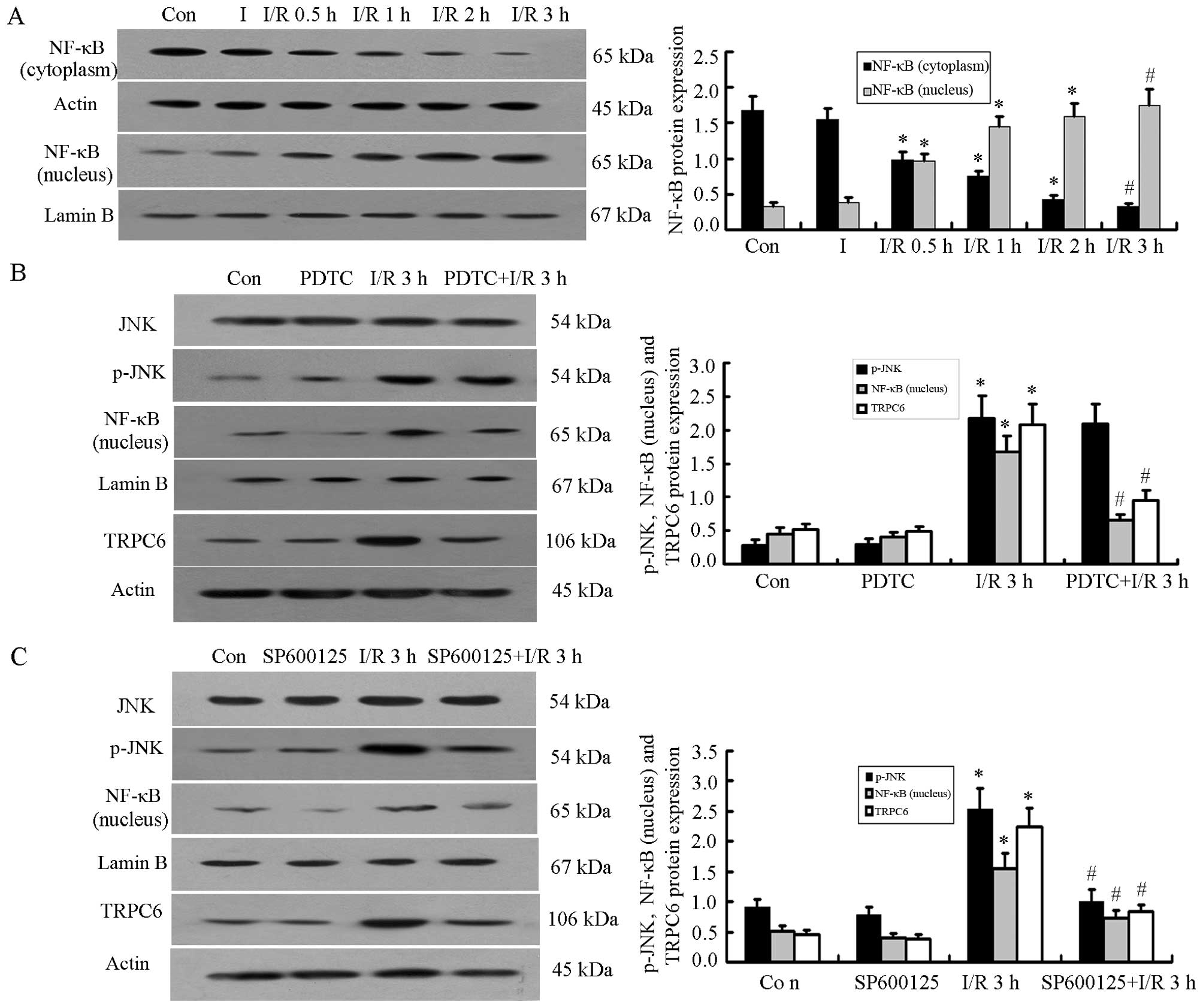
NF-κB p65 is activated during the I/R
process and translocates from the cytoplasm into the nucleus
Western blot analysis demonstrated that the protein
expression of NF-κB p65 in the nucleus increased significantly when
the H9c2 cells were subjected to I/R injury, with the highest level
observed in the I/R 3 h group. At the same time, the protein
content of NF-κB p65 in the cytoplasm decreased, with the level
decreasing gradually during reperfusion; the protein level of NF-κB
p65 in the cytoplasm was the lowest in the I/R 3 h group (P<0.05
vs. Con or I group; P<0.01 vs. Con or I group; n=5; Fig. 4A).
Inhibition of NF-κB reduces NF-κB and
TRPC6 protein expression
To further examine the association between p-JNK,
NF-κB and TRPC6 during I/R in H9c2 cells, the NF-κB pathway
inhibitor, PDTC (10 μM), was added 30 min before the H9c2
cells were subjected to I/R injury. The results revealed that the
protein expression of p-JNK and NF-κB in the nucleus and the
expression of TRPC6 increased significantly in the I/R 3 h group
compared with the Con or PDTC groups (P<0.05, n=5); the JNK
levels were not altered significantly between the groups.
Subsequently, when the cells were treated with PDTC prior to being
subjected to I/R injury, the protein expression of NF-κB in the
nucleus and the expression of TRPC6 decreased significantly
(P<0.05, n=5), whereas JNK and p-JNK protein expression were not
altered significantly following treatment with PDTC compared to the
I/R 3 h group (Fig. 4B).
Inhibition of p-JNK affects NF-κB
translocation and TRPC6 activation
We then examined the role of p-JNK in the
trans-location of NF-κB and the activation of TRPC6. We treated the
H9c2 cells for 30 min with the p-JNK inhibitor, SP600125 (10
μM), prior to the induction of I/R injury; this reduced the
increase in the protein expression of p-JNK, NF-κB (in the nucleus)
and TRPC6. We noted significant differences between the I/R 3 h
group and the Con or SP600125 group (P<0.05), while the levels
in the I/R 3 h and SP600125+I/R 3 h groups also differed
significantly (P<0.05, n=5; Fig.
4C).
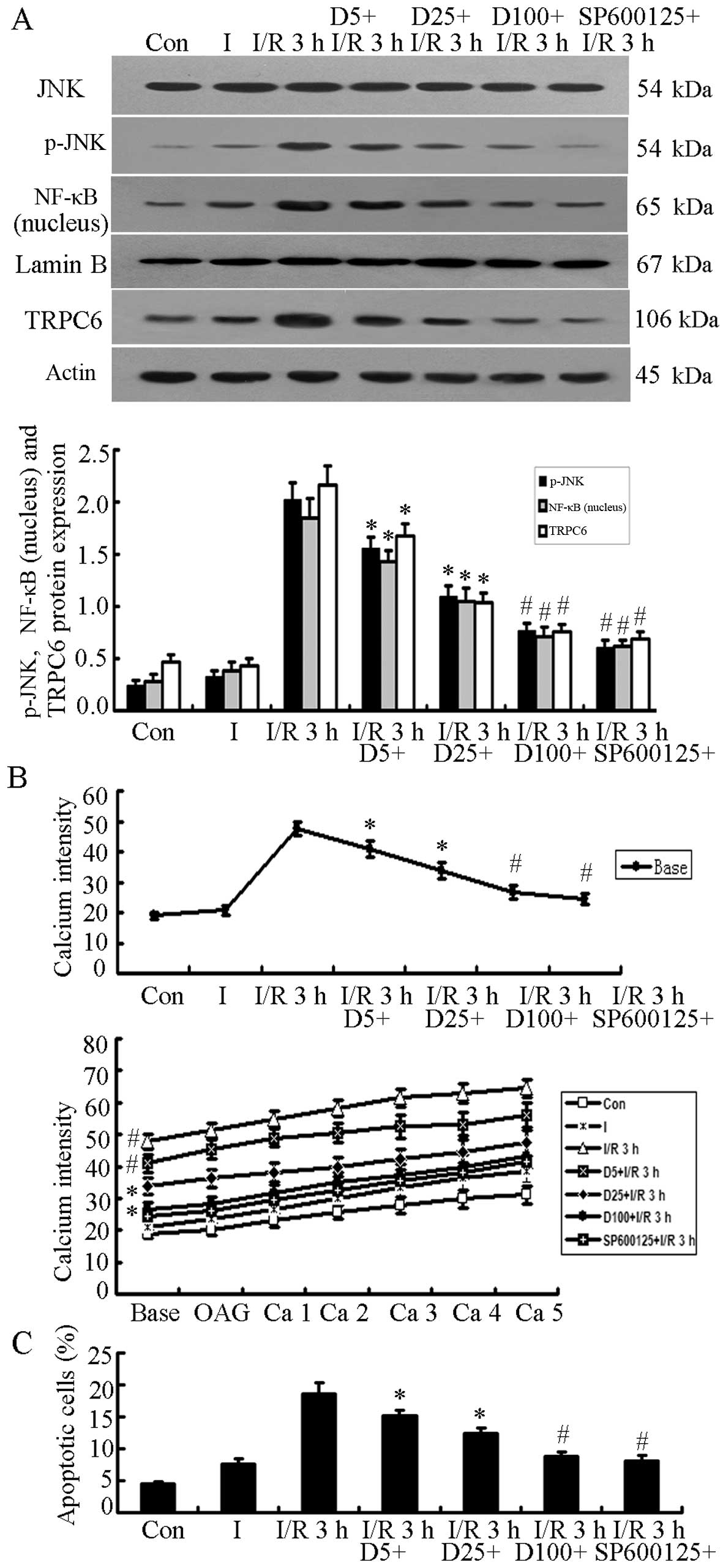
H9c2 cells treated with DSS or SP600125
prior to the inductoin of I/R injury
The protein expression of JNK, p-JNK, NF-κB (in the
nucleus) and TRPC6 was detected when the H9c2 cells were treated
with DSS (5, 25 and 100 mg/l) for 2 h or with SP600125 for 30 min
prior to the induction of I/R injury. p-JNK, NF-κB (nucleus) and
TRPC6 protein expression decreased significantly in a
dose-dependent manner in the cells treated with DSS compared with
the cells in the I/R 3 h group (P<0.05), while the cells in the
D100+I/R 3 h and SP600125+I/R 3 h groups exhibited the most
significant differences compared with the cells in the I/R 3 h
group (P<0.01, n=5; Fig. 5A).
Similarly, the [Ca2+]i level and the cell
apoptotic rate decreased significantly in a dose-dependent manner
when the H9c2 cells were cultured with DSS or SP600125 prior to
being subjected to I/R injury (P<0.05 or P<0.01 vs. the I/R 3
h group; n=5; Fig. 5B and C).
Discussion
Calcium influx leads to the development of various
diseases of the cardiovascular system (33,34); the discovery of mammalian TRP
channels was a new starting point for examinations of the molecular
basis of Ca2+ entry, and the discovery greatly promoted
the development of cardiovascular disease research (35–37). Previous studies have indicatd that
TRPC6 is widely expressed in the cardiovascular system and
participates in cardiac hypertrophy and remodeling (20,38), arrhythmia (39), heart failure (40) and pulmonary arterial hypertension
(41). TRPC6 has previously been
shown to accelerate the deterioration of cardiac function (42); however, the role of TRPC6 in
myocardial I/R injury remains unclear.
It is well known that calcium overload is an
important event for cellular apoptosis (6–8).
The TRP superfamily is highly permeable to calcium. Previous
studies have confirmed that the TRPC subfamily is involved in
calcium overload and apoptosis. It has been previously shown that
the overexpression of TRPC3 and the increased Ca2+
influx due to TRPC3 resulted in the apoptosis of mouse cells
(43). Satoh et al
confirmed that TRPC7 acts as a Ca2+ channel, leading to
myocardial apoptosis induced by AngII (44). Similarly, enhancing the expression
of TRPC6 has also been shown to increase Ca2+ influx
mediated by TRPC6 channels and contribute to podocyte apoptosis
(45). These data suggest that
the calcium overload resulting from the transient change in the
calcium level is an important event for apoptosis. However, the
association between myocardial I/R injury, the TRPC6 protein
content, calcium influx and apoptosis was not clear until this
study was undertaken.
In this study, our results suggested that TRPC6 was
expressed in H9c2 cells, and during I/R it increased over the
course of reperfusion. At the same time, the apoptotic rate and the
[Ca2+]i level also increased, which is
similar to the results of Shen et al (6) and Assayag et al (8). We noted an increase in the basal
[Ca2+]i level, and we also noted that the
continuous Ca2+ influx increased slightly following
stimulation with OAG, an activator of TRPC6, following the addition
of CaCl2. This suggested that the Ca2+ influx
mediated by TRPC6 was involved in I/R-induced cell injury, but not
all TRPC6 channels were activated in the I/R process in H9c2
cells.
Traditional Chinese medicine (TCM) has been
practiced for thousands of years; however, the specific mechanisms
involved are not so well known, thus restricting its clinical use.
DSS, the water-soluble active component of danshen, is abundant and
accessible and is known for its cardioprotective properties. Thus,
in this study, we used a series of experiments to examine the
protective effects of DSS against I/R injury and to elucidate the
potential mechanisms involved, in order to provide a sufficient
basis for its clinical use.
In the present study, H9c2 cells were treated with
various concentrations of DSS for 2 h prior to the induction of I/R
injury. This study confirmed that pre-treatment with DSS reduced
H9c2 cell apoptosis in a dose-dependent manner. These data
indicated that DSS exerted cardioprotective effects on myocardial
I/R injury in vitro, and that pre-treatment with 100 mg/l
DSS exerted the most prominent protective effect. Further studies
are required to further explore the underlying mechanisms. In this
study, we also showed that the TRPC6 protein and mRNA levels both
decreased significantly in a dose-dependent manner when the H9c2
cells were treated with DSS prior to being subjected to I/R injury,
which suggests that DSS exerts protective effects against I/R
injury by reducing TRPC6 expression.
Studies have demonstrated that JNK and NF-κB are
closely related to myocardial I/R injury (19,22,23). The p65 and p50 subunits of NF-κB
are activated in response to numerous stimuli and translocate into
the nucleus by the degradation of the inhibitor protein IκB. In
previous research, it has been demonstrated that NF-κB participates
in I/R injury. The activation of NF-κB induces the upregulation of
fibrinogen-like 2 (FGL2) expression through tumor necrosis factor
(TNF)-α during myocardial I/R (46). The levels of hepatic c-JUN, NF-κB
expression and the apoptotic rate have been shown to be decreased
during I/R in Toll-like receptor 4 (TLR4)-deficient mice compared
with wild-type mice (47).
In this study, the specific knockdown of JNK prior
to the induction of I/R injury using siRNA reduced JNK, p-JNK and
TRPC6 protein expression; p-JNK and TRPC6 protein expression
increased significantly in the H9c2 cells following I/R injury,
which suggests that the increase in TRPC6 protein expression is
mediated by the JNK signaling pathway. NF-κB p65 expression in the
nucleus increased significantly following I/R injury, whereas the
NF-κB protein content in the cytoplasm decreased. This suggests
that more and more NF-κB was activated and then transferred to the
nucleus during I/R. Previous studies have demonstrated that NF-κB
regulates the expression of the TRPC family. Hai et al
reported that NF-κB acts as an important positive transcriptional
regulator of TNF-α-induced COX-2-dependent prostaglandin E2 (PGE2)
production downstream of TRPC1-associated Ca2+ influx in
colonic myofibroblasts. Inhibitors of NF-κB (curcumin and SN-50)
attenuated the TNF-α-induced enhancement of TRPC1 expression and
store-dependent Ca2+ influx (48). Yu et al also provided
evidence that transforming growth factor (TGF)-β1 induces podocyte
damage by upregulating TRPC6 protein expression most likely through
the Smad3-ERK-NF-κB pathway (49). In the present study, we found that
the I/R-induced TRPC6 upregulation was reduced when the cells were
pretreated with PDTC, an NF-κB pathway inhibitor, but JNK and p-JNK
protein expression was not altered significantly. Thus, NF-κB may
be an essential upstream pathway that is able to block the
expression of TRPC6 protein but that of p-JNK during I/R.
We further examined the association between the
p-JNK signaling pathway, NF-κB and TRPC6. We found that the protein
expression of NF-κB in the nucleus and TRPC6 was suppressed when
the H9c2 cells were treated with the p-JNK inhibitor, SP600125,
prior to being subjected to I/R injury. Our results revealed that
p-JNK was involved in an upstream pathway that activates NF-κB and
results in its translocation into the nucleus, and increased TRPC6
protein expression in H9c2 cells during I/R. Previous research
supports this hypothesis: Pan et al reported that MAPKs,
particularly JNK, play important roles in the high glucose
(HG)-induced NF-κB activation in NRK-52E cells. The administration
of the JNK specific inhibitor, SP600125, markedly decreased NF-κB
activation, which emphasizes that JNK is a critical upstream
protein of NF-κB and plays an important role in HG-induced renal
inflammation (50).
In this study, the H9c2 cells were cultured with or
without DSS (5, 25 and 100 mg/l) for 2 h or SP600125 for 30 min
prior to the induction of I/R injury, and the results revealed that
p-JNK, NF-κB and TRPC6 protein expression decreased markedly in the
cells treated with DSS compared with the cells in the I/R 3 h
group. Similarly, the cell apoptotic rate and the
[Ca2+]i level significantly decreased in a
dose-dependent manner when H9c2 cells were treated with DSS or
SP600125 prior to being subjected to I/R. All these results suggest
that the DSS-induced cardioprotective effects are mediated through
the p-JNK, NF-κB and TRPC6 signaling pathways.
In conclusion, our data demonstrate that DSS exerts
significant protective effects against myocardial I/R injury,
possibly by inhibiting the phosphorylation of JNK, reducing the
production of p-JNK, inhibiting the NF-κB translocation into the
nucleus, and decreasing the protein expression of TRPC6, as well as
decreasing Ca2+ influx and reducing cell apoptosis.
Ultimately, DSS is an important component of Danshensu, a widely
used herb in traditional Chinese medicine, and it may prove to be a
promising therapeutic agent for reducing myocardial I/R injury in
clinical settings.
Acknowledgments
We would like to thank Dr Liu Shangquan, and the
laboratory technician, Yuan Yuan, for assisting with the
experiments at the Central Laboratory of Hefei Binhu Hospital. This
study was supported by the National Natural Science Foundation of
China (11040606 M201) and the Natural Science Foundation of Anhui
Province (11040606 M201).
References
|
1
|
Hausenloy DJ, Boston-Griffiths E and
Yellon DM: Cardioprotection during cardiac surgery. Cardiovasc Res.
94:253–265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Powers SK, Quindry JC and Kavazis AN:
Exercise-induced cardioprotection against myocardial
ischemia-reperfusion injury. Free Radic Biol Med. 44:193–201. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim TH and Lee SM: The effects of ginseng
total saponin, panaxadiol and panaxatriol on ischemia/reperfusion
injury in isolated rat heart. Food Chem Toxicol. 48:1516–1520.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ferdinandy P, Schulz R and Baxter GF:
Interaction of cardiovascular risk factors with myocardial
ischemia/reperfusion injury, preconditioning, and postconditioning.
Pharmacol Rev. 59:418–458. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jennings RB, Sommers HM, Smyth GA, Flack
HA and Linn H: Myocardial necrosis induced by temporary occlusion
of a coronary artery in the dog. Arch Pathol. 70:68–78.
1960.PubMed/NCBI
|
|
6
|
Shen AC and Jennings RB: Kinetics of
calcium accumulation in acute myocardial ischemic injury. Am J
Pathol. 67:441–452. 1972.PubMed/NCBI
|
|
7
|
Ma HJ, Li Q, Ma HJ, Guan Y, Shi M, Yang J,
Li DP and Zhang Y: Chronic intermittent hypobaric hypoxia
ameliorates ischemia/reperfusion-induced calcium overload in heart
via Na/Ca2+ exchanger in developing rats. Cell Physiol
Biochem. 34:313–324. 2014. View Article : Google Scholar
|
|
8
|
Assayag M, Saada A, Gerstenblith G,
Canaana H, Shlomai R and Horowitz M: Mitochondrial performance in
heat acclimation - a lesson from ischemia/reperfusion and calcium
overload insults in the heart. Am J Physiol Regul Integr Comp
Physiol. 303:R870–R881. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Santos EB, Koff WJ, Grezzana Filho TJ, De
Rossi SD, Treis L, Bona SR, Pêgas KL, Katz B, Meyer FS, Marroni NA
and Corso CO: Oxidative stress evaluation of ischemia and
reperfusion in kidneys under various degrees of hypothermia in
rats. Acta Cir Bras. 28:568–573. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Benhabbouche S, Crola da Silva C, Abrial M
and Ferrera R: The basis of ischemia-reperfusion and myocardial
protection. Ann Fr Anesth Reanim. 30(Suppl 1): S2–S16. 2011.In
French. View Article : Google Scholar
|
|
11
|
Sirvinskas E, Kinderyte A, Trumbeckaite S,
Lenkutis T, Raliene L, Giedraitis S, Macas A and Borutaite V:
Effects of sevoflurane vs. propofol on mitochondrial functional
activity after ischemia-reperfusion injury and the influence on
clinical parameters in patients undergoing CABG surgery with
cardiopulmonary bypass. Perfusion. Feb 16–2015.Epub ahead of print.
pii: 0267659115571174
|
|
12
|
Fauconnier J, Roberge S, Saint N and
Lacampagne A: Type 2 ryanodine receptor: a novel therapeutic target
in myocardial ischemia/reperfusion. Pharmacol Ther. 138:323–332.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nilius B and Owsianik G: The transient
receptor potential family of ion channels. Genome Biol. 12:2182011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dietrich A, Kalwa H, Fuchs B, Grimminger
F, Weissmann N and Gudermann T: In vivo TRPC functions in the
cardiopulmonary vasculature. Cell Calcium. 42:233–244. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Watanabe H, Murakami M, Ohba T, Takahashi
Y and Ito H: TRP channel and cardiovascular disease. Pharmacol
Ther. 118:337–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang J, Mao X, Zhou T, Cheng X and Lin Y:
IL-17A contributes to brain ischemia reperfusion injury through
calpain-TRPC6 pathway in mice. Neuroscience. 274:419–428. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weissmann N, Sydykov A, Kalwa H, Storch U,
Fuchs B, Mederos y Schnitzler M, Brandes RP, Grimminger F, Meissner
M, Freichel M, et al: Activation of TRPC6 channels is essential for
lung ischaemia-reperfusion induced oedema in mice. Nat Commun.
3:6492012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jones WK, Brown M, Ren X, He S and
McGuinness M: NF-kappaB as an integrator of diverse signaling
pathways: the heart of myocardial signaling? Cardiovasc Toxicol.
3:229–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chandrasekar B and Freeman GL: Induction
of nuclear factor kappaB and activation protein 1 in postischemic
myocardium. FEBS Lett. 401:30–34. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuwahara K, Wang Y, McAnally J, Richardson
JA, Bassel-Duby R, Hill JA and Olson EN: TRPC6 fulfills a
calcineurin signaling circuit during pathologic cardiac remodeling.
J Clin Invest. 116:3114–3126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hamid R and Newman JH: Evidence for
inflammatory signaling in idiopathic pulmonary artery hypertension.
Circulation. 119:2297–2298. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Laderoute KR and Webster KA:
Hypoxia/reoxygenation stimulates Jun kinase activity through redox
signaling in cardiac myocytes. Circ Res. 80:336–344. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kaiser RA, Liang Q, Bueno O, Huang Y,
Lackey T, Klevitsky R, Hewett TE and Molkentin JD: Genetic
inhibition or activation of JNK1/2 protects the myocardium from
ischemia-reperfusion-induced cell death in vivo. J Biol Chem.
280:32602–32608. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shimizu N, Yoshiyama M, Omura T, Hanatani
A, Kim S, Takeuchi K, Iwao H and Yoshikawa J: Activation of
mitogen-activated protein kinases and activator protein-1 in
myocardial infarction in rats. Cardiovasc Res. 38:116–124. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang D, Fan G, Wang Y, Liu H, Wang B, Dong
J, Zhang P, Zhang B, Karas RH, Gao X and Zhu Y: Vascular reactivity
screen of Chinese medicine danhong injection identifies Danshensu
as a NO-independent but PGI2-mediated relaxation factor. J
Cardiovasc Pharmacol. 62:457–465. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yin Y, Guan Y, Duan J, Wei G, Zhu Y, Quan
W, Guo C, Zhou D, Wang Y, Xi M and Wen A: Cardioprotective effect
of Danshensu against myocardial ischemia/reperfusion injury and
inhibits apoptosis of H9c2 cardiomyocytes via Akt and ERK1/2
phosphorylation. Eur J Pharmacol. 699:219–226. 2013. View Article : Google Scholar
|
|
27
|
Pan LL, Liu XH, Jia YL, Wu D, Xiong QH,
Gong QH, Wang Y and Zhu YZ: A novel compound derived from danshensu
inhibits apoptosis via upregulation of heme oxygenase-1 expression
in SH-SY5Y cells. Biochim Biophys Acta. 1830:2861–2871. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li H, Xie YH, Yang Q, Wang SW, Zhang BL,
Wang JB, Cao W, Bi LL, Sun JY, Miao S, et al: Cardioprotective
effect of paeonol and danshensu combination on
isoproterenol-induced myocardial injury in rats. PLoS One.
7:e488722012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu H, Tian A, Wu J, Yang C, Xing R, Jia P,
Yang L, Zhang Y, Zheng X and Li Z: Danshensu inhibits β-adrenergic
receptors-mediated cardiac fibrosis by ROS/p38 MAPK axis. Biol
Pharm Bull. 37:961–967. 2014. View Article : Google Scholar
|
|
30
|
Liu XH, Pan LL, Jia YL, Wu D, Xiong QH,
Wang Y and Zhu YZ: A novel compound DSC suppresses
lipopolysaccharide-induced inflammatory responses by inhibition of
Akt/NF-κB signalling in macrophages. Eur J Pharmacol. 708:8–13.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang T, Fu F, Han B, Zhang L and Zhang X:
Danshensu ameliorates the cognitive decline in
streptozotocin-induced diabetic mice by attenuating advanced
glycation end product-mediated neuroinflammation. J Neuroimmunol.
245:79–86. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu ZH, Dai LL, Yu BB and Li D: Effect of
danshensu on JNK and NF-kappaB signal transduction of rat hepatic
stellate cells induced by interleukin-1beta. Nan Fang Yi Ke Da Xue
Xue Bao. 29:914–917. 9212009.In Chinese.
|
|
33
|
Mandegar M, Remillard CV and Yuan JX: Ion
channels in pulmonary arterial hypertension. Prog Cardiovasc Dis.
45:81–114. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Varro A, Nanasi PP, Acsai K, Virag L and
Papp JG: Cardiac sarcolemmal ion channels and transporters as
possible targets for antiarrhythmic and positive inotropic drugs:
Strategies of the past - perspectives of the future. Curr Pharm
Des. 10:2411–2427. 2004. View Article : Google Scholar
|
|
35
|
Seo K, Rainer PP, Lee DI, Hao S, Bedja D,
Birnbaumer L, Cingolani OH and Kass DA: Hyperactive adverse
mechanical stress responses in dystrophic heart are coupled to
transient receptor potential canonical 6 and blocked by
cGMP-protein kinase G modulation. Circ Res. 114:823–832. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Malczyk M, Veith C, Fuchs B, Hofmann K,
Storch U, Schermuly RT, Witzenrath M, Ahlbrecht K, Fecher-Trost C,
Flockerzi V, et al: Classical transient receptor potential channel
1 in hypoxia-induced pulmonary hypertension. Am J Respir Crit Care
Med. 188:1451–1459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stiber JA, Tang Y, Li T and Rosenberg PB:
Cytoskeletal regulation of TRPC channels in the cardiorenal system.
Curr Hypertens Rep. 14:492–497. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kiso H, Ohba T, Iino K, Sato K, Terata Y,
Murakami M, Ono K, Watanabe H and Ito H: Sildenafil prevents the
up-regulation of transient receptor potential canonical channels in
the development of cardiomyocyte hypertrophy. Biochem Biophys Res
Commun. 436:514–518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Watanabe H, Iino K, Ohba T and Ito H:
Possible involvement of TRP channels in cardiac hypertrophy and
arrhythmia. Curr Top Med Chem. 13:283–294. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kuwahara K and Nakao K: New molecular
mechanisms for cardiovascular disease:transcriptional pathways and
novel therapeutic targets in heart failure. J Pharmacol Sci.
116:337–342. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yu Y, Keller SH, Remillard CV, Safrina O,
Nicholson A, Zhang SL, Jiang W, Vangala N, Landsberg JW, Wang JY,
et al: A functional single-nucleotide polymorphism in the TRPC6
gene promoter associated with idiopathic pulmonary arterial
hypertension. Circulation. 119:2313–2322. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Minamino T: Cardioprotection from
ischemia/reperfusion injury: basic and translational research. Circ
J. 76:1074–1082. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shan D, Marchase RB and Chatham JC:
Overexpression of TRPC3 increases apoptosis but not necrosis in
response to ischemia-reperfusion in adult mouse cardiomyocytes. Am
J Physiol Cell Physiol. 294:C833–C841. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Satoh S, Tanaka H, Ueda Y, Oyama J, Sugano
M, Sumimoto H, Mori Y and Makino N: Transient receptor potential
(TRP) protein 7 acts as a G protein-activated Ca2+
channel mediating angiotensin II-induced myocardial apoptosis. Mol
Cell Biochem. 294:205–215. 2007. View Article : Google Scholar
|
|
45
|
Zhang H, Ding J, Fan Q and Liu S: TRPC6
up-regulation in Ang II-induced podocyte apoptosis might result
from ERK activation and NF-kappaB translocation. Exp Biol Med
(Maywood). 234:1029–1036. 2009. View Article : Google Scholar
|
|
46
|
Jia P, Wang J, Wang L, Chen X, Chen Y, Li
WZ, Long R, Chen J, Shu YW, Liu K and Wang ZH: TNF-α upregulates
Fgl2 expression in rat myocardial ischemia/reperfusion injury.
Microcirculation. 20:524–533. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ben-Ari Z, Avlas O, Fallach R,
Schmilovitz-Weiss H, Chepurko Y, Pappo O and Hochhauser E: Ischemia
and reperfusion liver injury is reduced in the absence of Toll-like
receptor 4. Cell Physiol Biochem. 30:489–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hai L, Kawarabayashi Y, Imai Y, Honda A
and Inoue R: Counteracting effect of TRPC1-associated
Ca2+ influx on TNF-α-induced COX-2-dependent
prostaglandin E2 production in human colonic myofibroblasts. Am J
Physiol Gastrointest Liver Physiol. 301:G356–G367. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yu L, Lin Q, Liao H, Feng J, Dong X and Ye
J: TGF-β1 induces podocyte injury through Smad3-ERK-NF-κB pathway
and Fyn-dependent TRPC6 phosphorylation. Cell Physiol Biochem.
26:869–878. 2010. View Article : Google Scholar
|
|
50
|
Pan Y, Zhang X, Wang Y, Cai L, Ren L, Tang
L, Wang J, Zhao Y, Wang Y, Liu Q, et al: Targeting JNK by a new
curcumin analog to inhibit NF-κB-mediated expression of cell
adhesion molecules attenuates renal macrophage infiltration and
injury in diabetic mice. PLoS One. 8:e790842013. View Article : Google Scholar
|