Introduction
As the principal cation in extracellular fluid,
sodium (Na+) is an essential nutrient for the
maintenance of normal cell function. However, a high-sodium diet
(HSD) has been widely be implicated in the development of
hypertension (1–3) and cardiovascular diseases,
particularly fatal coronary heart diseases (1,4)
and stroke (1).
Under physiological conditions, cells, such as inner
medullary collecting duct (IMCD) cells of the collecting duct and
vascular smooth muscle cells (VSMCs) of renal capillaries in the
renal medulla, are normally exposed to variably high concentrations
of urea and sodium chloride (NaCl), as a consequence of the urine
concentrating mechanism (5).
Under antidiuretic conditions, NaCl and urea are the most prevalent
solutes in the medullary interstitium (6). In vitro studies have reported
that when IMCD3 cells are exposed to culture medium to which
extreme high concentrations of NaCl are added, this may lead to DNA
damage (212.5 mM NaCl added) (7),
oxidative stress (300 mM NaCl added) (8) and cell cycle arrest (100 mM NaCl
added) (9). Therefore, the
mechanisms responsible for the adaptation of cells such as IMCD3
and VSMCs to various concentrations of Na+ remain poorly
understood and thus warrant further investigations.
Apart from the renal medulla, the interstitium
containing large amounts of glycosaminoglycans is considered to be
a separately regulated space for Na+ homeostasis
(10,11). Long-term balance studies on humans
have confirmed that considerable amounts of Na+
accumulate in the interstitium due to excessive NaCl consumption
(12–14). The skin Na+
concentration due to HSD can be as high as 180 to 190 mM in rats
(15). The study by Machnik et
al further demonstrated that the interstitial accumulation of
Na+ in skin results in hyperplasia of the lymph
capillary network (10).
Therefore, apart from physiological conditions in the renal
medulla, it is necessary to further address whether HSD can lead to
excess Na+ accumulation in different tissues and whether
this Na+ retention may be associated with any adverse
effects.
It should be noted that salt restriction further
improves blood pressure control in patients treated with a
combination of an angiotensin-converting enzyme (ACE) inhibitor and
a diuretic (16). Matsushita
et al also found that the combination of HSD with bilateral
oophorectomy significantly increased the body Na+/water
ratio, and increased cerebral aneurysm formation irrespective of
hypertension (17). The abnormal
proliferation of VSMCs is considered responsible for the
physiological and pathophysiological changes taking place in the
vascular wall (18,19). In this study, we aimed to assess
whether Na+ per se directly affects the proliferation of
VSMCs at relatively higher concentrations and to elucidate the
underlying mechanisms. This may firstly shed light on the
mechanisms responsible for the adaptation of VSMCs to high
concentrations of Na+, and secondly, it may reveal the
possible direct pathogenic effect of the excessive consumption of
Na+, which is independent of pressure, the
renin-angiotensin-aldosterone system (RAAS) (20,21) and endothelial function (22).
Materials and methods
Reagents, kits and antibodies
Dulbecco's modified Eagle's medium (DMEM)/high
glucose and phenol red-free DMEM were purchased from HyClone
(Logan, UT, USA). Charcoal stripped fetal bovine serum (FBS) was
obtained from Gibco-BRL (Grand Island, NY, USA). Rabbit monoclonal
antibodies against proliferating cell nuclear antigen (PCNA;
1:1,000; #13110), phosphorylated c-Jun amino N-terminal kinases
(p-JNK; 1:1,000; #4668) and phosphorylated extracellular
signal-regulated kinase 1/2 (p-ERK1/2; 1:2,000; #4370) were
provided by Cell Signaling Technology (Beverly, MA, USA). Rabbit
polyclonal antibody against phosphorylated p38 mitogen-activated
protein kinase (p-p38 MAPK) was supplied by Signalway Antibody LLC
(College Park, MD, USA). Rabbit monoclonal antibody against β-actin
(1:10,000; JC-PA-βA1) and horseradish peroxidase-conjugated
goat-anti-rabbit antibody (1:4,000; JC-PC012-1h) were purchased
from Geneshare (Xi'an, Shannxi, China). NaCl, choline chloride,
sorbital, mouse monoclonal anti-actin, α-smooth muscle-FITC
antibody (anti-α-SM-actin antibody; F3777) and DAPI were obtained
from Sigma-Aldrich (St. Louis, MO, USA). The Cell-Light™
5-ethynyl-2′-deoxyuridine (EdU) imaging detecting kit was purchased
from RiboBio (Guangzhou, China). SP600125 (a JNK inhibitor) was
provided by Cell Signaling Technology. PD98059 (an ERK1/2
inhibitor), SB203580 (a p38 MAPK inhibitor) and anisomycin (a p38
MAPK activator) were supplied from Santa Cruz Biotechnology
(Dallas, TX, USA). In addition,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltet-razolium bromide (MTT)
was purchased from Amresco (Solon, OH, USA).
Cell culture and treatment
Rat VSMCs were purchased from CHI Scientific, Inc.
(Jiangsu, China) and cultured in DMEM supplemented with 10% FBS at
37°C under 5% CO2/95% air in a humidified incubator.
Cells at passages 3 to 8 with a purity of >95% (determined by
immunofluorescence staining for α-SM-actin) were used in the
experiments. In order to obtain quiescent VSMCs, the cells were
incubated in serum-free medium for 24 h. Subsequently, standard
DMEM supplemented with 5% FBS was used as the control culture
medium, in which the Na+ concentration was approximately
156 mM. High-sodium medium was prepared by the addition of NaCl
(10, 20, 30, 40 and 50 mM,respectively; additional to the levels
already present in the medium) to the control culture medium.
Choline chloride and sorbital were used to examine the effects of
chloridion (Cl−) and osmotic pressure on the
proliferation of the VSMCs. High-Cl− medium was prepared
by the addition of choline chloride (10, 20, 30, and 50 mM,
respectively; additional to the levels already present in the
medium) to the control culture medium. High-osmotic pressure medium
was prepared by the addition of sorbital (20, 40, 60, 80 and 100
mM, respectively; additional to the levels already present in the
medium) to the control culture medium. For MTT assay, the VSMCs
were treated for 12, 24 or 48 h. For EdU incorporation assay, the
VSMCs were treated for 24 h. PCNA expression at the mRNA level was
detected following treatment for 3, 6, 9 and 12 h. PCNA expression
at the protein level was detected following treatment for 6, 15, or
24 h. Phosphorylation levels were detected after 15, 30, 60, 90 and
120 min of intervention. To examine the effects of 3 MAPK members
on the proliferation of VSMCs induced by additional NaCl, the cells
were pre-treated wtih SP600125 (a JNK inhibitor), PD98059 (an
ERK1/2 inhibitor), SB203580 (a p38 MAPK inhibitor) and anisomycin
(a p38 MAPK activator) for 30 min.
Immunofluorescence staining
The VSMCs were cultured on sterile glass cover slips
in 12-well plates. Following fixation with 4% paraformaldehyde, the
VSMCs were permeabilized by 0.1% Triton X-100 for 30 min at room
temperature and blocked with goat serum for 1 h at 37°C. The cells
then covered with anti-α-SM-actin antibody were incubated at 4°C in
a dark humidified chamber overnight. The samples were
counter-stained with DAPI at room temperature for 10 min. The
images were captured using NIS-Elements imaging software (Nikon,
Tokyo, Japan).
MTT assay
Cell proliferation was firstly investigated by MTT
assay according to published literature (23). Briefly, the VSMCs were treated
with additional NaCl (10–50 mM), choline chloride (10–50 mM) or
sorbital (20–100 mM) in DMEM supplemented with 5% FBS for 12, 24 or
48 h. The cells were then incubated with MTT solution (0.1 mg/ml)
for 4 h. The formed formazan crystals were dissolved in 150
μl/well dimethyl sulfoxide (DMSO). The absorbance was
recorded at a wavelength of 490 nm using a microplate reader
(Bio-Rad, Hercules, CA, USA). All experiments were performed at
least 3 times. The relative proliferation of the cells was
calculated as the absorbance of treated cells/control cells
×100%.
EdU incorporation assay
Following synchronization with serum-free medium for
24 h, the cells were treated with or without additional NaCl (30
mM) in DMEM supplemented with 5% FBS for 24 h. The EdU
incorporation assay was performed according to the manufacturer's
instructions (RiboBio). In brief, the VSMCs were incubated with 50
μM EdU for 2 h. Following fixation with 4% paraformaldehyde
and permeabilization in 1.0% Triton X-100, the cells underwent EdU
staining. The cell nuclei were counterstained with Hoechst 33342.
EdU-positive nuclei were determined under a fluorescence microscope
(Olympus BX51; Olympus, Tokyo, Japan). The cell proliferation rate
was calculated as the proportion of nucleated cells incorporating
EdU in 5 high-power fields per well.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Quantification was carried out as previously
described (24). Briefly, total
RNA was extracted from the cells using TRIzol reagent and reverse
transcribed using the cDNA synthesis kit (Fermentas, Burlington,
CA, USA). Quantitative (real-time) PCR (qPCR) was performed using
SYBR® select Master Mix on an iQ5 Multicolor Real-Time
PCR Detection system (Bio-Rad). The primer sequences used for PCNA
were as follows: sense, 5′-ACCTCACCAGC ATGTCCAA-3′ and antisense,
5′-CATAGTCTGAAACTTTC TCTTGATTTG-3′. Beta-glucuronidase (GUSB) was
used as a housekeeping gene and the primer sequences were: sense,
5′-CTCTGGTGGCCTTACCTGAT-3′ and antisense, 5′-CAGA CTCAGGTGTTGTCA
TCG-3′. The relative expression level of the target gene was then
determined using a comparative method (2−ΔΔCT).
Western blot analysis
Western blot analysis was performed as previously
described (24). Briefly, the
VSMCs were lysed in RIPA buffer supplemented with protease and
phosphatase inhibitors. The protein content was determined using a
BCA protein assay kit (Pierce, Rockford, IL, USA). Equivalent
amounts of protein were subjected to 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for
electrophoresis and then transferred onto PVDF membranes (Bio-Rad).
The blots were incubated with primary antibodies against PCNA,
p-JNK, p-ERK1/2, p-p38 MAPK and β-actin, and then with horseradish
peroxidase-conjugated goat-anti-rabbit antibody. The protein
signals were detected using chemiluminescence. All densitometric
data for the target genes were corrected with β-actin as a loading
control.
Statistical analysis
Data are expressed as the means ± standard deviation
(SD). Comparisons among 3 or more groups were analyzed by one-way
analysis of variance (ANOVA), whereas the Student's t-test was used
for comparisons between 2 groups. A value of P<0.05 was
considered to indicate a statistically significant difference.
Statistical analysis was performed using SPSS 16.0 software (SPSS,
Inc., Chicago, IL, USA).
Results
High sodium levels rather than high
Cl− levels or osmotic pressure promote the proliferation
of VSMCs
In the present study, we used α-SM-actin as a marker
of contractile VSMCs to identify the phenotype and purity of the
commercially available VSMCs (25). The cultured VSMCs exhibited a
spindle-like shape. Immunofluorescence staining revealed an
abundance of green myonemes in the cytoplasm. The purity of the
obtained cells was >95% (data not shown).
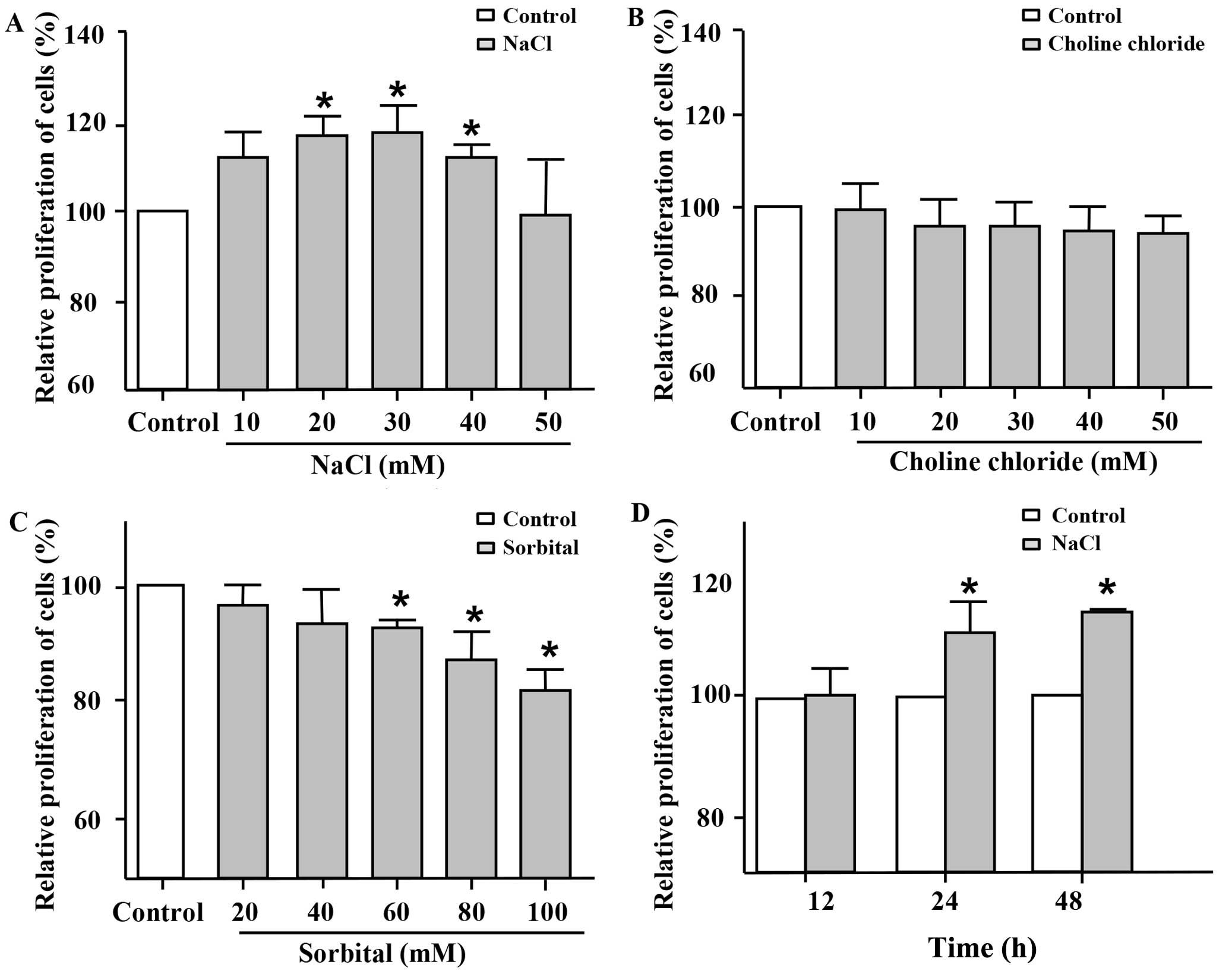
Additional NaCl was added to the basal medium in
order to verify whether Na+ directly promotes the
proliferation of VSMCs at higher concentrations. First, MTT assay
was used to evaluate cell proliferation. Following incubation for
24 h, the addition of 20–40 mM NaCl to the cell medium markedly
promoted the proliferation of the VSMCs compared with the untreated
control group (Fig. 1A).
Furthermore, this induction of cell proliferation by the high
sodium levels was the most significant when 30 mM NaCl were added.
To further clarify whether the proliferative effects of NaCl on
VSMCs are due to Na+ itself, choline chloride and
sorbital were also employed in the present study. Choline chloride
at various concentrations, which was used as Cl−
intervention, did not increase the proliferation of the VSMCs
(Fig. 1B). Sorbital, which was
used as osmotic intervention, exhibited no significant effect on
the proliferation of the VSMCs (Fig.
1C) at low concentrations (20 and 40 mM). Indeed, sorbital
inhibited the proliferation of the VSMCs when used at a
concentration of >60 mM. As shown in Fig. 1D, the addition of NaCl (30 mM;
additional to the levels already in medium) to the cell medium
promoted the proliferation of the VSMCs following incubation for 24
and 48 h, but not for 12 h. These results indicated that
Na+ at relatively high concentrations per se, rather
than Cl− or osmotic pressure promoted the proliferation
of VSMCs.
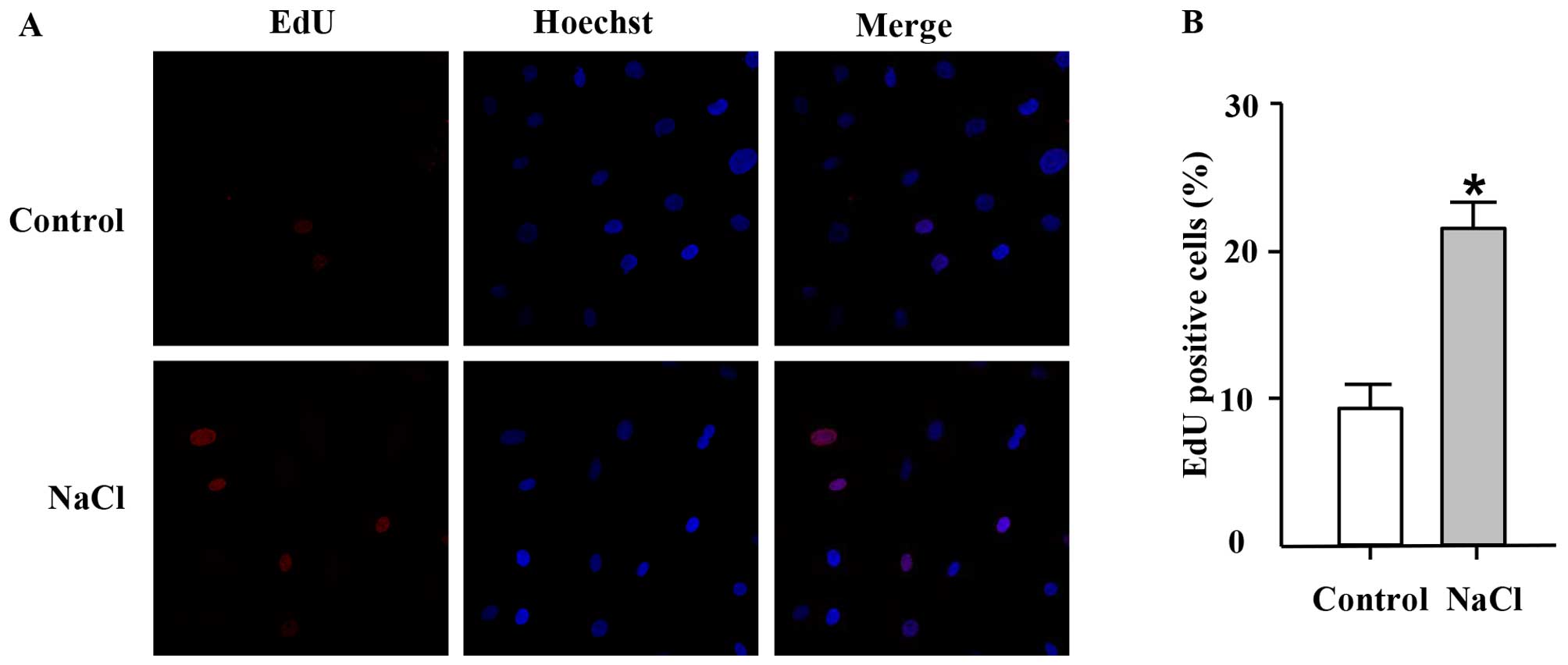
EdU, a thymidine analogue, is incorporated into
cellular DNA during cell proliferation (26). Thus, in this study, we used the
EdU incorporation assay to further confirm the effects of high
sodium levels on the proliferation of VSMCs. In accordance with the
results of MTT assay, EdU incorporation assay revealed that
addition of NaCl (30 mM; additional to the levels already in
medium) to the cell culture medium for 24 h increased the
percentage of EdU-positive nuclei compared with the control group
(Fig. 2), thus indicating an
increase in DNA synthesis in the ultured VSMCs which was induced by
NaCl.
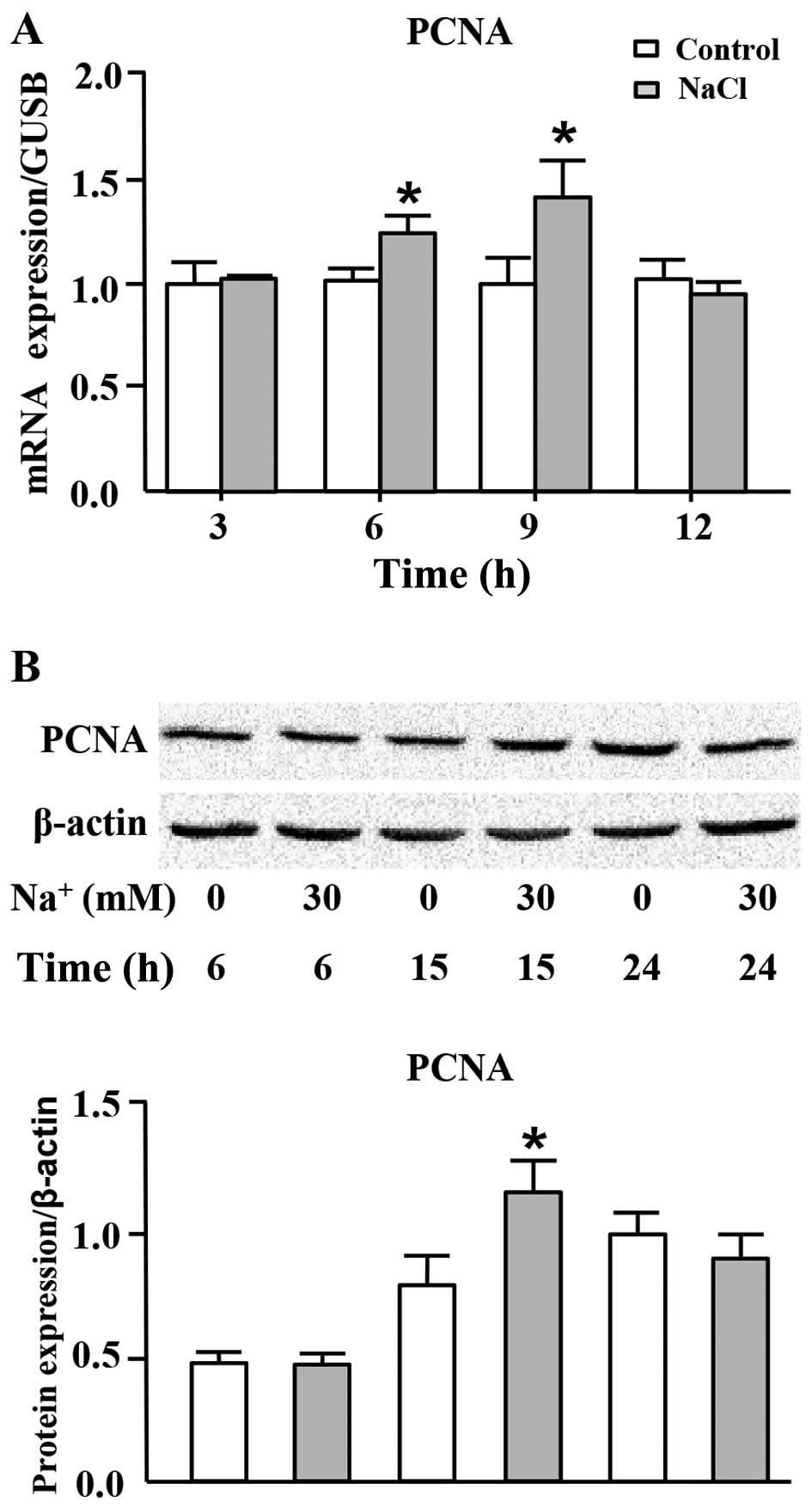
High sodium level increases PCNA
expression at both the mRNA and protein levels
PCNA, the eukaryotic DNA sliding clamp, confers high
processivity to replicative DNA polymerase, which is recognized as
a marker of cell proliferation (27,28). We thus measured the mRNA and
protein expression levels of PCNA in the cells cultured in medium
with the addition of high level of sodium. As shown in Fig. 3A, the mRNA expression of PCNA was
significantly increased following the addition of 30 mM
Na+ to the cell culture medium (additional to the levels
already in medium) for 6 and 9 h. In addition, the protein
expression of PCNA was transiently, but markedly increased
following the addition of Na+ to the medium for 15 h
(Fig. 3B).
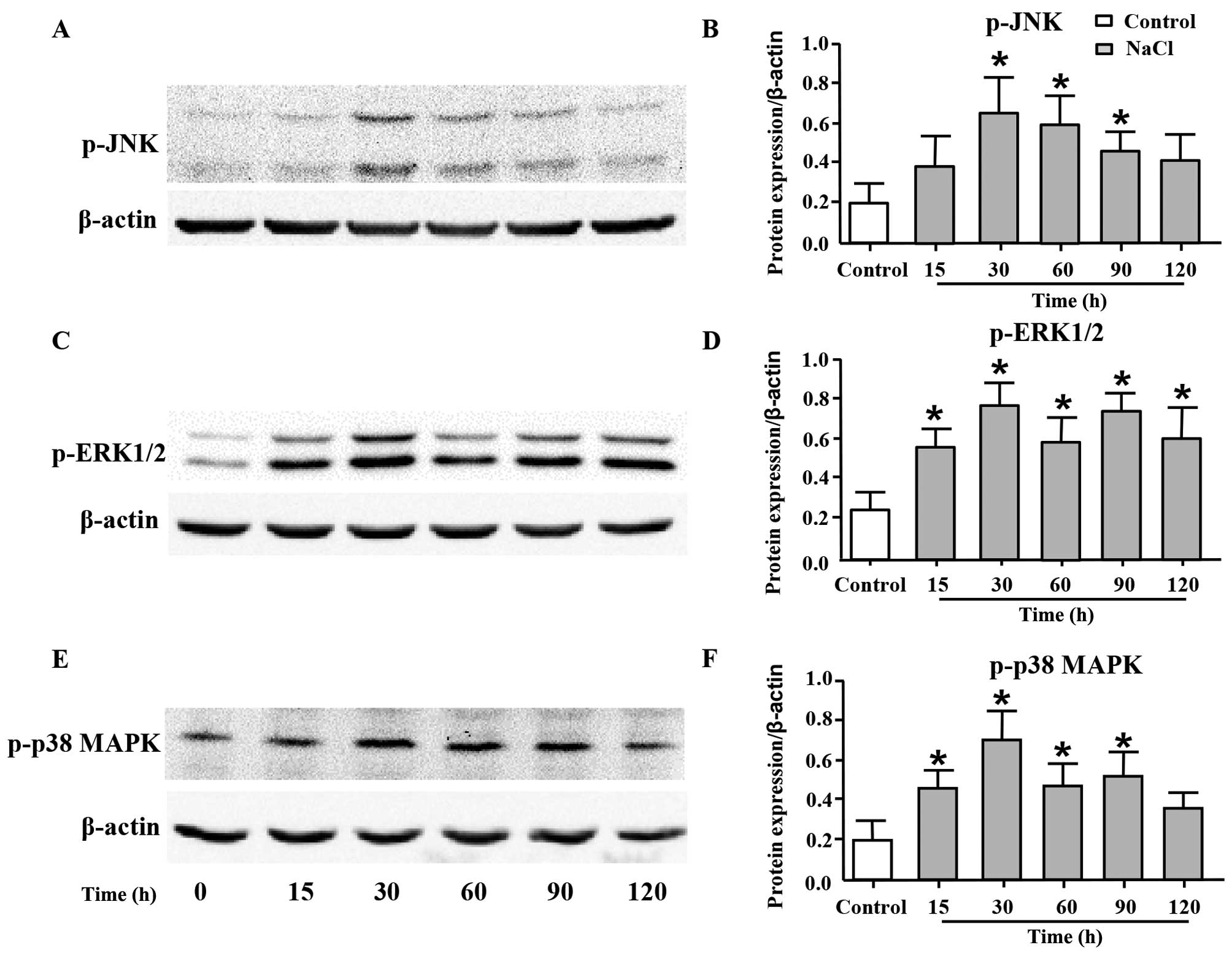
High sodium level increases the
phosphorylation levels of JNK, ERK1/2 and p38 MAPK
JNK, ERK1/2 and p38 MAPK are the members of MAPK
family, and they play important roles in cell proliferation
(29,30). Thus, in order to elucidate the
underlying mechanisms responsible for the proliferative effects of
additional Na+ on VSMCs, we measured the phosphorylation
levels of JNK, ERK1/2 and p38 MAPK by western blot analysis. As
shown in Fig. 4, the expression
levels of p-JNK (Fig. 4A and B),
p-ERK1/2 (Fig. 4C and D) and
p-p38 MAPK (Fig. 4E and F) were
significantly increased following the addition of 30 mM
Na+ to the cell culture medium (additional to the levels
already in medium) for 30 min, and such an increase was maintained
until 120 min post-treatment.
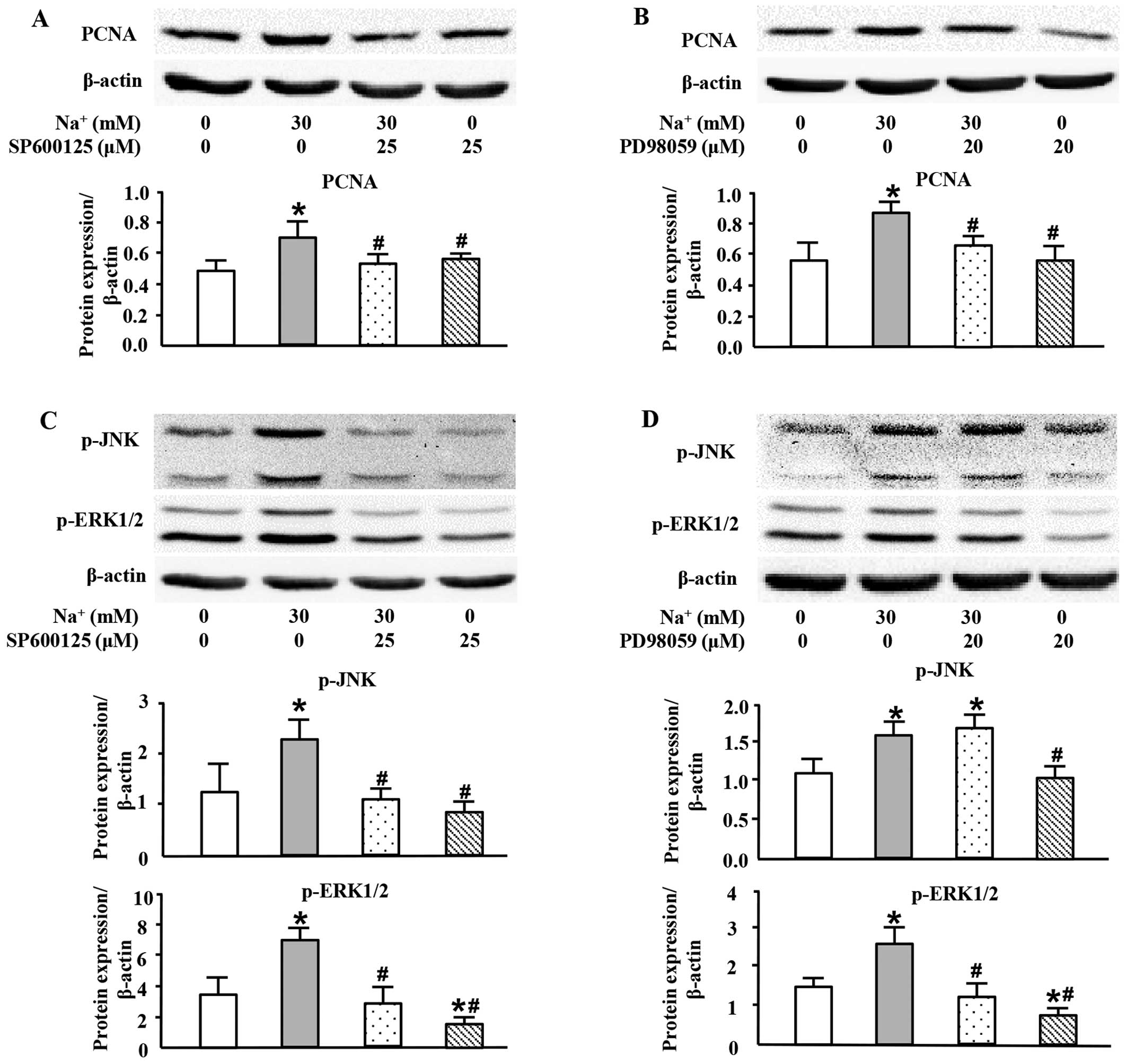
High sodium level increases PCNA
expression at the protein level through the JNK/ERK1/2 pathway
To further investigate the exact roles of MAPK
members in the high-sodium induced proliferation of VSMCs, specific
inhibitors of JNK and ERK1/2 were used in this study. As shown in
Fig. 5, both SP600125, a JNK
inhibitor (Fig. 5A), and PD98059,
an ERK1/2 inhibitor (Fig. 5B),
almost completely inhibited PCNA expression induced by the addition
of Na+ (30 mM; additional to the levels already in
medium). The JNK inhibitor, SP600125, also decreased the expression
of p-ERK1/2 which was induced by the addition of Na+ (30
mM; additional to the levels already in medium) (Fig. 5C). However, the ERK1/2 inhibitor,
PD98059, did not significantly affect the phosphorylation level of
JNK (Fig. 5D). These results
indicated that high sodium levels induced the expression of PCNA
through JNK/ERK1/2 pathway, and that JNK was located upstream of
ERK1/2.
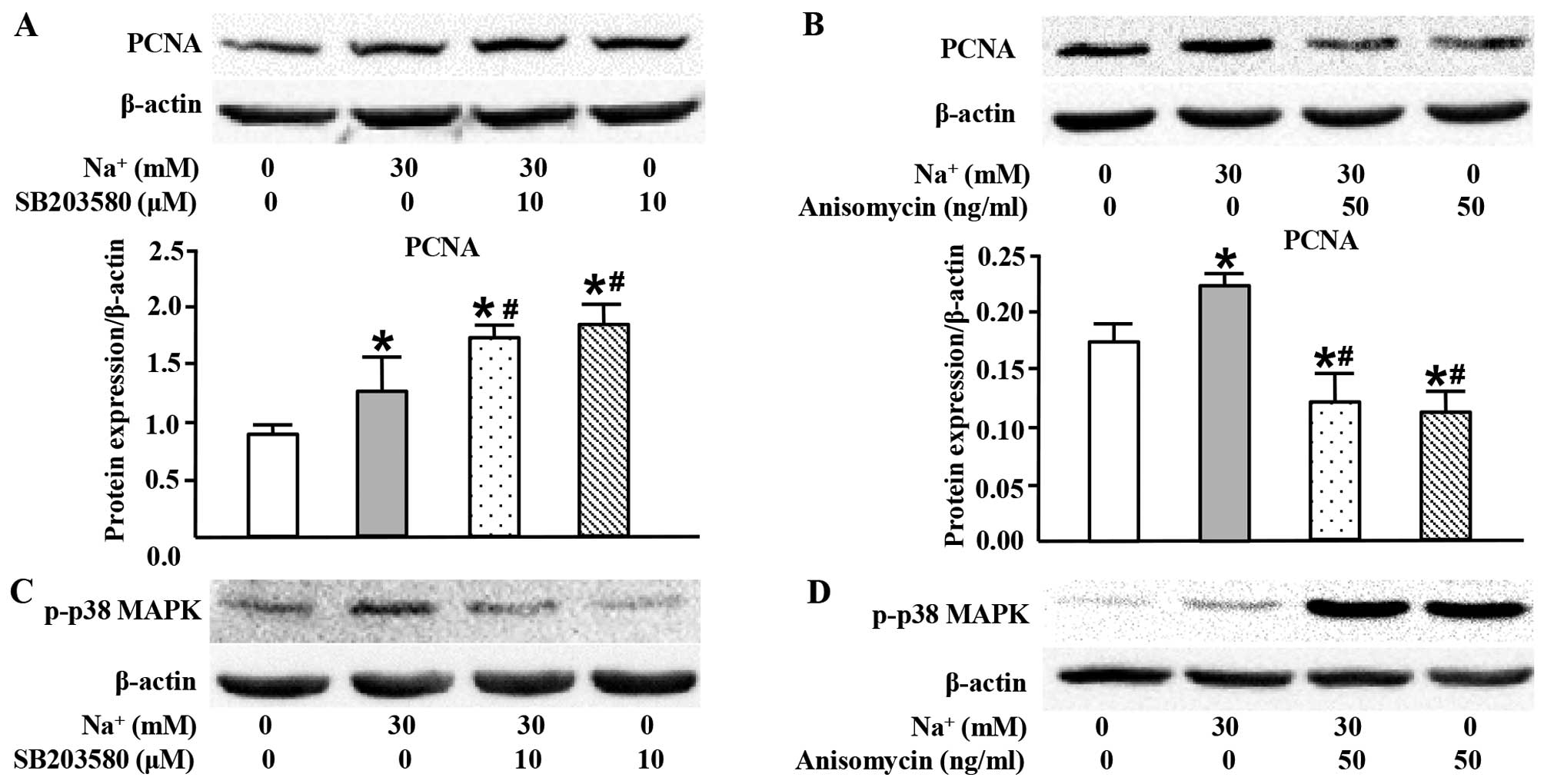
High sodium level simultaneously inhibits
PCNA expression through p38 MAPK
In the present study, we also used SB203580 (a p38
MAPK inhibitor) in order to examine the role of p38 MAPK in the
high-sodium induced proliferation of VSMCs. Surprisingly, SB203580
markedly increased PCNA expression at the protein level (Fig. 6A). Conversely, anisomycin, the
activator of p38 MAPK, inhibited PCNA expression (Fig. 6B). These results suggested that
activated p38 MAPK played an opposite role to JNK and ERK1/2 in the
high-sodium induced proliferation of VSMCs.
Discussion
As the major risk factor for cardiovascular diseases
(1–4,31),
increasing attention has been paid to the physiological and
pathophysiological effects of excess Na+ consumption.
The present study demonstrated two major findings: i) Additional
Na+ per se increased the proliferation of VSMCs weakly
(10%), but directly at relatively higher concentrations; ii) the
addition of Na+ activated the JNK/ERK1/2/PCNA pathway to
promote the proliferation of VSMCs, and this abnormal proliferative
effect was limited by simultaneously activated p38 MAPK.
In the present study, we mainly focused on the
direct effects of additional NaCl on the proliferation of VSMCs to
avoid the confounding impact from other factors, as shown in in
vivo studies, such as RAAS (20,21), endothelial function (22) and ouabain (32). We demonstrated that additional
Na+ itself directly increased the proliferation of VSMCs
at a concentration ranging from 20 to 40 mM (Figs. 1Figure 2–3). Since the basic culture medium of
VSMCs is DMEM, in which the Na+ concentration is
approximately 156 mM, the final Na+ concentrations thus
ranged from 176 to 196 mM in the present study. This level of
Na+ concentration could appear both in the renal medulla
under physiological conditions (5) and in the skin interstitium due to
HSD (10). Although
Na+ increased the proliferation of VSMCs by only
approximately 10% compared with the control group, and this effect
is not as prominent as that of other factors, such as angiotensin
II (20), its proliferative
effects were still significant since i) the proliferation of the
VSMCs was maintained at a low level when the cells were exposed to
relatively higher concentrations of Na+. This
interesting response of VSMCs to high concentrations of
Na+ may indicate a possible mechanism which plays a role
in the adaptation of tissues exposed to fluctuant concentrations of
Na+ in the renal medulla; ii) aside from the increase in
blood pressure, the activation of RAAS and endothelial dysfunction,
the increase in the proliferation of the VSMCs by 10% induced by
the interstitial Na+ accumulation itself can still be
considered as an important pathogenic factor for hypertension,
cardiovascular disease and stroke; iii) as previously described by
Singer et al (16),
medication against RAAS and other possible treatments cannot solve
all the health issues and sodium restriction itself is a simple,
but significant prevention or even therapeutic approach.
As an extracellular stimuli, it has been proven that
NaCl can affect MAPKs in several cell types, such as endothelial
cells (33), monocytes (34) and HEK293 cells (35). For cell cycle progression, it is
considered that the activation of JNK and ERK1/2 can accelerate
cell proliferation (36,37), whereas p38 MAPK plays an opposite
role (38–40). In the present study,
Na+ increased the phosphorylation of JNK (Fig. 4A) and ERK1/2 (Fig. 4C) and increased PCNA expression
(Fig. 5), and these results were
consistent with those of the above-mentioned studies. Moreover, p38
MAPK was phosphorylated (Fig. 4E)
at the same time, but played an opposite role (Fig. 6). Since Na+ is an
essential nutrient for normal cell function, the findings of our
study indicate that high sodium levels may simultaneously activate
p38 MAPK, a negative regulatory mechanism, in order to limit
abnormal proliferation, maintaining cell proliferation at a
relatively low level.
There were several limitations to the present study
as follows: i) in spite of the abundance of existing connective
tissue, to the best of our knowledge, no studies to date have
demonstrated the exact amount of Na+ which is
accumulated in the vascular wall. Therefore, it is necessary to
develop a novel, but feasible method with which to determine the
accumulation of Na+ and measure its exact concentrations
in the vascular walls due to HSD; ii) the detailed mechanisms and
the cross- talk between JNK, ERK1/2 and p38 MAPK warrants further
investigations.
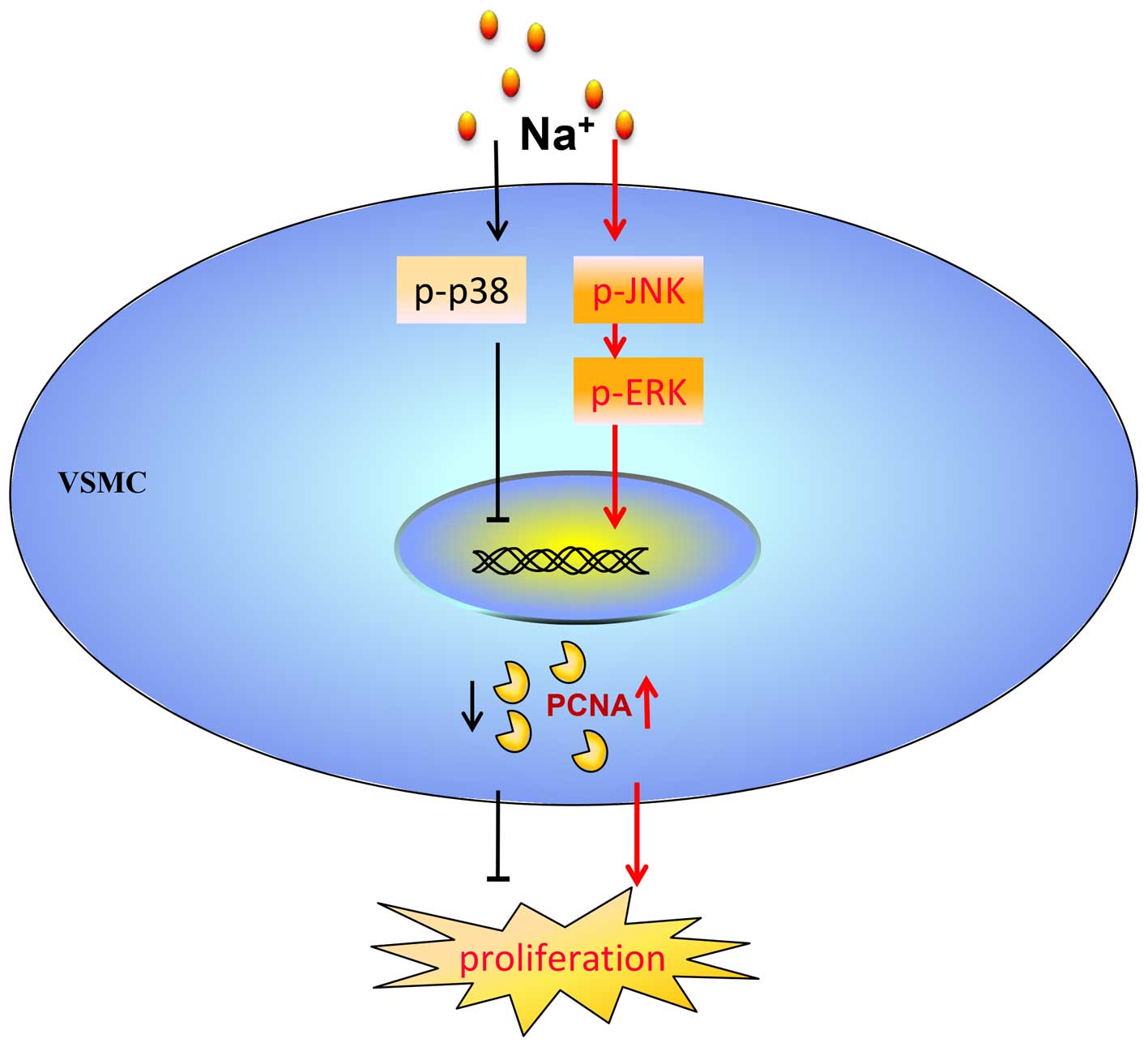
In conclusion, the findings of this study
demonstrated that additional Na+ per se directly
promoted the proliferation of VSMCs through the JNK/ERK1/2/PCNA
pathway. At the same time, the proliferation of the VSMCs which was
induced by additional Na+ was maintained at a low level
via the activation of p38 MAPK (Fig.
7).
Acknowledgments
This study was financially supported by the National
Natural Science Fund (nos. 91339116 and 81102843) and the National
Basic Research Program of China ('973 Project' no.
2012CB517804).
References
|
1
|
Aburto NJ, Ziolkovska A, Hooper L, Elliott
P, Cappuccio FP and Meerpohl JJ: Effect of lower sodium intake on
health: Systematic review and meta-analyses. BMJ. 346:f13262013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Graudal NA, Hubeck-Graudal T and Jürgens
G: Effects of low-sodium diet vs. high-sodium diet on blood
pressure, renin, aldosterone, catecholamines, cholesterol, and
triglyceride (Cochrane Review). Am J Hypertens. 25:1–15. 2012.
View Article : Google Scholar
|
|
3
|
He FJ, Li J and Macgregor GA: Effect of
longer term modest salt reduction on blood pressure: Cochrane
systematic review and meta-analysis of randomised trials. BMJ.
346:f13252013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Taylor RS, Ashton KE, Moxham T, Hooper L
and Ebrahim S: Reduced dietary salt for the prevention of
cardiovascular disease: A meta-analysis of randomized controlled
trials (Cochrane review). Am J Hypertens. 24:843–853. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Neuhofer W and Beck FX: Cell survival in
the hostile environment of the renal medulla. Annu Rev Physiol.
67:531–555. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cha JH, Woo SK, Han KH, Kim YH, Handler
JS, Kim J and Kwon HM: Hydration status affects nuclear
distribution of transcription factor tonicity responsive enhancer
binding protein in rat kidney. J Am Soc Nephrol. 12:2221–2230.
2001.PubMed/NCBI
|
|
7
|
Kültz D and Chakravarty D: Hyperosmolality
in the form of elevated NaCl but not urea causes DNA damage in
murine kidney cells. Proc Natl Acad Sci USA. 98:1999–2004. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Z, Dmitrieva NI, Park JH, Levine RL
and Burg MB: High urea and NaCl carbonylate proteins in renal cells
in culture and in vivo, and high urea causes 8-oxoguanine lesions
in their DNA. Proc Natl Acad Sci USA. 101:9491–9496. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Santos BC, Chevaile A, Hébert MJ,
Zagajeski J and Gullans SR: A combination of NaCl and urea enhances
survival of IMCD cells to hyperosmolality. Am J Physiol.
274:F1167–F1173. 1998.PubMed/NCBI
|
|
10
|
Machnik A, Neuhofer W, Jantsch J, Dahlmann
A, Tammela T, Machura K, Park JK, Beck FX, Müller DN, Derer W, et
al: Macrophages regulate salt-dependent volume and blood pressure
by a vascular endothelial growth factor-C-dependent buffering
mechanism. Nat Med. 15:545–552. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nijst P, Verbrugge FH, Grieten L, Dupont
M, Steels P, Tang WH and Mullens W: The pathophysiological role of
interstitial sodium in heart failure. J Am Coll Cardiol.
65:378–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Heer M, Baisch F, Kropp J, Gerzer R and
Drummer C: High dietary sodium chloride consumption may not induce
body fluid retention in humans. Am J Physiol Renal Physiol.
278:F585–F595. 2000.PubMed/NCBI
|
|
13
|
Kirkendall AM, Connor WE, Abboud F,
Rastogi SP, Anderson TA and Fry M: The effect of dietary sodium
chloride on blood pressure, body fluids, electrolytes, renal
function, and serum lipids of normotensive man. J Lab Clin Med.
87:411–434. 1976.PubMed/NCBI
|
|
14
|
Palacios C, Wigertz K, Martin BR, Jackman
L, Pratt JH, Peacock M, McCabe G and Weaver CM: Sodium retention in
black and white female adolescents in response to salt intake. J
Clin Endocrinol Metab. 89:1858–1863. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Titze J, Shakibaei M, Schafflhuber M,
Schulze-Tanzil G, Porst M, Schwind KH, Dietsch P and Hilgers KF:
Glycosaminoglycan polymerization may enable osmotically inactive
Na+ storage in the skin. Am J Physiol Heart Circ
Physiol. 287:H203–H208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Singer DR, Markandu ND, Sugden AL, Miller
MA and MacGregor GA: Sodium restriction in hypertensive patients
treated with a converting enzyme inhibitor and a thiazide.
Hypertension. 17:798–803. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matsushita N, Kitazato KT, Tada Y,
Sumiyoshi M, Shimada K, Yagi K, Kanematsu Y, Satomi J and Nagahiro
S: Increase in body Na+/water ratio is associated with
cerebral aneurysm formation in oophorectomized rats. Hypertension.
60:1309–1315. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lacolley P, Regnault V, Nicoletti A, Li Z
and Michel JB: The vascular smooth muscle cell in arterial
pathology: A cell that can take on multiple roles. Cardiovasc Res.
95:194–204. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shi N and Chen SY: Mechanisms
simultaneously regulate smooth muscle proliferation and
differentiation. J Biomed Res. 28:40–46. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu G, Hitomi H, Rahman A, Nakano D, Mori
H, Masaki T, Ma H, Iwamoto T, Kobori H and Nishiyama A: High sodium
augments angiotensin II-induced vascular smooth muscle cell
proliferation through the ERK1/2-dependent pathway. Hypertens Res.
37:13–18. 2014. View Article : Google Scholar :
|
|
21
|
Makita S, Nakamura M, Yoshida H and
Hiramori K: Effect of angiotensin II receptor blocker on
angiotensin II stimulated DNA synthesis of cultured human aortic
smooth muscle cells. Life Sci. 56:PL383–PL388. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Komuro I, Kurihara H, Sugiyama T,
Yoshizumi M, Takaku F and Yazaki Y: Endothelin stimulates c-fos and
c-myc expression and proliferation of vascular smooth muscle cells.
FEBS Lett. 238:249–252. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu J, Ren Y, Kang L and Zhang L: Oxidized
low-density lipoprotein increases the proliferation and migration
of human coronary artery smooth muscle cells through the
upregulation of osteopontin. Int J Mol Med. 33:1341–1347.
2014.PubMed/NCBI
|
|
24
|
Wang H, Liu Y, Zhu L, Wang W, Wan Z, Chen
F, Wu Y, Zhou J and Yuan Z: 17β-estradiol promotes cholesterol
efflux from vascular smooth muscle cells through a liver X receptor
α-dependent pathway. Int J Mol Med. 33:550–558. 2014.PubMed/NCBI
|
|
25
|
Weissberg PL, Cary NR and Shanahan CM:
Gene expression and vascular smooth muscle cell phenotype. Blood
Press Suppl. 2:68–73. 1995.PubMed/NCBI
|
|
26
|
Salic A and Mitchison TJ: A chemical
method for fast and sensitive detection of DNA synthesis in vivo.
Proc Natl Acad Sci USA. 105:2415–2420. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guzińska-Ustymowicz K, Pryczynicz A,
Kemona A and Czyzewska J: Correlation between proliferation
markers: PCNA, Ki-67, MCM-2 and antiapoptotic protein Bcl-2 in
colorectal cancer. Anticancer Res. 29:3049–3052. 2009.
|
|
28
|
Liu D, Huang Y, Bu D, Liu AD, Holmberg L,
Jia Y, Tang C, Du J and Jin H: Sulfur dioxide inhibits vascular
smooth muscle cell proliferation via suppressing the Erk/MAP kinase
pathway mediated by cAMP/PKA signaling. Cell Death Dis.
5:e12512014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boutros T, Chevet E and Metrakos P:
Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase
regulation: Roles in cell growth, death, and cancer. Pharmacol Rev.
60:261–310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Powles J, Fahimi S, Micha R, Khatibzadeh
S, Shi P, Ezzati M, Engell RE, Lim SS, Danaei G, Mozaffarian D, et
al Global Burden of Diseases Nutrition and Chronic Diseases Expert
Group (NutriCoDE): Global, regional and national sodium intakes in
1990 and 2010: A systematic analysis of 24 h urinary sodium
excretion and dietary surveys worldwide. BMJ Open. 3:e0037332013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Blaustein MP, Leenen FHH, Chen L, Golovina
VA, Hamlyn JM, Pallone TL, Van Huysse JW, Zhang J and Wier WG: How
NaCl raises blood pressure: A new paradigm for the pathogenesis of
salt-dependent hypertension. Am J Physiol Heart Circ Physiol.
302:H1031–H1049. 2012. View Article : Google Scholar :
|
|
33
|
Duzgun SA, Rasque H, Kito H, Azuma N, Li
W, Basson MD, Gahtan V, Dudrick SJ and Sumpio BE: Mitogen-activated
protein phosphorylation in endothelial cells exposed to
hyperosmolar conditions. J Cell Biochem. 76:567–571. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kleinewietfeld M, Manzel A, Titze J,
Kvakan H, Yosef N, Linker RA, Muller DN and Hafler DA: Sodium
chloride drives autoimmune disease by the induction of pathogenic
TH17 cells. Nature. 496:518–522. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou X, Ferraris JD, Dmitrieva NI, Liu Y
and Burg MB: MKP-1 inhibits high NaCl-induced activation of p38 but
does not inhibit the activation of TonEBP/OREBP: Opposite roles of
p38alpha and p38delta. Proc Natl Acad Sci USA. 105:5620–5625. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jaeschke A, Karasarides M, Ventura JJ,
Ehrhardt A, Zhang C, Flavell RA, Shokat KM and Davis RJ: JNK2 is a
positive regulator of the cJun transcription factor. Mol Cell.
23:899–911. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Meloche S and Pouysségur J: The ERK1/2
mitogen-activated protein kinase pathway as a master regulator of
the G1- to S-phase transition. Oncogene. 26:3227–3239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cuadrado A and Nebreda AR: Mechanisms and
functions of p38 MAPK signalling. Biochem J. 429:403–417. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mikhailov A, Shinohara M and Rieder CL:
The p38-mediated stress-activated checkpoint. A rapid response
system for delaying progression through antephase and entry into
mitosis. Cell Cycle. 4:57–62. 2005. View Article : Google Scholar
|
|
40
|
Perdiguero E, Ruiz-Bonilla V, Serrano AL
and Muñoz-Cánoves P: Genetic deficiency of p38alpha reveals its
critical role in myoblast cell cycle exit: The p38alpha-JNK
connection. Cell Cycle. 6:1298–1303. 2007. View Article : Google Scholar : PubMed/NCBI
|