Introduction
Stroke, also known as cerebrovascular disease, is
the second leading cause of mortality, accounting for hundreds of
thousands of mortalities annually worldwide, and is a major cause
of disability in adults (1).
Ischemic stroke due to the lack of blood flow is more prevalent
than hemorrhagic stroke due to bleeding. Within the ischemic core
of the brain, where blood flow is most severely restricted,
excitotoxic and necrotic cell death occurs within minutes. In cases
of prolonged ischemia, hypoxanthine is formed as a product of
adenosine triphosphate (ATP) metabolism (2). However, rapid revascularization of
the occluded vessels and the restoration of blood flow in cases of
acute ischemic stroke may cause ischemia/reperfusion (I/R) injury
(3). An imbalance in the energy
supply and demand within the ischemic brain tissue results in
surrounding tissue hypoxia and dysfunction. Subsequent reperfusion
further enhances the activation of innate and adaptive immune
responses and cell death programs (4).
The classic forms of cell death include necrosis,
apoptosis and autophagy (6)
Autophagy, or cellular self-digestion, is an evolutionarily highly
conserved catabolic process that is important for balancing sources
of energy at critical time points in development and in response to
nutrient stress, and has been proposed as the third type of cell
death (5–7). Autophagy is constitutively active in
the central nervous system (CNS), and protects against neuronal
injury and neurodegeneration following ischemic stroke (8,9).
In early cerebral ischemia, autophagy can degrade damaged
organelles, eliminate I/R-induced damaged components and toxic
metabolites or provide the nutrient source required to maintain
metabolism, ATP levels, cellular homeostasis and survival (10–12).
Macroautophagy is the main pathway involved in
autophagy, and it is used mainly to eradicate damaged cell
organelles or unused proteins (13). In canonical macroautophagy, a
small part of the cytoplasm is sequestered by a membrane sac, the
so-called isolation membrane (also termed the phagophore), which
results in the formation of a double-membrane structure, the
autophagosome. The autophagosome matures as it fuses with endosomes
and then finally fuses with lysosomes. Following fusion, the inner
membrane and enclosed cytoplasmic materials are degraded by
lysosomal enzymes (14). In the
canonical ischemia-induced pathway, autophagosome formation is
regulated by autophagy-related gene (Atg) 8 [also known as light
chain 3 (LC3)-phosphatidylethanolamine conjugate (LC3-II)], and the
mammalian target of rapamycin (mTOR) complex 1 (mTORC1) signaling
pathway (15–18). Under nutrient-rich conditions, the
upregulation of mTORC1 leads to the inactivation of the
Atg1/Unc-51-like kinase (ULK) complex that consists of ULK1, ULK2,
Atg13 and FIP200 in mammals (19). Under starvation conditions, the
ULK complex is involved in the initial step of autophagosome
formation, which is activated by inactivated mTORC1 (20,21). The ULK complex promotes autophagy
by targeting the autophagosome formation-specific class III
phosphoinositide 3-kinase (PI3K) complex which consists of class
III PI3K, Atg14L (also known as Atg14 and Barkor), Beclin1 (Atg6
homolog), hVps15 and hVps34 (22,23).
However, it has been previously noted that the
excessive activation of autophagy may promote cell autophagic death
(24). The overactivation of
autophagy causes neuronal cells to self-digest their own necessary
components or interact with the apoptotic cascade, thus promoting
nerve cell death in the ischemic surrounding zones in cerebral
ischemia (25–27). We, as well as others have
demonstrated that electroacupuncture (EA) at the LI11 or ST36
acupoints activats the class I PI3K/Akt signaling pathway (28–30). However, activated Akt leads to the
activation of downstream mTORC1 in the modulation of autophagy
(31,32). Therefore, we concluded that EA at
the LI11 and ST36 acupoints induced the expression of mTORC1 in
ischemic stroke. Thereby, a study hypothesis was proposed that EA
protects against ischemic stroke through the mTORC1-ULK1
complex-Beclin1 pathway-mediated regulation of autophagosome
formation and autophagy in the peri-infarct cortex following
cerebral middle cerebral artery occlusion and reperfusion (MCAO/R)
injury.
Materials and methods
Experimental animals and groups
Healthy male Sprague-Dawley (SD) rats, weighting
280±20 g, were provided by Shanghai Laboratory Animal Co., Ltd.,
(SLAC) Shanghai, China (license no. SCXK 2014-0005) and housed
under pathogen-free conditions with a 12-h light/dark cycle, at
23±2°C and 60–70% humidity. The rats were allowed free access to
food and water. All experimental protocols were approved by the
Committee on Animal Care and Usage of Fujian University of
Traditional Chinese Medicine (FUTCM), and all the principles in the
Chinese Specifications for the Care and Use of the Laboratory
Animals (SPF animal laboratory) were taken into account. The
animals were subjected to adaptive feeding at the Laboratory Animal
Center of FUTCM [license no. SYXK(min) 2014-007]. All efforts were
made to minimize animal suffering.
According to the random number table method, 45 SD
rats were randomly and evenly divided into 3 groups (n=15) as
follows: i) the sham-operated control group (sham group); ii) the
MCAO/R control group; and iii) the MCAO/R + EA treatment group
(MCAO/R + EA group).
Establishment of the rat model of MCAO/R
injury
The rat model of MCAO/R injury was established using
the suture-occluded method. The detailed procedure has previously
been described in the study by Longa et al (33). Briefly, the rats were anesthetized
with 10% chloral hydrate (300 mg/kg body weight) by intraperitoneal
injection. MCAO on the left side was performed using an occluding
suture with an embolus (Jia Ling embolus; Jia Ling Biological
Technology Ltd., Guangzhou, China) for 2 h, and after 2 h of
MCAO-induced cerebral ischemia, the suture was slowly withdrawn to
allow for reperfusion. The ipsilateral cerebral blood flow (CBF)
was measured by Laser Doppler Flowmetry (Biopac Systems, Goleta,
CA, USA). The MCAO model was considered successful (inclusion) only
when CBF dropped to become equal or >80% of the baseline during
occlusion. The rectal temperature of the rats was maintained at
37°C throughout the surgical procedures. In the sham group, the
artery was isolated, but no embolus was introduced. After surgery
and recovery, the animals were placed in an environment at room
temperature, and they resumed a normal diet.
Assessment of neurological deficit
scores
Neurological function was assessed in each rat 2 h
after MCAO/R injury and 3 days after EA treatment in a blinded
manner using a previously described method (33). Detailed scores are as follows:
score 0, no neurological deficit; score 1, the tail was raised and
adduction (not able to fully extend) of the right forelimb was
observed; score 2, spontaneous circling to the right when walking;
score 3, the body was slanted to the right when walking; score 4,
not able to walk spontaneously along with possible loss of
consciousness. Scores of 0 and 4 indicated modeling failure, and
rats with these scores were excluded.
Treatment with EA
The rats in the MCAO/R + EA group received EA
treatment. Stainless acupuncture needles (0.3 mm in diameter,
Huatuo acupuncture needle; Suzhou Medical Appliance Factory,
Suzhou, China) were inserted 2–3 mm deep into the Quchi (LI11,
located in the depression lateral to the anterior aspect of the
radius joint of the forelimb) and Zusanli (ST36, located 5 mm below
the knee joint of the hind-limb and 2 mm lateral to the anterior
tubercle of the tibia) acupoints on the right paralyzed limb.
Stimulation was then generated by an electroacupuncture instrument
[model G6805; Shanghai Marine Instrument General Factory (SMIF),
Shanghai, China], and the stimulation parameters were set as dense
disperse waves of 1–20 Hz (adjusted to the muscle twitch
threshold); peak voltage of 6V, 0.2 mA intensity for 30 min/day,
once a day. EA treatment was administered 24 h after MCAO/R and
continued until the animals were sacrificed with 10% chloral
hydrate (Fujian Academy of Integrative Medicine, FUTCM) anesthesia
after EA treatment, which ran consecutively for 3 days.
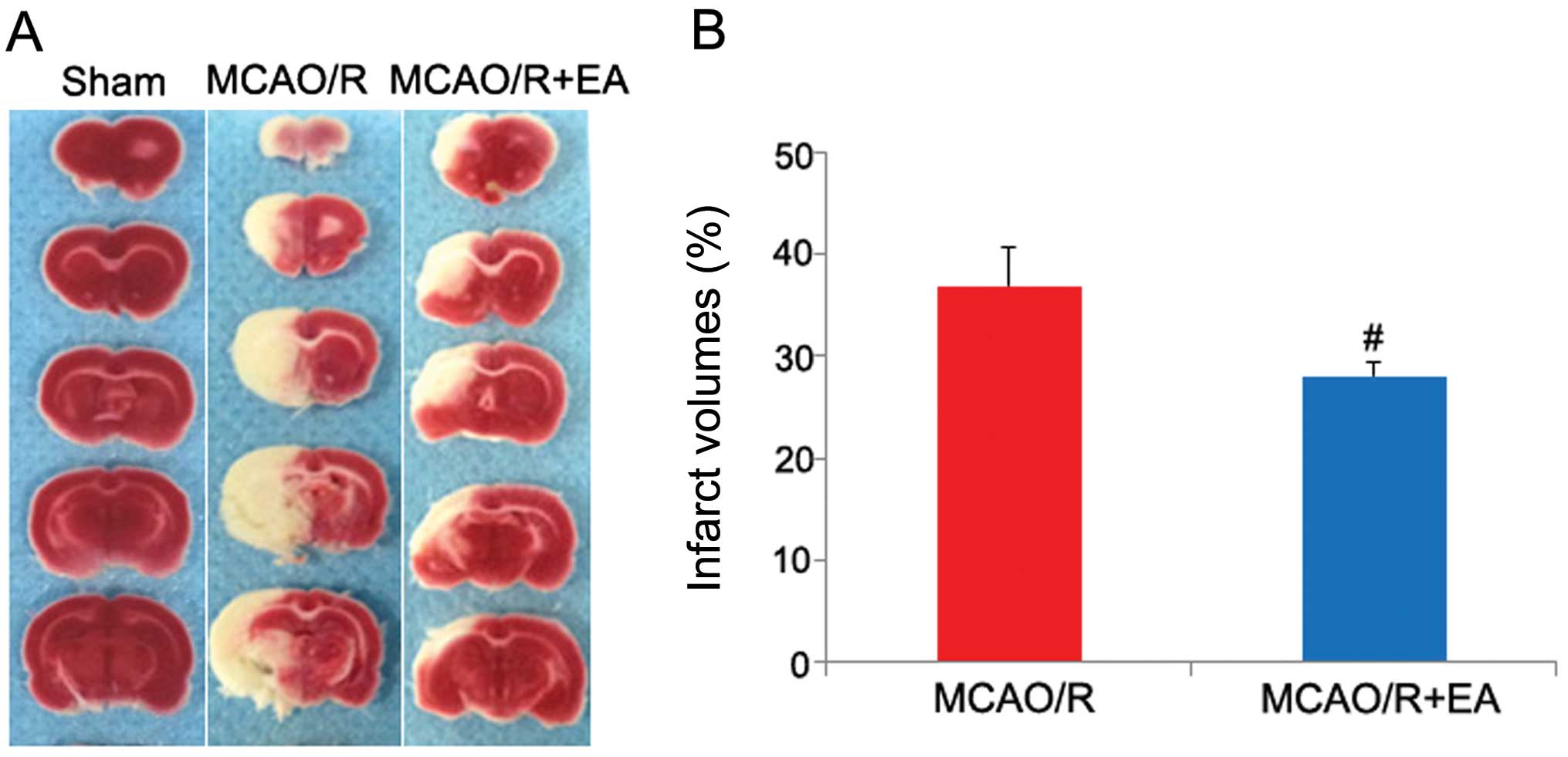
Measurement of the infarct volume
At the end of the neurological examination, the
brains were quickly removed to measure the infarct volume, as
previously described (34). The
rat brains were rapidly dissected after the were anesthetized using
10% chloral hydrate (Fujian Academy of Integrative Medicine,
FUTCM), fresh brain tissue was placed in a freezer at −80°C for 20
min and 2-mm coronal sections were cut. The fresh slices were
immediately immersed in 2% 2,3,5-triphenyltetrazolium chloride
(Sigma-Aldrich, St. Louis, MO, USA). The TTC-stained sections were
photographed using a high-resolution digital camera (Canon SX20),
and the infarct area was determined with computerized image
analysis software (Motic Med 6.0 system). The total infarct volume
was analyzed using 6 slices of 2-mm coronal sections from each
brain and calculated using the following formula: infarct volume
(%) = (infarct volume/total volume) ×100. The percentage brain
infarct volume was used for subsequent statistical analysis.
Computer-assisted method for gait
analysis
We used CatWalk (Noldus Information Technology,
Wageningen, The Netherlands), a quantitative and automated gait
analysis system, which measures numerous parameters of gait.
Previous studies using CatWalk have identified multiple
MCAO/R-sensitive gait parameters which can be utilized to assess
the gait and motor function in murine models of MCAO/R injury
(35–37). The 'CatWalk' apparatus is made of
a 1.3-m-long glass platform which is illuminated from the long edge
with green fluorescent lights which are completely internally
reflected. When the rat walks along the walkway with paws in
contact with the glass surface, the light is reflected downward and
the images of the paw prints are captured by a high-speed video
camera under the walkway. Images and data were analyzed using
CatWalk XT 10.0 software. The animals received 7 days of training 6
times each day to familiarize them with the walkway prior to the
operation, as previously described (38). There were 2 basic criteria: i)
rats had to run across the walkway without any interruption,
walking backwards or rearing during the run, and ii) at least 3
correct crossings per rat were required (39). All rats were subjected to gait
assessment following treatment with EA for 3 days using the CatWalk
automated gait analysis system.
Transmission electron microscopic
examination
Following treatment with EA for 3 days, the rats
were anesthetized and then perfused transcardially with pre-cooled
phosphate-buffered saline (PBS; pH 7.4) followed by PBS containing
4% paraformaldehyde and 0.25% glutaraldehyde. The following day,
the rat brains were sectioned with a vibratome (Leica Microsystems
Ltd., Milton Keynes, UK) into 500-µm-thick slices. The
parietal lobe cortex of the peri-infarct cortical tissue and the
corresponding area of the sham-operated rats were selected for
analysis, and selected areas were processed by post-fixation in 1%
osmium tetroxide and 1.5% potassium ferrocyanide for 2 h, and they
were then washed 3 times with PBS, dehydrated in graded ethanol and
then embedded in epoxy resin (E51-618). Polymerization was
performed at 37°C for 24 h. Blocks were sectioned using a PowerTome
PC ultramicrotome (RMC, Inc., Tucson, AZ, USA) into ultra-thin
sections (80 nm), which were post-stained with uranyl acetate and
lead citrate, and visualized using a Hitachi H-7650 electron
microscope (Hitachi, Tokyo, Japan) at 80 kV. For quantitative
analysis of the number of mitochondria, autophagosomes,
autolysosomes and lysosomes, 3 rats per group and 10 random fields
of vision for each rat were counted by a blinded observer.
Western blot analysis
Total proteins were extracted from the peri-infarct
cortical tissue supplied by the rats which had been subjected to
MCAO/R on the left side and the corresponding area of sham-operated
rats. Western blot analysis was carried out with 10–12% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Equal amounts of protein (25 µg) were loaded onto the gels
for western blot analysis. Following electrophoresis, proteins were
transferred onto polyvinylidene fluoride (PVDF) membranes (pore
size, 0.45 µm; Millipore, Billerica, MA, USA), which were
blocked with 5% skim milk for 2 h at room temperature. The mmbranes
were washed in TBS with 0.25% Tween-20 (TBST), then probed with
primary antibodies against LC3B (1:1,000 dilution; No. 3868, Cell
Signaling Technology, Danvers, MA, USA), Beclin1 (ser14) (1:2,000
dilution; ab183335), Atg13 (1:10,000 dilution; ab105392), ULK1
(1:10,000 dilution; ab128859) (all from Abcam, Cambridge, MA, USA),
mTORC1 (1:1,000 dilution; No. 2587, Cell Signaling Technology) and
β-actin (1:8,000 dilution; HC201-02, TransGen Biotech, Beijing,
China) at 4°C overnight, followed by incubation with the
appropriate HRP-conjugated secondary antibody (1:5,000 dilution;
No. 58203 and No. 7077, Cell Signaling Technology) for 2 h at room
temperature. The membranes were then washed again in TBST. The
antibody-bound protein bands were detected with an enhanced
chemiluminescence kit, and images were analyzed using a Bio-Image
Analysis System (Bio-Rad, Hercules, CA, USA). β-actin was used as a
loading control. The ratio of grayscale values of the target
protein to the control was used to measure and stored for
subsequent analysis.
Statistical analysis
The data are expressed as the means ± standard error
of mean (SEM) and analyzed using SPSS 18.0 software. Comparisons
between 2 groups were performed using the independent two-sample
t-test or Mann-Whitney U test, and comparisons between multiple
groups were performed with one-way ANOVA. A p-value <0.05 was
considered to indicate a statistically significant difference.
Results
Effects of EA on neurological deficits
and infarct volumes
The neuroprotective effects of EA at the LI11 and
ST36 acupoints were evaluated by neurological behavioral assessment
and measurement of the cerebral infarct volume. As shown in
Table I and Fig. 1, rats in the MCAO/R + EA group and
the MCAO/R group all exhibited manifestations of neurological
deficits and cerebral infarction, whereas the rats in the sham
group did not exhibit any signs of cerebral injury, which indicated
that the rat model of cerebral MCAO/R injury was successfully
established. The neurological behavioral scores between the rats in
the MCAO/R + EA group and those in the MCAO/R group at 2 h after
MCAO/R injury (prior to EA treatment) did not differ significantly
(Table I, p>0.05). However,
following treatment with EA for 3 days, the neurological behavioral
scores and cerebral infarct volumes were all reduced significantly
(Table I and Fig. 1; p<0.05). These results
demonstrated that EA protected the rat brains against cerebral
MCAO/R injury.
 | Table INeurological deficit scores. |
Table I
Neurological deficit scores.
| Group | n | 2 h after
MCAO/R | 3 days after EA
treatment |
|---|
| Sham | 15 | 0 | 0 |
| MCAO/R | 15 | 2.17±0.17 | 1.75±0.18 |
| MCAO/R + EA | 15 | 2.08±0.19 | 1.42±0.19a |
Effects of EA on motor function
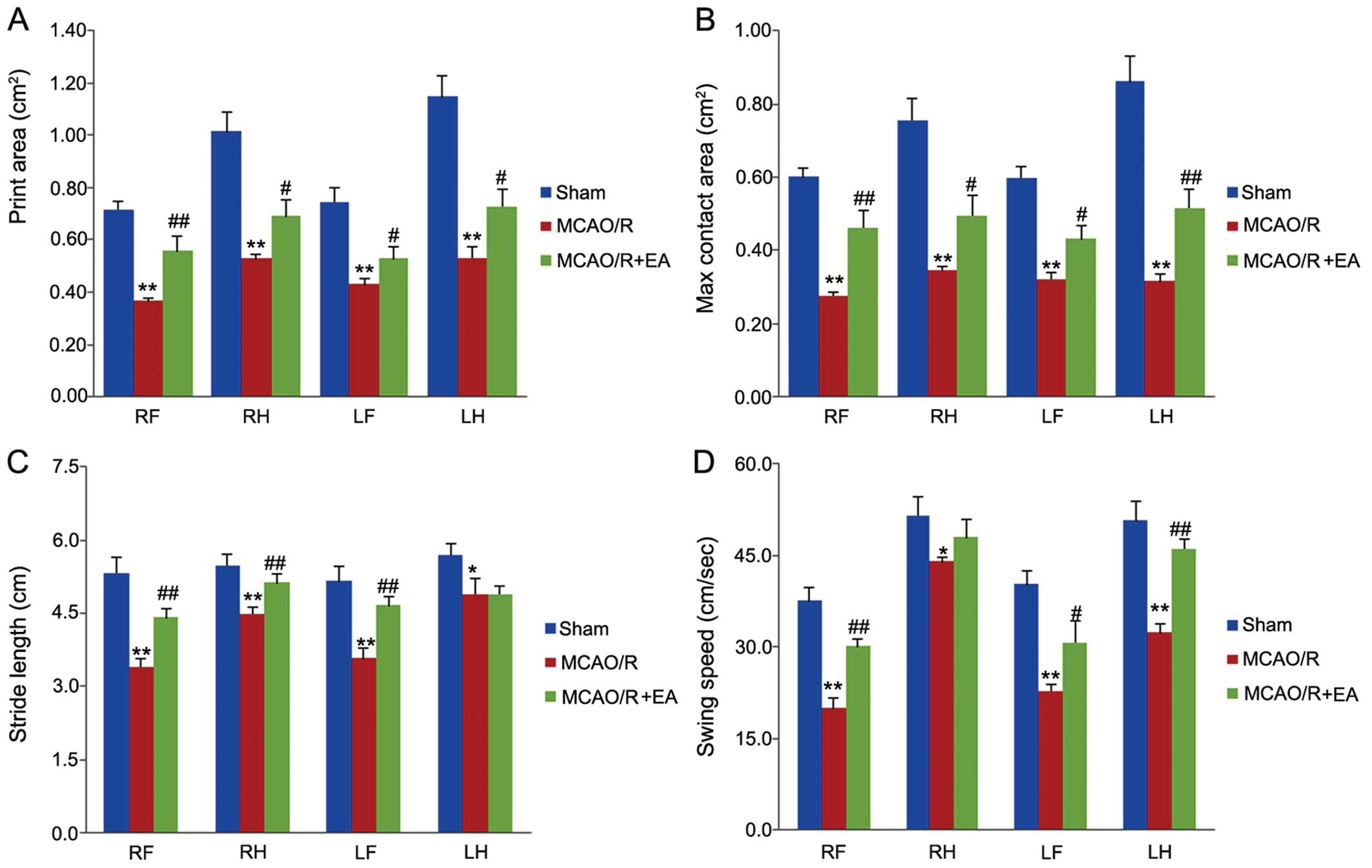
To examine the effects of EA treatment on motor
function, the CatWalk automated gait analysis system was used
following treatment with EA for 3 days. As shown in Fig. 2, following cerebral MCAO/R injury,
in the rats in the MCAO/R group, the print area, maximum contact
area, stride length and swing speed of their 4 limbs were all
decreased compared to those of the rats in the sham group
(p<0.05 or p<0.01). Following treatment with EA for 3 days,
in the rats in the MCAO/R + EA group, the print area and maximum
contact area of their 4 limbs, the stride length of their right
fore (RF), right hind (RH) and left fore (LF) limbs, and the swing
speed of their RF, LF and left hind (LH) limbs were increased
significantly, compared to the rats in the MCAO/R group (p<0.05
or p<0.01. However, the stride length of their LH limbs, and the
swing speed of their RH limbs in both groups were not altered
significantly (p>0.05). These results suggest that EA at the
LI11 and ST36 acupoints improves motor dysfunction in rats with
cerebral MCAO/R injury.
Effects of EA on the morphological
ultrastructural changes in the peri-infarct cortex
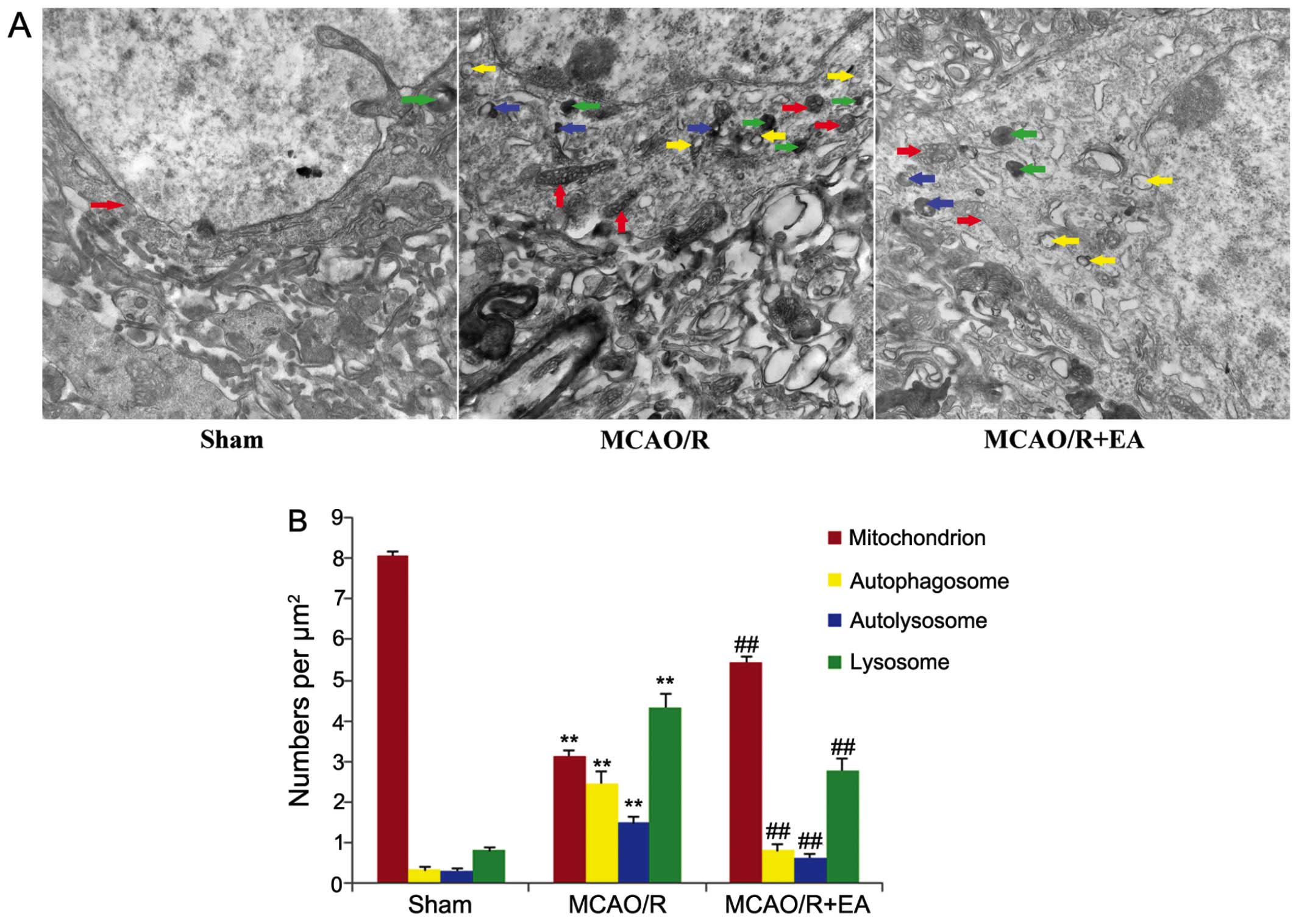
Transmission electron microscopy (TEM) is considered
the gold standard for the detection of autophagic structures
(40,41). In order to further examine the
effects of EA treatment on autophagy, we used TEM to observe the
morphological ultrastructural changes of peri-infarct cortical
neurons in each group. The neurons in the cerebral cortex of the
rats in the sham group appeared to be relatively healthy, with
normal polyribosomes, endoplasmic reticula, Golgi apparatus,
mitochondria, lysosomes, nuclei and fine granular chromatin evenly
distributed (Fig. 3A, sham
group). By contrast, the cortical neurons in the peri-infarct area
in the rats in the MCAO/R displayed morphological changes typical
of infarction: the neurons exhibited a dilated endoplasmic
reticulum, swollen and balloon-like mitochondria, mitochondrial
crests which had fused or disappeared, and the formation of
numerous vacuoles in the cytoplasm and numerous darkened lysosomes
and autophagosomes (C-shaped double-membrane structures) were also
observed (Fig. 3A, MCAO/R group).
The number of normal mitochondria was decreased in the rats in the
MCAO/R and MCAO/R + EA groups compared to those of the sham group,
whereas the numbers of autophagosomes, autolysosomes and lysosomes
were increased in both these groups compared to the sham group
(p<0.01; Fig. 3B). However,
following treatment with EA for 3 days, in the MCAO/R + EA group,
only mild injury to the neurons was visible. Nonetheless, a number
of normal organelles and nuclei were still observed. Chromatin was
distributed relatively evenly. The autophagosomes, autolysosomes
and lysosomes remained present in the cortical neurons in the
peri-infact are (Fig. 3A, MCAO/R
+ EA group). The number of normal mitochondria was increased,
whereas the numbers of autophagosomes, autolysosomes and lysosomes
were decreased in the rats in the MCAO/R + EA group compared to
those in the MCAO/R group (p<0.01; Fig. 3B).
Effects of EA on autophagosome membran
protein levels in the peri-infarct cortex
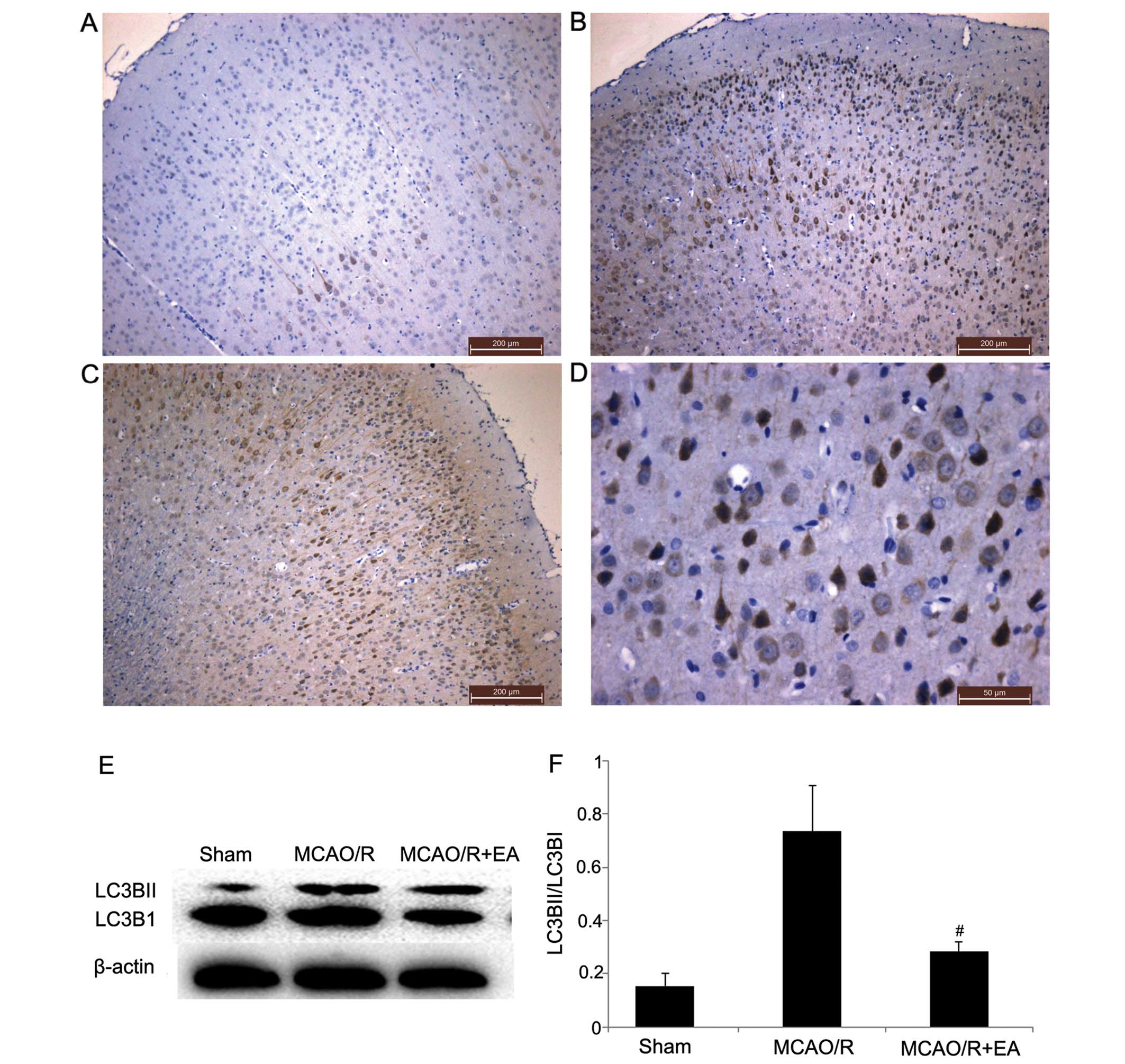
Microtubule-associated protein 1A/B LC3 is important
for the transport and maturation of autophagosomes. During
autophagosome formation, a cytosolic form of LC3 (LC3-I) is
conjugated to phosphatidylethanolamine to form LC3-II, which is
recruited to autophagosomal membranes. Therefore, LC3-II/LC3-I is
considered a biomarker of autophagosome formation and autophagy
(42,43). As shown in Fig. 4A–D, in the rats in each group,
there was a range of shapes of LC3B brown-yellow-positive cells
located in the cytoplasm, such as round-, oval and cone-shaped
cells, which were present in the cortical and/or cortical
peri-infarct area of granule cells and pyramidal cells. The level
of LC3BII/LC3BI was significantly increased in the MCAO/R group
compared to the sham group (p<0.01; Fig. 4E and F). Following treatment with
EA for 3 days, the level of LC3BII/LC3BI was significantly
decreased in the MCAO/R + EA group compared to the MCAO/R group
(P<0.05; Fig. 4E and F). The
abovementioned results suggest that EA at the LI11 and ST36
acupoints suppresses autophagosome formation and autophagy in
cortical neurons in the peri-infarct area.
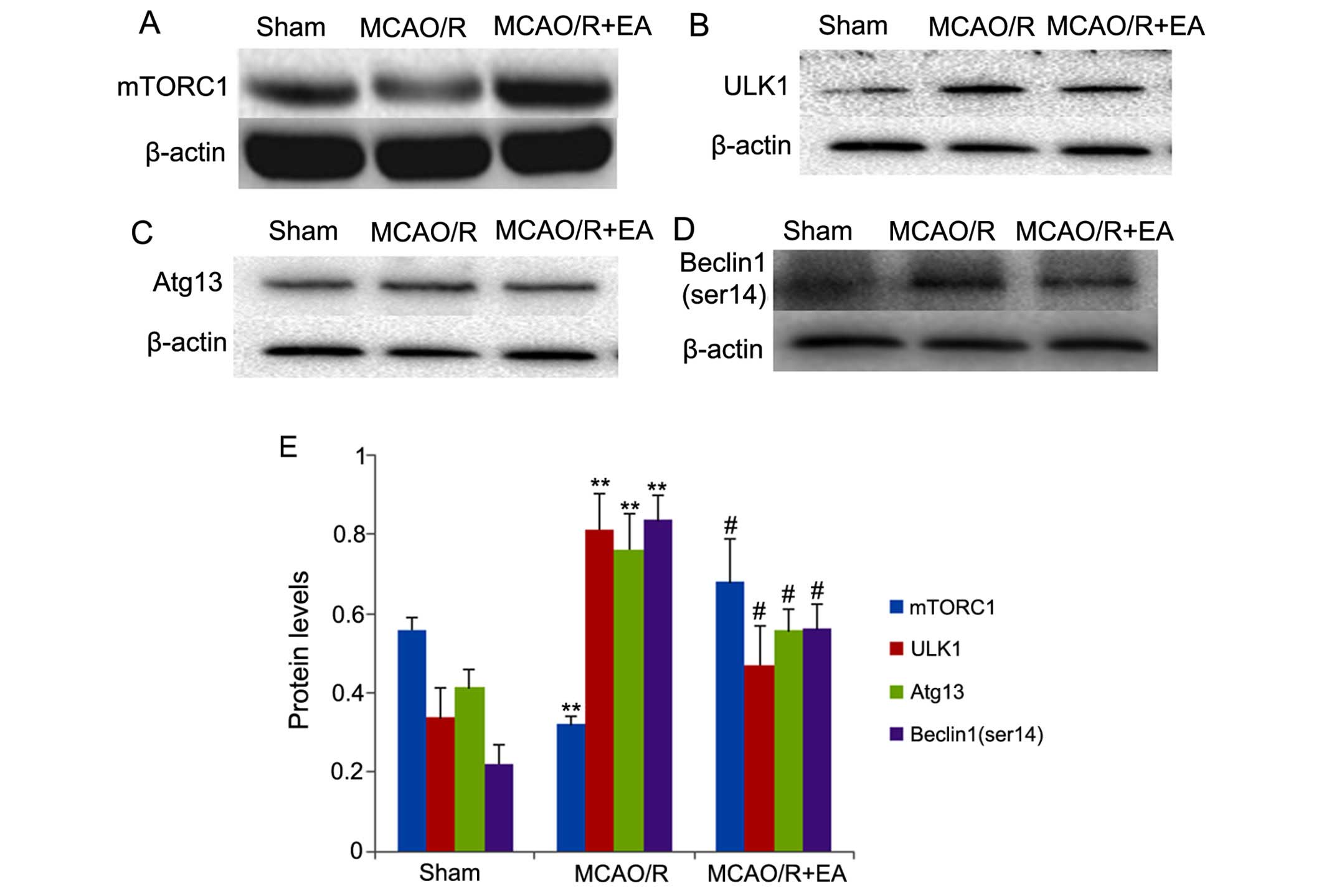
Effects of EA on the mTORC1-ULK1
complex-Beclin1 pathway
In order to examine the mechanisms involved in the
inhibition of autophagosome formation and autophagy by EA, we
examined the expression of mTORC1-ULK1 complex-Beclin1 signaling
pathway-related proteins. The results are shown in Fig. 5. In the rats in the MCAO/R group,
the expression of mTORC1 was significantly decreased compared with
that in the rats in the sham group (p<0.01; Fig. 5), whereas the levels of ULK1,
Atg13 and Beclin1 (ser14) were significantly increased compared
with those in the rats in the sham group (p<0.01; Fig. 5B–E). However, in the MCAO/R + EA
group, treatment with EA significantly increased the expression of
mTORC1 compared with that in the rats in the MCAO/R group
(p<0.05; Fig. 5A and E),
whereas treatment with EA significantly decreased the levels of
ULK1, Atg13 and Beclin1 (ser14) compared with those in the MCAO/R
group (p<0.01; Fig. 5B–E). Our
results indicated that EA at the LI11 and ST36 acupoints markedly
increased the expression of mTORC1, leading to the inactivation of
the ULK complex and inhibiting the phosphorylation of Beclin1
(ser14) in the peri-infarct cortex following MCAO/R injury.
Therefore, the abovementioned results support the
hypothesis that EA at the LI11 and ST36 acupoints protects against
the harmful effects of ischemic stroke through inhibition of
autophagosome formation and autophagy, and these effects are
mediated through the mTORC1-ULK complex-Beclin1 pathway.
Discussion
Thrombosis, embolisms and systemic hypoperfusion can
all cause an ischemic stroke. EA, electric stimulation to the
acupoints through acupuncture needles, is one of the safest and
most effective therapies for ischemic stroke patients, and is a
novel therapy based on acupuncture of traditional Chinese medicine
(TCM) combined with modern electrotherapy (44–47). EA has served as a complementary
therapy and has been noted to improve motor functional outcomes in
cases of ischemic stroke (45,48). While reviewing ancient Chinese
documents regarding acupuncture and motor impairment, we discovered
the LI11 and ST36 were the most frequently selected acupoints in
motor rehabilitation treatment in China (49). Animal experimental studies have
demonstrated that treatment with EA exerts neuroprotective effects
following ischemic stroke (50,51). Consistent with the results of
these studies, the results of the present study demonstrated that
treatment with EA at the LI11 and ST36 acupoints for 3 consecutive
days in rats with MCAO effectively reduced the cerebral infarct
volumes and improved neurological deficits. The CatWalk automated
gait analysis system has previously been noted to be a sensitive
tool for the evaluation of motor function in rat models of ischemic
stroke (52,53). Significant brain damage has
previously been observed in regions controlling the contralateral
limbs, leading to a reduction in the paw print area, and intensity
and maximum contact area by day 4 post-injury (54). In the present study, using the
CatWalk automated gait analysis system, we found that treatment
with EA improved the print area and maximum contact area of their
four limbs, the stride length of their RF, RH and LF limbs, and the
swing speed of their RF, LF and LH limbs at the 4 days after MCAO/R
injury, and these results were accompanied by improved neurological
deficit outcomes and infarct volumes.
Previous studies have demonstrated that autophagy
plays an important role in focal cerebral ischemia (55,56). Published data have shown that
cerebral ischemia induces autophagosome formation and autophagy
through the activation of the autophagy-related proteins, LC3 and
Beclin1 (57,58). Moreover, autophagy has emerged as
a potential therapeutic target for the treatment of stroke
(59). In a previous study,
3-methyladenine (3-MA) treatment significantly reduced LC3-II
conversion and autophagosome formation, which attenuates secondary
thalamic damage after focal cerebral infarction (57). Pre-treatment with EA has been
shown to promote tolerance against cerebral ischemic injury through
the inhibition of LC3-II and Beclin1 expression (60). In the present study, we
demonstrated that treatment with EA reduced the number of
autophagosomes, autolysosomes and lysosomes, and simultaneously
decreased the ratio of LC3BII/LC3BI in the peri-infarct cortex.
These results indicate that EA treatment suppresses autophagy
following ischemic stroke, which is associated with recovery
following brain damage.
In the present study, we further investigated the
possible mechanisms involved in the inhibition of autophagy.
Increasing evidence suggests that mTOR signaling plays an important
role in autophagosome formation and autophagy (19–22). By interacting with other partners,
the mTOR kinase forms two functionally distinct complexes, termed
mTORC1 and mTORC2. mTORC1 is a central regulator of autophagosome
formation and autophagy (61). A
number of upstream signaling molecules, including PI3K/Akt,
converge on the mTORC1 complex (62). However, inactivated mTORC1 induces
autophagy through the coordinated phosphorylation of the ULK
complex (ULK1, ULK2 and Atg13) at specific sites (63). Furthermore, previous research has
demonstrated that the ULK complex induces autophagy by
phosphorylating Beclin1 (26).
Importantly, Beclin1 interacts with phosphatidylinositol 3-kinase,
class III (PI3KC3), forming Beclin1-PI3KC3 complexes which consist
of the mammalian orthologues of class III PI3K, Atg14, Beclin1,
hVps15 and hVps34, which are key signaling complexes required for
autophagosome formation (64). A
marked elevation in Beclin1 levels in the ischemic penumbra of rats
following cerebral ischemia has previously been noted 65). The
upregulation of Beclin1 together with the increased number of
autophagosomes in the ipsilateral hemisphere is likely a response
to MCAO reperfusion (66). It has
previously been suggested that Beclin1 is important for autophagy
in the area surrounding the ischemic core and may be a target for
repairing ischemic injury following stroke (67). The results of the present study
demonstrated that treatment with EA at the LI11 and ST36 acupoints
markedly increased the mTORC1 levels, leading to the inactivation
of the ULK complex, and the inhibition of the phosphorylation of
Beclin1 (ser14) in the peri-infarct cortex following MCAO/R injury,
suggesting that EA at the LI11 and ST36 acupoints protected against
the damaging effects of ischemic stroke through the inhibition of
autophagosome formation and autophagy; these effects were mediated
through the mTORC1-ULK complex-Beclin1 pathway.
However, it is still unknown whether increasing, or
inhibiting autophagy exerts a neuroprotective effect against
cerebral infarction and neurological deficits following ischemic
stroke (68). Although autophagy
has been examined using animal models of brain ischemia, the
majority of the studies have been inconclusive, or have produced
contradictory outcomes (69,70). This lack of consensus on the role
of autophagy in stroke-induced injury may largely be due to
methodological approaches through which autophagy has been either
'activated' or 'inhibited'. In addition, the activation or the
inhibition of autophagy is associated with the type of ischemic
models and the stage of ischemic stroke. The present study
suggested that the inhibition of autophagy in the peri-infarct
cortex contributes to the neuroprotective effects induced by EA
against focal cerebral ischemia.
In conclusion, our data suggested that EA at the
LI11 and ST36 acupoints improved motor dysfunction, and this was
accompanied by improved neurological deficit outcomes and decreased
infarct volumes following cerebral ischemia and reperfusion. EA at
the LI11 and ST36 acupoints protected against the damaning effects
of ischemic stroke through the inhibition of autophagosome
formation and autophagy, and these effects were mediated through
the mTORC1-ULK complex-Beclin1 pathway.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (nos. 81273835, 81373778 and
81403450).
References
|
1
|
Donnan GA, Fisher M, Macleod M and Davis
SM: Stroke. Lancet. 371:1612–1623. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nour M, Scalzo F and Liebeskind DS:
Ischemia-reperfusion injury in stroke. Interv Neurol. 1:185–199.
2013. View Article : Google Scholar
|
|
3
|
Zhao H, Sapolsky RM and Steinberg GK:
Interrupting reperfusion as a stroke therapy: ischemic
postconditioning reduces infarct size after focal ischemia in rats.
J Cereb Blood Flow Metab. 26:1114–1121. 2006.PubMed/NCBI
|
|
4
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion - from mechanism to translation. Nat Med. 17:1391–1401.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Glick D, Barth S and Macleod KF:
Autophagy: cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hotchkiss RS, Strasser A, McDunn JE and
Swanson PE: Cell death. N Engl J Med. 361:1570–1583. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martínez-Borra J and López-Larrea C:
Autophagy and self-defense. Adv Exp Med Biol. 738:169–184. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tan CC, Yu JT, Tan MS, Jiang T, Zhu XC and
Tan L: Autophagy in aging and neurodegenerative diseases:
implications for pathogenesis and therapy. Neurobiol Aging.
35:941–957. 2014. View Article : Google Scholar
|
|
9
|
Wen YD, Sheng R, Zhang LS, Han R, Zhang X,
Zhang XD, Han F, Fukunaga K and Qin ZH: Neuronal injury in rat
model of permanent focal cerebral ischemia is associated with
activation of autophagic and lysosomal pathways. Autophagy.
4:762–769. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi R, Weng J, Zhao L, Li XM, Gao TM and
Kong J: Excessive autophagy contributes to neuron death in cerebral
ischemia. CNS Neurosci Ther. 18:250–260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang JY, Xia Q, Chu KT, Pan J, Sun LN,
Zeng B, Zhu YJ, Wang Q, Wang K and Luo BY: Severe global cerebral
ischemia-induced programmed necrosis of hippocampal CA1 neurons in
rat is prevented by 3-methyladenine: a widely used inhibitor of
autophagy. J Neuropathol Exp Neurol. 70:314–322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nah J, Yuan J and Jung YK: Autophagy in
neurodegenerative diseases: from mechanism to therapeutic approach.
Mol Cells. 38:381–389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mochida K, Oikawa Y, Kimura Y, Kirisako H,
Hirano H, Ohsumi Y and Nakatogawa H: Receptor-mediated selective
autophagy degrades the endoplasmic reticulum and the nucleus.
Nature. 522:359–362. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tanida I: Autophagosome formation and
molecular mechanism of autophagy. Antioxid Redox Signal.
14:2201–2214. 2011. View Article : Google Scholar
|
|
15
|
Tanida I, Ueno T and Kominami E: LC3 and
Autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mizushima N, Yoshimori T and Ohsumi Y: The
role of Atg proteins in autophagosome formation. Annu Rev Cell Dev
Biol. 27:107–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dooley HC, Razi M, Polson HE, Girardin SE,
Wilson MI and Tooze SA: WIPI2 links LC3 conjugation with PI3P,
autophagosome formation, and pathogen clearance by recruiting
Atg12-5-16L1. Mol Cell. 55:238–252. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim YM, Jung CH, Seo M, Kim EK, Park JM,
Bae SS and Kim DH: mTORC1 phosphorylates UVRAG to negatively
regulate autophagosome and endosome maturation. Mol Cell.
57:207–218. 2015. View Article : Google Scholar
|
|
19
|
Hosokawa N, Hara T, Kaizuka T, Kishi C,
Takamura A, Miura Y, Iemura S, Natsume T, Takehana K and Yamada N:
Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200
complex required for autophagy. Mol Biol Cell. 20:1981–1991. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Szymańska P, Martin KR, MacKeigan JP,
Hlavacek WS and Lipniacki T: Computational analysis of an
autophagy/translation switch based on mutual inhibition of MTORC1
and ULK1. PLoS One. 10:e01165502015. View Article : Google Scholar
|
|
21
|
Denton D, Nicolson S and Kumar S: Cell
death by autophagy: facts and apparent artefacts. Cell Death
Differ. 19:87–95. 2012. View Article : Google Scholar :
|
|
22
|
Puyal J, Vaslin A, Mottier V and Clarke
PG: Postischemic treatment of neonatal cerebral ischemia should
target autophagy. Ann Neurol. 66:378–389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wei K, Wang P and Miao CY: A double-edged
sword with therapeutic potential: an updated role of autophagy in
ischemic cerebral injury. CNS Neurosci Ther. 18:879–886. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rami A and Kögel D: Apoptosis meets
autophagy-like cell death in the ischemic penumbra: two sides of
the same coin? Autophagy. 4:422–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tanida I: Autophagy basics. Microbiol
Immunol. 55:1–11. 2011. View Article : Google Scholar
|
|
26
|
Russell RC, Tian Y, Yuan H, Park HW, Chang
YY, Kim J, Kim H, Neufeld TP, Dillin A and Guan KL: ULK1 induces
autophagy by phosphorylating Beclin-1 and activating VPS34 lipid
kinase. Nat Cell Biol. 15:741–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nazarko VY and Zhong Q: ULK1 targets
Beclin-1 in autophagy. Nat Cell Biol. 15:727–728. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen A, Lin Z, Lan L, Xie G, Huang J, Lin
J, Peng J, Tao J and Chen L: Electroacupuncture at the Quchi and
Zusanli acupoints exerts neuroprotective role in cerebral
ischemia-reperfusion injured rats via activation of the PI3K/Akt
pathway. Int J Mol Med. 30:791–796. 2012.PubMed/NCBI
|
|
29
|
Xue X, You Y, Tao J, Ye X, Huang J, Yang
S, Lin Z, Hong Z, Peng J and Chen L: Electro-acupuncture at points
of Zusanli and Quchi exerts anti-apoptotic effect through the
modulation of PI3K/Akt signaling pathway. Neurosci Lett. 558:14–19.
2014. View Article : Google Scholar
|
|
30
|
Du F and Liu S: Electroacupuncture with
high frequency at acupoint ST-36 induces regeneration of lost
enteric neurons in diabetic rats via GDNF and PI3k/Akt signal
pathway. Am J Physiol Regul Integr Comp Physiol. 309:R109–R118.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu YT, Tan HL, Huang Q, Ong CN and Shen
HM: Activation of the PI3K-Akt-mTOR signaling pathway promotes
necrotic cell death via suppression of autophagy. Autophagy.
5:824–834. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Heras-Sandoval D, Pérez-Rojas JM,
Hernández-Damián J and Pedraza-Chaverri J: The role of
PI3K/AKT/mTOR pathway in the modulation of autophagy and the
clearance of protein aggregates in neurodegeneration. Cell Signal.
26:2694–2701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang J, Ye X, You Y, Liu W, Gao Y, Yang
S, Peng J, Hong Z, Tao J and Chen L: Electroacupuncture promotes
neural cell proliferation in vivo through activation of the ERK1/2
signaling pathway. Int J Mol Med. 33:1547–1553. 2014.PubMed/NCBI
|
|
35
|
Neumann M, Wang Y, Kim S, Hong SM, Jeng L,
Bilgen M and Liu J: Assessing gait impairment following
experimental traumatic brain injury in mice. J Neurosci Methods.
176:34–44. 2009. View Article : Google Scholar :
|
|
36
|
Encarnacion A, Horie N, Keren-Gill H,
Bliss TM, Steinberg GK and Shamloo M: Long-term behavioral
assessment of function in an experimental model for ischemic
stroke. J Neurosci Methods. 196:247–257. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Y, Yoshimura R, Manabe H, Schretter
C, Clarke R, Cai Y, Fitzgerald M and Lee KS: Trans-sodium
crocetinate improves outcomes in rodent models of occlusive and
hemorrhagic stroke. Brain Res. 1583:245–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu Y, Ao LJ, Lu G, Leong E, Liu Q, Wang
XH, Zhu XL, Sun TF, Fei Z, Jiu T, et al: Quantitative gait analysis
of long-term locomotion deficits in classical unilateral striatal
intracerebral hemorrhage rat model. Behav Brain Res. 257:166–177.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Koopmans GC, Deumens R, Honig WM, Hamers
FP, Steinbusch HW and Joosten EA: The assessment of locomotor
function in spinal cord injured rats: the importance of objective
analysis of coordination. J Neurotrauma. 22:214–225. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dupont N, Orhon I, Bauvy C and Codogno P:
Autophagy and autophagic flux in tumor cells. Methods Enzymol.
543:73–88. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mauvezin C, Ayala C, Braden CR, Kim J and
Neufeld TP: Assays to monitor autophagy in Drosophila. Methods.
68:134–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li YC, He SM, He ZX, Li M, Yang Y, Pang
JX, Zhang X, Chow K, Zhou Q and Duan W: Plumbagin induces apoptotic
and autophagic cell death through inhibition of the PI3K/Akt/mTOR
pathway in human non-small cell lung cancer cells. Cancer Lett.
344:239–259. 2014. View Article : Google Scholar
|
|
44
|
Li X, Luo P, Wang Q and Xiong L:
Electroacupuncture pretreatment as a novel avenue to protect brain
against ischemia and reperfusion injury. Evid Based Complement
Alternat Med. 2012:1953972012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu SY, Hsieh CL, Wei TS, Liu PT, Chang YJ
and Li TC: Acupuncture stimulation improves balance function in
stroke patients: a single-blinded controlled, randomized study. Am
J Chin Med. 37:483–494. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wayne PM, Krebs DE, Macklin EA, Schnyer R,
Kaptchuk TJ, Parker SW, Scarborough DM, McGibbon CA, Schaechter JD,
Stein J and Stason WB: Acupuncture for upper-extremity
rehabilitation in chronic stroke: a randomized sham-controlled
study. Arch Phys Med Rehabil. 86:2248–2255. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang GC, Fu WB, Xu NG, Liu JH, Zhu XP,
Liang ZH, Huang YF and Chen YF: Meta analysis of the curative
effect of acupuncture on post-stroke depression. J Tradit Chin Med.
32:6–11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lim SM, Yoo J, Lee E, Kim HJ, Shin S, Han
G and Ahn HS: Acupuncture for spasticity after stroke: a systematic
review and meta-analysis of randomized controlled trials. Evid
Based Complement Alternat Med. 2015:8703982015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yang HY, Liu TY, Wang YH, Ying SG, Zheng
CY, Kuai L and Gao M: Acupoint electrogymnastics therapy for
treatment of apoplectic hemiplegia: a multicenter randomized
control study. Zhongguo Zhen Jiu. 28:635–638. 2008.In Chinese.
PubMed/NCBI
|
|
50
|
Feng X, Yang S, Liu J, Huang J, Peng J,
Lin J, Tao J and Chen L: Electroacupuncture ameliorates cognitive
impairment through inhibition of NF-κB-mediated neuronal cell
apoptosis in cerebral ischemia-reperfusion injured rats. Mol Med
Rep. 7:1516–1522. 2013.PubMed/NCBI
|
|
51
|
Zhang TS, Yang L, Hu R, Qiao XL, Yang X
and Liu XG: Effect of electroacupuncture on the contents of
excitatory amino acids in cerebral tissue at different time courses
in rats with cerebral ischemia and reperfusion injury. Zhen Ci Yan
Jiu. 32:234–236. 2007.In Chinese. PubMed/NCBI
|
|
52
|
Vandeputte C, Taymans JM, Casteels C, Coun
F, Ni Y, Van Laere K and Baekelandt V: Automated quantitative gait
analysis in animal models of movement disorders. BMC Neurosci.
11:922010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lubjuhn J, Gastens A, von Wilpert G,
Bargiotas P, Herrmann O, Murikinati S, Rabie T, Marti HH, Amende I,
Hampton TG and Schwaninger M: Functional testing in a mouse stroke
model induced by occlusion of the distal middle cerebral artery. J
Neurosci Methods. 184:95–103. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang Y, Bontempi B, Hong SM, Mehta K,
Weinstein PR, Abrams GM and Liu J: A comprehensive analysis of gait
impairment after experimental stroke and the therapeutic effect of
environmental enrichment in rats. J Cereb Blood Flow Metab.
28:1936–1950. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xu M and Zhang HL: Death and survival of
neuronal and astrocytic cells in ischemic brain injury: a role of
autophagy. Acta Pharmacol Sin. 32:1089–1099. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xu F, Gu JH and Qin ZH: Neuronal autophagy
in cerebral ischemia. Neurosci Bull. 28:658–666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xing S, Zhang Y, Li J, Zhang J, Li Y, Dang
C, Li C, Fan Y, Yu J, Pei Z and Zeng J: Beclin 1 knockdown inhibits
autophagic activation and prevents the secondary neurodegenerative
damage in the ipsilateral thalamus following focal cerebral
infarction. Autophagy. 8:63–76. 2012. View Article : Google Scholar
|
|
58
|
Rami A, Langhagen A and Steiger S: Focal
cerebral ischemia induces upregulation of Beclin 1 and
autophagy-like cell death. Neurobiol Dis. 29:132–141. 2008.
View Article : Google Scholar
|
|
59
|
Levine B, Packer M and Codogno P:
Development of autophagy inducers in clinical medicine. J Clin
Invest. 125:14–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wu Z, Zou Z, Zou R, Zhou X and Cui S:
Electroacupuncture pretreatment induces tolerance against cerebral
ischemia/reperfusion injury through inhibition of the autophagy
pathway. Mol Med Rep. 11:4438–4446. 2015.PubMed/NCBI
|
|
61
|
Inoki K, Kim J and Guan KL: AMPK and mTOR
in cellular energy homeostasis and drug targets. Annu Rev Pharmacol
Toxicol. 52:381–400. 2012. View Article : Google Scholar
|
|
62
|
Leung EY, Askarian-Amiri M, Finlay GJ,
Rewcastle GW and Baguley BC: Potentiation of growth inhibitory
responses of the mTOR inhibitor everolimus by dual mTORC1/2
inhibitors in cultured breast cancer cell lines. PLoS One.
10:e01314002015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zha QB, Zhang XY, Lin QR, Xu LH, Zhao GX,
Pan H, Zhou D, Ouyang DY, Liu ZH and He XH: Cucurbitacin E induces
autophagy via downregulating mTORC1 signaling and upregulating AMPK
activity. PLoS One. 10:e01243552015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wirth M, Joachim J and Tooze SA:
Autophagosome formation - the role of ULK1 and Beclin1-PI3KC3
complexes in setting the stage. Semin Cancer Biol. 23:301–309.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Rami A: Upregulation of Beclin 1 in the
ischemic penumbra. Autophagy. 4:227–229. 2008. View Article : Google Scholar
|
|
66
|
Zheng YQ, Liu JX, Li XZ, Xu L and Xu YG:
RNA interference-mediated downregulation of Beclin1 attenuates
cerebral ischemic injury in rats. Acta Pharmacol Sin. 30:919–927.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L,
Su L and Zhang Y: Inhibition of autophagy contributes to ischemic
postconditioning-induced neuroprotection against focal cerebral
ischemia in rats. PLoS One. 7:e460922012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Buckley KM, Hess DL, Sazonova IY,
Periyasamy-Thandavan S, Barrett JR, Kirks R, Grace H, Kondrikova G,
Johnson MH, Hess DC, et al: Rapamycin up-regulation of autophagy
reduces infarct size and improves outcomes in both permanent MCAL,
and embolic MCAO, murine models of stroke. Exp Transl Stroke Med.
6:82014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zheng Y, Hou J, Liu J, Yao M, Li L, Zhang
B, Zhu H and Wang Z: Inhibition of autophagy contributes to
melatonin-mediated neuroprotection against transient focal cerebral
ischemia in rats. J Pharmacol Sci. 124:354–364. 2014. View Article : Google Scholar : PubMed/NCBI
|